

Synopsis
Liver cirrhosis is caused by iterative cycles of tissue injury, inflammation and repair. Although most causes of acute hepatitis resolve without scarring, chronic hepatitis is associated with persistent inflammation and matrix remodelling which leads to fibrosis and eventually cirrhosis. The mechanisms that govern wound healing are complex and involve interactions between both the innate and adaptive immune systems with stromal cells within a microenvironment comprised of cytokines, growth factors and modified matricellular proteins. As our understanding of the processes that govern inflammation and fibrosis improves it has become clear that the immune system plays a central role in the regulation of fibrosis, tissue repair and recovery that are vital for the maintenance of tissue homeostasis. Chronic inflammation and fibrosis are inextricably linked and the cellular interactions between immune effector cells, local fibroblasts and tissue macrophages at sites of scar formation determine the outcome of liver injury and the development of scarring.
Keywords: Liver, Inflammation, Stroma, Lymphocytes, Fibrosis, Hepatic Stellate Cell
Introduction
Far from being a static process chronic inflammation is a dynamic aggregate of lymphocytes, macrophages and stromal cells held together by autocrine and paracrine interactions. Consequently the role of leukocytes in wound healing must be considered within the context of these organ-specific stromal microenvironments. This is particularly the case in the liver where interactions between stromal cells and infiltrating leukocytes are critical in determining the outcome of liver injury as demonstrated by the observations that animals deficient in macrophages, T-cells or B-cells all show reduced fibrotic responses. The ability of leukocyte-derived cytokines to modulate the behaviour of liver fibroblasts is widely accepted but it has become clear that fibroblasts also contribute to the processes of leukocyte recruitment, positioning and survival at sites of chronic inflammation1. Neutrophil recruitment in the immediate/early response to liver injury is preceded by rapid upregulation of adhesion molecules on stellate cells within the sub-endothelial space whilst in animals recovering from liver fibrosis resolution of liver inflammation is prefaced by apoptosis of activated liver myofibroblasts2,3. Thus the induction, maintenance and resolution of liver inflammation and fibrosis is determined by interactions between infiltrating leucocytes and activated fibroblasts within the liver stroma4,5. This review will discuss the immunological mechanisms that drive liver fibrosis and in particular the complex interactions that exist between cellular components of the stroma (macrophages and fibroblasts particularly) and the innate and adaptive immune systems.
Stromal microenvironments in chronic inflammatory disease
A characteristic feature of chronic liver disease is the presence of a persistent inflammatory infiltrate associated with regions of fibrosis and matrix remodelling. Whilst macrophages play an important role in the initiation and resolution of fibrosis fibroblasts are the most numerous cells within the liver stroma and are primarily responsible for the synthesis of the extracellular matrix6. Fibroblasts provide an architectural framework for tissues and regulate important homeostatic and developmental functions7. Until recently resident stromal cells were not thought to make significant contributions to immune responses within the liver, but it has become clear that fibroblasts and macrophages within the liver stroma play critical roles in regulating the inflammatory as well as the fibrotic reaction at sites of tissue injury1,8,9. Disruption of these processes results in abnormal wound healing, scarring and perpetuation of inflammation10. This is particularly the case in human liver disease where reiterative cycles of tissue injury perpetuate stromal cell activation and provoke a switch from acute reversible inflammation to chronic inflammatory disease.
Monocytes demonstrate considerable plasticity and can differentiate into macrophages, dendritic cells (DC) and stromal cell lineages in response to microenvironmental activation signals11,12. In humans CD16+ monocytes expressing high levels of the fractalkine receptor CX3CR1 are recruited to tissues where they undergo further site specific differentiation into resident populations of cells such as Kupffer cells and interstitial DCs13 whereas CD16low cells are recruited during inflammation (Figure 1). Most ‘inflammatory’ monocytes differentiate into macrophages in tissue and mediate clearance of pathogens and the resolution of inflammation, but some CCR7+ cells can emigrate from tissues via lymphatics to the draining lymph nodes where they acquire DC function. In the absence of inflammation, CD16+CX3CR1high monocytes enter the liver to replenish kupffer cells and DC populations12. In addition a population of adherent CD14+ monocytes can be induced to differentiate along fibroblast lineages when cultured with T cells. The resulting cells known as fibrocytes, express both haematopoietic and mesenchymal markers (collagen-1+, CD11a/b, CD45+, CD34+). When recruited to inflamed tissues and exposed to cytokines such as TGFβ111,14 they can differentiate into αSMA+ collagen-secreting contractile cells with characteristics of myofibroblasts. Fibrocytes are implicated in wound healing and inflammation in a variety of diseases including chronic liver disease15,16 and exemplify the cellular plasticity and divergence exhibited within the stroma.
Figure 1.
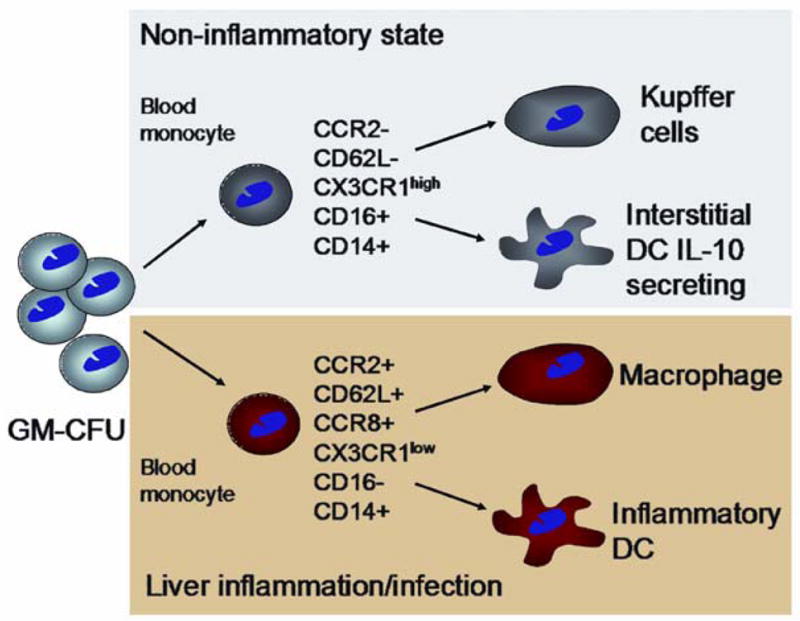
Based on work in other tissues the figure shows the two main monocyte subsets in blood and how they can give rise to functionally diverse subsets of kupffer cells, macrophages and myeloid DCs after recruitment into the liver. Two distinct subpopulations of monocytes can be identified in the human circulation which show differences in expression of chemokine receptors and adhesion molecules involved in recruitment through sinusoidal endothelium. Adapted from 12.
Transcriptional profiling of human fibroblasts from different anatomical sites has demonstrated patterns of gene expression as divergent as those seen amongst the different leukocyte lineages17,18 and this biological diversity is reflected in differential patterns of growth factor and matrix expression19. The liver contains several phenotypically distinct fibroblasts20,21 which may be derived from distinct progenitor cells22. The intrinsic variation in phenotype and function of macrophages and fibroblasts within tissues may be an important factor that predisposes tissues to scarring, and could explain why relapses in chronic inflammatory disease are often tissue and site-specific.
Macrophages regulate the induction and resolution of liver fibrosis
Many of the cells that play an important role in the immediate innate immune response, including neutrophils and mast cells, make little contribution to tissue fibrosis and NK-cells actually suppress fibrosis by killing activated myofibroblasts possibly by TRAIL dependent mechanisms23–25. This may explain why many acute inflammatory reactions in the liver resolve without scarring26,27. On the other hand macrophages are critical for fibrosis as demonstrated by depletion studies in animals which implicate hepatic macrophages in both the progression and resolution of fibrosis. Activation of fibroblasts by macrophage–derived TGFβ1 or insulin-like growth factor is an early feature of fibrogenesis which promotes a switch in fibroblast gene expression to initiate matrix remodelling28 and macrophage-derived cytokines including IL-6 and TGFb maintain the proliferation and differentiation of liver fibroblasts29,30.
The divergent roles played by macrophages within the liver are a consequence of distinct pathways of activation and differentiation determined by local cytokines and interactions with other liver cells31. Pathogen-associated molecular patterns (PAMP-s) are conserved sequences in microbes that stimulate TLRs and other pattern recognition receptors on macrophages and thereby activate innate immune responses. Classical activation resulting in so-called M1 macrophages is driven by IFN-γ and characterised by a signature of proinflammatory genes associated with Th1 lymphocyte responses and cell-mediated immunity. An alternative activation profile results in M2 macrophages associated with a Th2-type polarised response characterised by STAT-6 signalling, IL-4 and IL-13 secretion and upregulation of endocytic lectin receptors including the mannose receptor. Alternative activation is characteristic of parasitic infections, allergy, humoral immunity and fibrosis. The deactivation of macrophages is also important to promote resolution and this can be triggered by exposure to apoptotic cells, IL-10 and other regulatory mediators and glucocorticoids. Although there is a clear distinction in vitro between pro-inflammatory matri-lytic M1 and profibrotic M2 macrophages32 the situation in-vivo is more complex with the presence of both classical and alternatively differentiated macrophages in chronically inflamed liver tissue33.
The importance of macrophages in liver fibrosis has been demonstrated by animal studies where macrophages were selectively depleted at different stages of fibrosis. Macrophage depletion at the induction of fibrosis, either by inhibiting CCL2-dependent recruitment or selective depletion of tissue macrophage populations with gadolinium chloride or diphtheria toxin, reduced scarring and resulted in fewer activated (αSMA+) HSC4,34,35. By contrast depleting macrophages during the immediate/early phase of tissue recovery delayed matrix degradation and prolonged fibrosis. These divergent roles suggest that either different populations of macrophages are involved at each stage or more likely that macrophage function switches during fibrogenesis in response to cytokines in the microenvironment.
Whether macrophages revert to a quiescent phenotype following tissue recovery or undergo unidirectional activation followed by apopotic death is unknown. The IkappaB kinase (IKK) complex which controls the activation of NF-kappaB transcription factors plays a pivotal role in switching off macrophage activation and the resolution of inflammation36,37 and in vitro macrophage deactivation can be induced by a variety of signals including IL-10 and TGFβ, or ligation of the inhibitory receptors CD172 and CD200 which lead to anti-inflammatory cytokine production and a reduction in MHC class II expression12.
Cross-talk between macrophages and liver fibroblasts leads to changes in function and in the patterns of cytokines secreted by both cell types38,39. Survival of activated HSC in the fibrotic liver is dependent on expression of TIMPs (which prevent degradation of matrix proteins) and survival signals from accumulated fibrillar Type 1 and 3 collagens. The failure to degrade collagen-1 critically impairs HSC apoptosis thereby perpetuating fibrosis40. Recovery from liver fibrosis is associated with the secretion of macrophage-derived proteases that degrade cross-linked fibrillar collagens allowing other more promiscuous MMPs to accelerate matrilysis and thereby to remove the survival signals on which activated HSC depend 4,41. In addition macrophages may actively promote apoptosis of HSC by expressing TRAIL and other apoptotic stimuli that accelerate clearance of myofibroblasts at sites of scar formation42.
Thus the resident populations of macrophages within the liver direct different stromal responses to injury at different times but whether this reflects functionally distinct populations of cells derived from distinct blood monocyte precursors or is the result of a local intrahepatic switch in function is unknown.
Stromal cells modulate the differentiation and function of antigen presenting cells
We have previously reported that non-inflamed liver contains a subset of myeloid DCs which preferentially secrete IL-10 and induce IL-10 secretion in responding T cells43,44. These DCs may play an important role in suppressing damaging immune responses in non-inflammatory states. In normal human liver most DCs are CD16+ and express high levels of CX3CL1. CD16+ circulating monocytes can differentiate into DCs during the process of transendothelial migration45 and this process may be enhanced by interactions with fibroblasts 46. The interaction between stroma and leucocytes in human tissues has received relatively little attention although there is accumulating evidence to suggest that HSC and extracellular matrix provide differentiation signals to monocytes during recruitment into the liver. Human spleen-derived myofibroblasts express IL-15 that drives CD34+ blood cells to differentiate into activated natural killer (NK) cells and myeloid DCs after 3–4 weeks in culture demonstrating how stromal cells in tissues can shape the innate immune system47. Such interactions will also determine the nature of the DCs generated. Murine splenic DCs can be generated from haematopoetic precursors by culture on splenic stromal cells46 and these DCs have regulatory properties similar to those seen in human liver44,48. Maturation was dependent on fibronectin-mediated adhesion and CXCL12 secreted by the fibroblasts. The DCs generated on the stromal cells mediated suppression of T cell proliferation in response to antigen presentation by mature DCs. These two studies show how different fibroblast-derived cytokines, IL-15 in the former study and CXCL12 in the latter may drive different differentiation pathways of interacting DCs and thereby shape the subsequent immune response.
Similar processes have been described in the skin where interactions between blood monocytes and human dermal fibroblasts mediated by β2 integrins, ICAM-1 and Thy-1 (CD90) induce the maturation of DC with potent T cell activating capabilities49. The importance of the local microenvironment is emphasised by our finding that culturing human monocyte-derived DCs in liver-conditioned media induced a regulatory phenotype similar to that seen in DCs isolated directly from the liver whereas culturing in skin-conditioned media had no such effect44. IL-10 is the critical cytokine mediating this effect as defective hepatic DC function could be restored by inhibiting IL-10 but the precise role played by HSC in modulating these responses has not been determined50. Thus the outcome of interactions between DC precursors and fibroblasts are likely to be tissue-specific responses modulated by local inflammatory signals that regulate DC differentiation and function.
Recent studies have demonstrated that HSC can themselves act as professional antigen-presenting cells (APC) by efficiently presenting antigens to MHC-I and MHC-II-restricted T cells and lipid antigens to CD1-restricted natural killer (NK) T cells. HSC share many phenotypic similarities with follicular dendritic cells, a specialised antigen presenting cell located in secondary lymphoid tissue51. Unlike other resident liver APCs, notably sinusoidal endothelium and hepatocytes, the outcome of presentation by HSC appears to be full effector responses rather than tolerance. Thus HSC not only modulate the differentiation of other APC but can also directly stimulate immune responses. As immune responses develop and HSC differentiate into activated myofibroblasts their role may shift from direct antigen presentation to indirect effects on monocyte differentiation resulting in complex effects on intrahepatic immune responses52. Gene array studies have shown that as HSC mature they switch gene transcription from a fibrotic to a pro-inflammatory phenotype providing evidence that senescent HSC continue to modulate local inflammatory responses long after they have stopped remodelling the ECM in response to liver injury53.
HSC activation in the liver promotes chronic inflammation
Liver myofibroblasts contribute to leukocyte recruitment to the injured liver by secreting cytokines, chemokines and other chemoattractants such as platelet activating factor and complement proteins54–56. In most circumstances inflammation resolves once the initiating stimulus has been removed but in some conditions inflammation persists resulting in chronic hepatitis and fibrogenesis57. The switch from acute resolving to chronic persistent hepatitis is associated with the development of a specialised microenvironmental niche that maintains leukocytes, particularly lymphocytes in close proximity with scar-associated fibroblasts at sites of tissue injury5. Chronic inflammation in other tissues including the kidney, lung and eye is also associated with the presence of closely associated lymphocytes and activated fibroblasts indicating that these links are a generic feature of fibrotic diseases1,58–60.
To date the role of lymphocyte-fibroblast interactions in driving chronic inflammation has been most closely studied in rheumatoid arthritis but it is likely that general mechanisms defined in the joint will apply to chronic inflammation in the liver. Fibroblasts in rheumatoid joints shape the inflammatory infiltrate in several ways61. Fibroblast-derived chemokines can be presented on the endothelium of vessels in the joint thereby contributing to inflammatory cell recruitment form blood. In addition the local secretion of CXCL12 activates integrin-mediated adhesion of lymphocytes to fibroblast VCAM-1 thereby retaining lymphocytes in the joint62,63. Other cytokines secreted by fibroblasts, particularly type-1 interferons provide survival signals for activated T cells and prevent them dying by apoptosis64. Thus fibroblasts contribute to the persistence of chronic joint inflammation by promoting lymphocyte recruitment from blood, retaining infiltrating lymphocytes and by providing them with survival signals that prevent death by apoptosis, a critical component of immune resolution1.
The interactions between fibroblasts and lymphocytes are bidirectional and T cell-derived cytokines are potent activators of fibroblasts in many chronic inflammatory diseases65–67. The outcome of fibroblast activation will be determined by the activation of both disease-specific and tissue-specific signalling pathways and in humans polymorphisms in regulatory genes can result in major differences in response to particular stimuli68. Thus LPS-mediated NFκB activation and CXCL8 secretion by liver myofibroblasts vary between subjects with liver disease69,70. Failure to regulate activation of NFkB leads to sustained inflammation and a tendency towards a Th1 polarised immune response36,37 which may in part be driven by uncontrolled fibroblast activation. Thus mice with a selective deletion of RelB (a transcriptional regulator that stabilises the NFκB inhibitor IκB) in fibroblasts develop an overwhelming systemic inflammatory response to endotoxin which is characterised by persistent chemokine induction and leukocyte recruitment. Transfecting the wild type gene back into these animals attenuates inflammation by restoring Rel-B mediated stromal regulation71. More recently it has been shown that mice lacking NFkB1 (p50) treated with repeated injections of carbon tetrachloride develop a more severe inflammatory and fibrotic reaction than wild type controls. This is associated with increased stellate cell expression of TNF-α in areas of inflammation, suggesting that the p50 subunit of NF-kB may protect the injured liver by limiting TNF-α dependent inflammatory cell recruitment72.
Thus iterative cycles of fibroblast activation at sites of chronic injury result in the development of a stromal phenotype that perpetuates inflammation and leukocyte recruitment resulting in dysregulation of the normal processes of tissue repair10.
HSC activation is associated with leukocyte recruitment
Quiescent HSC are specialised pericytes found immediately adjacent to the sinusoids in the space of Disse. They maintain endothelial integrity by secreting angiogenic peptides secreted in response to bi-directional interactions with the overlying endothelium73. The expression of markers associated with stem cells such as CD133, Thy-1 (CD90), nanog and OCT-4 and their ability to trans-differentiate into hepatocytes and endothelial-like cells suggest that HSC should also be regarded as progenitor cells73,74. Quiescent HSC undergo a phenotypic switch in response to liver injury, becoming spindle-shaped and secreting pro-inflammatory cytokines and upregulating programmes of genes that result in excess deposition of fibrillar collagens6,75. Whilst there is increasing evidence that human liver fibrosis is the product of both intra and extra-hepatic populations of fibroblasts, activated hepatic HSC are still regarded as the principle collagen producing cells in the fibrotic liver76.
In liver fibrosis HSC-like cells are seen immediately adjacent to fibro-vascular membranes at the margins of fibrotic septa associated with the inflammatory infiltrate77–79. Thus while paracrine interactions with quiescent HSC maintain the overlying endothelium HSC activation results in fibrogenesis associated with an inflammatory infiltrate. HSC activation is precipitated in a variety of ways; reactive oxygen intermediates released from injured cells activate quiescent HSC whilst activation signals are generated within the stroma as latent TGFβ1 is activated via plasmin released by injured endothelial cells6,80. Perpetuation of HSC activation is associated with increased sensitivity to local cytokine signals (particularly PDGF) and the effects of remodelling the existing ECM81. These enhanced cytokine responses typify activated HSC and are principally mediated by upregulation of receptor tyrosine kinases (RTKs) 80,82.
Migration and positioning of T-cells is determined by patterns of cytokine and chemokine secretion within the stroma
The liver responds to injury by recruiting leukocytes from the circulation, expanding populations of scar-forming fibroblasts and positioning the inflammatory infiltrate within the modified neo-matrix. An effective hepatic immune response requires that leukocytes are recruited to the liver and then appropriately positioned at sites of injury57. Leukocyte adhesion to endothelial cells is a prerequisite for recruitment into tissue and is regulated by a sequence of interactions in which the leukocyte is captured from the flowing blood by carbohydrate-dependent mechanisms; activated by chemokines presented on the endothelial glycocalyx resulting in integrin-activation and arrest on the vessel wall allowing the cell to then migrate through endothelium into tissue by as yet poorly understood mechanisms. To trigger leukocyte recruitment chemokines must be presented at the endothelium where they can be delivered by transcytosis from underlying cells and then presented bound to proteoglycans in the endothelial glycocalyx83. Thus the chemokines present on sinusoidal endothelium during liver inflammation may be derived from paracrine secretion by kupffer cells, other infiltrating leukocytes, hepatocytes and activated HSC. The situation of the latter immediately beneath the endothelium in close association with endothelium places them in an ideal site to “post” chemokines onto the overlying endothelium.
As a result of the complex microvascular anatomy of the liver, leukocytes can enter the liver via endothelial cells at three anatomical sites, in the portal tract, the sinusoids or the terminal hepatic veins84. Although some branches of the hepatic artery and portal veins terminate in post-capillary venules, most blood is diverted to the liver parenchyma via the sinusoids before returning to the systemic circulation via hepatic veins and the sinusoidal bed provides the major route of entry into the parenchyma. Recent studies show that recirculating lymphocytes enter the liver via sinusoids and, if not retained, then migrate via the space of Disse to portal tracts before moving on to draining lymph nodes. Similarly dendritic cells, which enter via the sinusoids as monocyte precursors, take the same route via portal tracts to draining lymph nodes after activation85. The lumen of the sinusoids is narrow and blood flow is regulated in part by stellate cells acting as pericytes which facilitates the attachment of flowing leukocytes86–88. Leukocytes interact with sinusoids in a modified version of the adhesion cascade in which the capture phase is attenuated and does not involve selectins which capture leukocytes in other tissues89,90. A different adhesion molecule, vascular adhesion protein 1 (VAP-1) which is constitutively expressed on hepatic endothelium mediates capture and transmigration of leukocytes on hepatic endothelium91,92 Once captured and firmly adherent a lymphocyte must breach the endothelial barrier to enter tissue and under physiological shear stress VAP-1 and ICAM-1 promote lymphocyte transendothelial migration through sinsuoidal endothelium92. Selection at the endothelial level is only one aspect of the processes that govern the development of an inflammatory infiltrate cells and retention of leukocytes within tissue is equally important although much less studied. Based on studies in other organs, particularly the thymus and bone marrow it is very likely that stromal cells will be integral in this process10,93.
HSC accumulate at sites of tissue injury in response to PDGF and CCL294 and once activated secrete proinflammatory cytokines and chemokines that upregulate adhesion molecules such as ICAM-1, VCAM-1 and CXC3CR1 which facilitate interactions with local leukocytes3,5,49,95. HSC can modulate leukocyte recruitment in at least three ways 1) by paracrine interactions which prime overlying sinusoidal endothelial cells to recruit leukocytes 2) by secreting matrix which provides a substrate through which infiltrating leukocytes can migrate or be retained 3) by direct interactions with leukocytes to modulate their migratory behaviour or retain them at sites of injury.
Leukocytes entering the sub-endothelial space interact with stromal cells and extracellular matrix via cell surface integrins which not only provide anchorage but also mediate ‘outside-in’ signals that direct cellular activation and differentiation10. Injury to the liver stimulates vigorous infiltration of leukocytes as a result of cytokines and chemokines displayed on the endothelium and within the subendothelial stroma96. Once in the sub-endothelial space lymphocytes undergo directed migration within the stroma by responding to signals displayed within the ECM. The distribution and spatial organisation of the hepatic infiltrate varies according to the inflammatory stimulus and reflects the expression of chemokines and adhesion molecules within the tissue57. Thus in inflamed liver adhesion molecules and chemokines are increased and in the sinusoids both endothelial cells and activated HSC secrete the potent lymphocyte recruiting chemokines CXCL9 and CXCL10 in response to TNFα and IFNγ97.
Simply recruiting inflammatory cells is not enough to maintain an effective immune response because chronic inflammation requires that effector cells are retained in tissues. Adhesion molecules and chemokines including CXCL16 and Fractalkine (CX3CR1) are expressed by target epithelial cells and stromal cells at sites of injury where they localise infiltrating T cells98–101. Thus stromal expression of chemokines determines the position and shape of the resulting inflammatory infiltrate, and serves to localise leukocytes to areas of fibrosis.
Activated HSC modulate recruitment and positioning of leukocytes within the liver
By being situated between the endothelium and parenchyma HSC can participate in the recruitment of leukocytes into the liver. The first evidence that activated liver myofibroblasts contributed to liver inflammation came from the observation that CCL2 in HSC conditioned media promoted vigorous monocyte chemotaxis54. CCL2 also recruits memory T cells, basophils and DCs suggesting it may be an important recruitment signal involved in chronic inflammation102,103. Studies in rat models of liver fibrosis revealed that nearly all of the CCL2 generated came from activated HSC104. Moreover activated HSC isolated from fibrotic human livers continue to secrete of very high concentrations of CCL2 in-vitro even in the absence of pro-inflammatory cytokines. Activated myofibroblasts secrete a wide variety of other chemokines, cytokines and matricellular proteins that regulate recruitment and subsequent differentiation of neutrophils, macrophages and lymphocytes (Figure 2). Our own unpublished data show that human hepatic myofibroblasts secrete not only CXCL8 and CCL2 but also CCL5, CCL11 and CCL3 and in response to INFγ and TNFα large amounts of CXCL9 and CXCL10 (A. Holt et al submitted). These factors in cell supernatants from liver fibroblasts exert a powerful chemotactic effect on human lymphocytes in real time chemotaxis studies (Figure 3) increasing both the rate and magnitude of lymphocyte migration and demonstrating the potential of liver fibroblasts to direct lymphocyte migration within the stroma.
Figure 2. Activated hepatic stellate cells control many aspects of tissue inflammation.
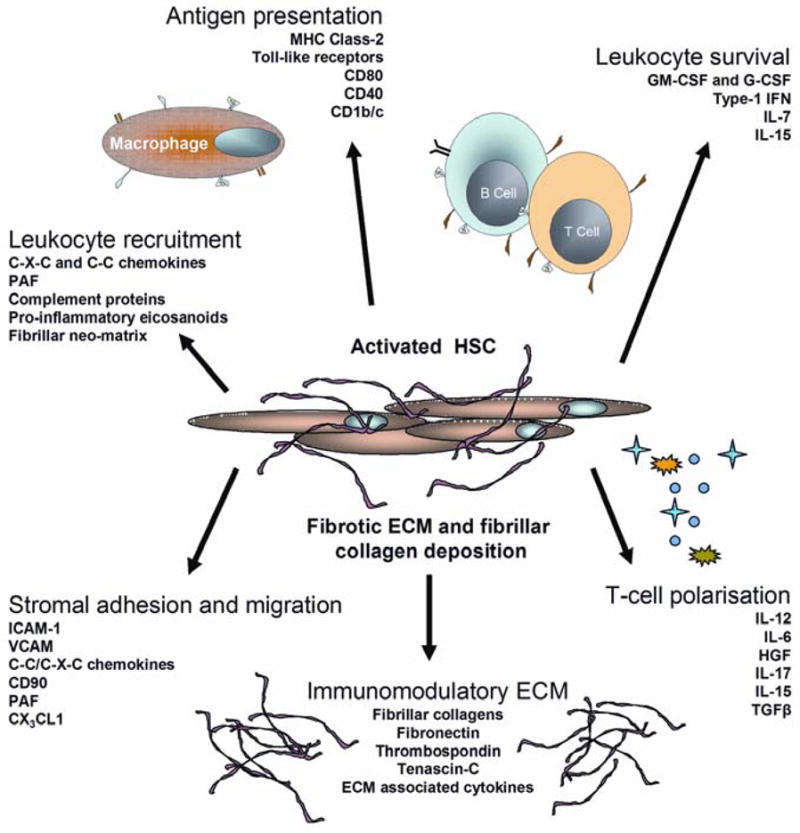
Following activation HSC undergo a phenotypic switch adopting a myofibroblast morphology and upregulating pro-inflammatory cytokines that regulate stromal immune responses as well as secreting fibrillar collagens and neo-matricellular proteins that generate tissue scarring. See text for further discussion.
Figure 3. Real-time lymphocyte chemotaxis to liver fibroblast conditioned media.

Directed real-time migration of fluorescently labelled human lymphocytes towards liver fibroblast conditioned media147. Conditioned supernatant taken from human liver fibroblasts isolated from diseased livers contains soluble factors that promote rapid lymphocyte chemotaxis even in the absence of proinflammatory cytokine stimulation. Data represents mean ± SEM 3 replicate experiments.
The importance of lymphocytes in fibrosis is illustrated by the fact that animals with targeted disruptions to chemokines that promote lymphocyte recruitment show reduced fibrotic responses to experimentally induced liver injury, whereas inhibition of neutrophil chemotaxis has no effect on fibrosis27,35. Cytokine therapy with IL-10 antagonises pro-inflammatory chemokines and cytokines secreted by activated liver fibroblasts and reduces fibrosis in humans with chronic viral hepatitis and animals with experimentally induced hepatitis105,106. Fibroblasts secrete other cytokines that can promote lymphocyte migration in addition to chemokines. IL-6, HGF and TGFβ, all of which are secreted by αSMA+ liver myofibroblasts107 have lymphocyte chemotactic activity and could contribute to the recruitment and positioning of lymphocytes within the stroma108–111.
Chronic inflammation is associated with the accumulation of T cell, B cells, and DCs in ectopic or tertiary lymphoid structures that resemble lymph nodes112. Their development is a consequence of aberrant and sustained expression of cytokines that drive lymph node development including lymphotoxin and homeostatic chemokines. CCL21 is an important chemokine in lymph node development and function because it is critical for the recruitment of naïve T cells and dendritic cells to lymph nodes113. CCL21 is secreted by liver fibroblasts including those found associated with tertiary lymphoid structures in chronic inflammatory diseases including PBC, PSC and chronic hepatitis C99,114,115. These findings suggest that activated fibroblasts may be responsible for the development and persistence of lymphoid follicles providing another mechanism by which they can modulate the recruitment of lymphocytes and determine the nature of chronic hepatic inflammation.
Thus activated HSC and liver fibroblasts at sites of tissue injury contribute to leukocyte recruitment by secreting chemokines and non-chemokine chemotactic factors that drive Gi-associated and Gi-independent mechanisms respectively. These operate throughout the inflammatory response, playing a role in early leukocyte recruitment and then in the remodelling of the stromal microenvironment to promote the recruitment and survival of T cells, B cells and dendritic cells as part of the chronic inflammation that accompanies fibrogenesis. Because fibroblasts are not all the same, site specific differences between stromal cells may play an important role in determining distinct patterns of inflammation seen in particular diseases or tissues17,19.
Direct interactions with activated liver fibroblasts retain lymphocytes in tissue and prevent resolution of inflammation
The close temporal and spatial association between infiltrating lymphocytes and fibrosis has been recognised for many years, but the cellular interactions are poorly understood. In other tissues T-cells and B-cells rely on cellular elements within the stroma for structural anchorage and directed migration. Thus within secondary lymph nodes fibroblastic reticular cells provide the adhesive scaffold that facilitates lymphocyte migration within the lymph node and which maintains the zonal separation of T cells and B-cells116. These stromal cells bring together T-cells and dendritic cells by secreting chemokines that attract both cell types within the T zone in the medulla117,118. Similarly stromal cells play a critical role in recruiting and positioning thymocytes within the thymus during lymphocyte development. It is very likely that liver fibroblasts play a similar role in directing lymphocyte recruitment to the inflamed liver. In human liver disease infiltrating lymphocytes are closely associated with myofibroblasts at the margins of fibrotic septa and with αSMA+ myofibroblasts within portal tracts. In mice with experimentally induced liver fibrosis lymphocytes are only found in the proximity of activated HSC/aLMF suggesting that there is a direct interaction between liver myofibroblasts and infiltrating lymphocytes5. Similarly, NK cells are found attached to αSMA+ HSC following fibrosis induction along the interface zone of fibrotic septa119 suggesting that paracrine interactions between fibroblasts and lymphocytes at sites of tissue damage facilitate the matricellular changes associated with fibrosis and chronic inflammation.
Studies in animals indicate that the release of CCL2 and expression of ICAM-1 by stellate cells immediately precedes leukocyte infiltration in the early phase of liver injury, indicating that HSC activation promotes the recruitment of leukocytes into the parenchyma120. Part of this response is mediated by the secretion of cytokines and chemokines that promote recruitment through the overlying endothelium but recent evidence suggests that fibroblasts also modulate lymphocyte migration by direct contacts with the infiltrating cells. We have shown that if lymphocytes are placed in contact with myofibroblasts isolated from diseased human liver they show increased motility and migrate rapidly over and through the fibroblast monolayer in a process termed pseudoemperopolesis. Treatment of the fibroblasts with pro-inflammatory cytokines increases the kinetics of stromal transmigration which is dependent on increased expression of ICAM-1 and VCAM-1 95. Murine studies have demonstrated that HSC support lymphocyte adhesion in an ICAM-1 dependent manner and human HSC also express high levels of CD90 which supports adhesive interactions with dendritic cells49,95,121. Thus in response to tissue injury liver myofibroblasts provide an adhesive template that facilitates stromal migration and approximates lymphocytes and fibrotic HSC-like cells in regions of matrix remodelling thus perpetuating the inflammatory reaction by promoting leukocyte recruitment at sites of tissue damage.
The development of a stable chronic inflammatory infiltrate depends upon the recruitment of leukocytes from blood and their subsequent survival at the site of inflammation outweighing the mechanisms of resolution which include local death of inflammatory cells by apoptosis and emigration out of the inflammatory site by lymphatics1. Stromal cells modulate all of these processes including the survival of the inflammatory infiltrate as interactions with synovial fibroblasts induce expression of anti-apoptotic genes in T cells that prevent apoptosis. Fibroblasts can also keep neutrophils alive particularly by secreting cytokines including GM-CSF as well as type-1 interferons. These survival signals are amplified by adhesive interactions between neutrophil α9β1 integrin and VCAM-1 on stromal cells63,122–124. The cycles of tissue injury and repair associated with chronic liver disease leads to local secretion of cytokines including IL-6, IL-12 and TNFα by activated HSC, all of which can promote antigen-independent bystander T cell activation. Thus the cytokine milieu in the liver of patients with chronic hepatitis is shaped by activated HSC and favours not only lymphocyte survival but also bystander activation of T cells - promoting effector T-cell function even in the absence of the original stimulating antigen125,126. This amplifies antigen-specific responses within the tissue, but also increases the resistance of responder cells to apoptotic stimuli127. As a result infiltrating T cells become increasingly resistant to apoptosis whilst bystander activation increases their effector function thereby exacerbating tissue damage and fibrosis128.
Stromal interactions with infiltrating lymphocytes shape fibrogenesis in chronic hepatitis
Whilst interactions between macrophages and fibroblasts are critical for fibrosis induction and resolution129 the role of the adaptive immune system is less well defined (Figure 4). Nevertheless there is good evidence that T-cells and B-cells are implicated in sustaining fibrotic responses in chronic liver disease130. In animal models and diseased human liver lymphocytes are seen in close association with fibroblasts in inflamed portal tracts and fibrous septa suggesting the involvement of the adaptive immune response in fibrogenesis5,96. Functional support for this hypothesis comes from animal studies in which depletion of T-cells and B-cells protects animals from liver fibrosis105,131. Moreover mice deficient in both B-cells and T-cells (RAG2−/−) are resistant to fibrosis in acute and chronic models of liver injury131. The specific role of particular lymphocyte subsets during fibrogenesis is less clear.
Figure 4.
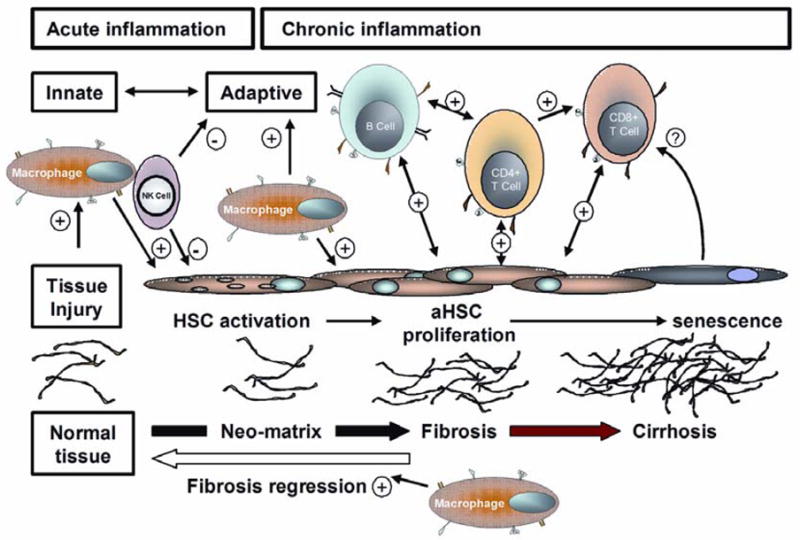
Liver fibrosis is the product of interactions between stromal cells and the innate and adaptive immune systems. For further discussion see text.
The contribution of intra-hepatic T-cell to fibrogenesis is accepted and adoptive transfer studies in a variety of murine models confirm the involvement of both CD4+ and CD8+ T-cells. CD4+ T-cells were originally identified as the source of a secreted ‘fibrotic factor’ in a TH2-polarised mouse model of liver fibrosis which was subsequently shown to be IL-13132,133. The polarity of the inflammatory response is critical for fibrogenesis, and different programmes of gene expression are induced when chronic inflammatory responses are dominated by TH1 or TH2 cytokines. Although TH1 cytokines characteristically generate an intense cellular response they cause little fibrosis134, whereas TH2 cytokines increase transcription of several genes involved in fibrogenesis including pro-collagen I and III, MMP2, MMP9 and TIMPs30, all of which are expressed in CCl4 liver injury suggesting that mechanisms of fibrosis may share a common cytokine profile. The importance of TH2 polarised T-cell subsets is confirmed by studies demonstrating that IL-13Rα2 blocking antibodies reduce liver fibrosis132 and inhibition of TH2 responses using targeted mutations of IL-4, STAT6, IL-4Rα attenuate fibrosis in a mouse model of scleroderma32. Other studies have demonstrated that transfer of CD8+ (but not CD4+) T-cells from mice with CCl4 induced liver fibrosis provokes significant liver injury in SCID-mice recipients105. Conversely fibrosis is attenuated in transgenic mice that over-express IL-10 in hepatocytes and in animals depleted of T-lymphocytes. Although CD4+ T-cell and CD8+ make important contributions to fibrogenesis, their precise role is unclear. CD4 T-cells can modulate the local cytokine response to favour fibrogenesis via direct effects on stromal macrophages and fibroblasts and CD8 T-cells may promote fibrosis by amplifying tissue injury as part of a bystander response driven by the local cytokine milieu.
The role of B-lymphocytes in hepatic fibrosis is poorly understood although their ability to activate T-cells at low antigen loads and to secrete cytokines suggests they could be involved in shaping and maintaining the inflammatory response. Regulatory B-cells can direct the polarity of the local inflammatory response away from a Th1-like phenotype in autoimmunity and may contribute to local production of TGFβ1135,136. A recent study demonstrated an antibody-independent role for B-cells in liver fibrosis induced by two different agents CCl4 or α-Naphthylisothiocyanate (ANIT). Surprisingly this study was unable to show any protective effect in CD4, CD8 or γδ T-cell deplete mice but found that mice lacking both T cells and B-cells were resistant to experimental fibrosis despite mounting a similar inflammatory response to wild type animals131. A role for antibody was excluded because mice that were unable to secrete immunoglobulin still showed reduced fibrosis when B cells were depleted131. The fact that T cell deficient mice showed normal fibrotic responses in this model argues against a role for B cells in antigen presentation. Therefore, it is most likely that B cells promote fibrosis by secreting cytokines or by contact-dependent interactions with other cells that favour a profibrotic microenvironment. Differentiation of naïve B-cells into Th1-like or Th2-like patterns of cytokine secretion is determined by similar cytokine microenvironments to those that regulate corresponding T-cell differentiation, and B-cells secrete the pro-fibrotic cytokines IL-4, IL-6, and IL-13 at levels similar to CD4+ T-cells137. Whilst BCR ligation is required for optimal cytokine secretion, repetitive BCR-independent stimulation is sufficient to stimulate naive B-cells to secrete cytokines in a polarised fashion138,139. Thus the repeated cycles of tissue injury seen in human liver disease could maintain B-cell activation and allow the process to become self-perpetuating. Interactions between NKT cells, macrophages and B-cells within this fibrotic environment will also reinforce local cellular responses and secretion of TGFβ140. Co-stimulatory signals provided by B cells to T-cells, NKTs and Kupffer cells may also be important.
CD40 is a strong candidate for a central role in these paracrine interactions. The ligand for CD40, CD154, is strongly expressed on lymphocytes and macrophages at sites of liver injury in several diseases31,141 and could activate B-cell CD40 resulting in cell survival, IL-6 secretion and paracrine activation of HSC proliferation and collagen synthesis. Dysregulated bystander activation of B-cells is usually prevented by activation of Fas-dependent apoptosis but survival signals such as BAFF/BLyS that are increased in the fibrotic microenvironment may inhibit Fas-mediated apoptosis allowing inappropriate survival of effector B-cells and amplification of fibrosis1. However other animal studies have failed to show a major role for B cells in liver fibrosis. B cell depletion in a TH2 dominated model of parasitic liver fibrosis did not provide any protection against fibrosis. This might be because the strongly polarised Th2 response in this model was unaffected by B cells. The example illustrates that different cellular mechanisms may be involved depending on the nature of liver injury and the pattern of inflammation142.
Recent interest has focussed the role of NK T cells in fibrogenesis. Invariant NKT cells are a subset of lymphocytes that recognize endogenous lipid ligands presented by CD1d. They regulate host responses to cell injury, tissue damage and viral infection and are potent secretors of cytokines. NKT cells are found in normal liver tissues but their numbers increase with necroinflammatory activity and in the context of chronic viral hepatitis they have been shown to secrete type 2 profibrotic cytokines including IL-4 and IL-13 which could drive progression of fibrosis. Part of this may be driven by increased expression of CD1d on APCs in chronically inflamed liver tissue 143. These studies are supported by animal experiments in which administration of the lipid β-glucosylceramide results in reduced intrahepatic NKT-cells associated with amelioration of fibrosis. 144. However there is likely to be further complexity because in other models IFNγ secreting NKT cells have an anti-fibrotic role145. A recent study in the double negative TGFβRII mouse model of primary biliary cirrhosis shows a marked increase of NKT cells within the liver associated with tissue injury. When the same mice were crossed onto a CD1−/− background, in which there are no CD1d-restricted NKT cells, liver inflammation and injury was markedly decreased suggesting NKT cells are involved in liver injury in this model. Furthermore they showed age-dependent differences in interferon-γ secretion by hepatic CD1d-restricted NKT cells suggesting that their precise role may change with ageing146. Thus it is the combinations of leukocytes recruited and the outcome of complex cell-cell crosstalk and cytokine expression within the stroma which will determine the nature and progression of fibrosis regardless of the specific lymphocytes in the inflammatory infiltrate.
Summary
Advances in our understanding of liver fibrosis have highlighted the importance of interactions between liver fibroblasts and the innate and adaptive arms of the immune system in determining the outcome following tissue injury. Whilst many acute injuries to the liver result in self-limiting non-fibrotic inflammatory reactions, chronic hepatitis is associated with persistent inflammation and scarring. This is because the development of a stromal microenvironment comprising modified neomatrix, activated fibroblasts and macrophages recruits, retains and maintains lymphocytes at the site of tissue injury resulting in chronic persistent inflammation. The lymphocytes in turn secrete cytokines and interact directly with cellular components of the stroma to maintain the local pro-fibrotic environment. Strategies that aim to restore liver function, either by preventing fibrosis progression or replenishing hepatocytes from stem cell precursors, must take these complex bi-directional interactions into account. Future anti-fibrotic interventions in liver disease may utilise the complex interactions between fibroblasts and T-cells to ameliorate the production of scar tissue and prevent inflammation pesrsisting. In this context the use of specific protein tyrosine-kinases inhibitors such as imatinib (Glivec) to treat fibrotic liver diseases may offer particular promise both as a means of preventing proliferation of fibroblasts in response to chronic hepatitis, but also inhibiting stromal patterns of cytokine secretion.
Acknowledgments
Grant Support: Dr Holt is supported by grants from the MRC, British Liver Trust, and CORE
Glossary
- HSC
hepatic stellate cell
- aLMF
activated liver myofibroblasts
- ECM
extra cellular matrix
- NK cell
natural killer cell
- MMP
matrix metalloproteinase
- TIMP
tissue inhibitor of metalloproteinase
- IL
interleukin
- CCl4
Carbon tetrachloride
- DC
Dendritic Cell
Reference List
- 1.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 2.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest. 1998;102:538–549. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knittel T, Dinter C, Kobold D, et al. Expression and regulation of cell adhesion molecules by hepatic stellate cells (HSC) of rat liver: involvement of HSC in recruitment of inflammatory cells during hepatic tissue repair. Am J Pathol. 1999;154:153–167. doi: 10.1016/s0002-9440(10)65262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muhanna N, Horani A, Doron S, Safadi R. Lymphocyte-hepatic stellate cell proximity suggests a direct interaction. Clin Exp Immunol. 2007;148:338–347. doi: 10.1111/j.1365-2249.2007.03353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 7.Filer A, Pitzalis C, Buckley CD. Targeting the stromal microenvironment in chronic inflammation. Curr Opin Pharmacol. 2006;6:393–400. doi: 10.1016/j.coph.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iredale JP. Hepatic stellate cell behavior during resolution of liver injury. Semin Liver Dis. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 10.Parsonage G, Filer AD, Haworth O, et al. A stromal address code defined by fibroblasts. Trends Immunol. 2005;26:150–156. doi: 10.1016/j.it.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–7562. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 12.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 13.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 14.Phillips RJ, Burdick MD, Hong K, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–446. doi: 10.1172/JCI20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quan TE, Cowper SE, Bucala R. The role of circulating fibrocytes in fibrosis. Curr Rheumatol Rep. 2006;8:145–150. doi: 10.1007/s11926-006-0055-x. [DOI] [PubMed] [Google Scholar]
- 16.Kisseleva T, Uchinami H, Feirt N, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Spanakis E, Brouty-Boye D. Discrimination of fibroblast subtypes by multivariate analysis of gene expression. Int J Cancer. 1997;71:402–409. doi: 10.1002/(sici)1097-0215(19970502)71:3<402::aid-ijc17>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 18.Chang HY, Chi JT, Dudoit S, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brouty-Boye D, Doucet C, Clay D, Bousse-Kerdiles MC, Lampidis TJ, Azzarone B. Phenotypic diversity in human fibroblasts from myelometaplasic and non-myelometaplasic hematopoietic tissues. Int J Cancer. 1998;76:767–773. doi: 10.1002/(sici)1097-0215(19980529)76:5<767::aid-ijc24>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Magness ST, Bataller R, Yang L, Brenner DA. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology. 2004;40:1151–1159. doi: 10.1002/hep.20427. [DOI] [PubMed] [Google Scholar]
- 21.Cassiman D, Roskams T. Beauty is in the eye of the beholder: emerging concepts and pitfalls in hepatic stellate cell research. J Hepatol. 2002;37:527–535. doi: 10.1016/s0168-8278(02)00263-5. [DOI] [PubMed] [Google Scholar]
- 22.Marinova-Mutafchieva L, Taylor P, Funa K, Maini RN, Zvaifler NJ. Mesenchymal cells expressing bone morphogenetic protein receptors are present in the rheumatoid arthritis joint. Arthritis Rheum. 2000;43:2046–2055. doi: 10.1002/1529-0131(200009)43:9<2046::AID-ANR16>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Dunn C, Brunetto M, Reynolds G, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204:667–680. doi: 10.1084/jem.20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melhem A, Muhanna N, Bishara A, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 25.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 26.Saito JM, Bostick MK, Campe CB, Xu J, Maher JJ. Infiltrating neutrophils in bile duct-ligated livers do not promote hepatic fibrosis. Hepatol Res. 2003;25:180–191. doi: 10.1016/s1386-6346(02)00247-4. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Lee G, Wang H, Vierling JM, Maher JJ. Limited role for CXC chemokines in the pathogenesis of alpha-naphthylisothiocyanate-induced liver injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G734–G741. doi: 10.1152/ajpgi.00300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- 29.Pauleau AL, Rutschman R, Lang R, Pernis A, Watowich SS, Murray PJ. Enhancer-mediated control of macrophage-specific arginase I expression. J Immunol. 2004;172:7565–7573. doi: 10.4049/jimmunol.172.12.7565. [DOI] [PubMed] [Google Scholar]
- 30.Sandler NG, Mentink-Kane MM, Cheever AW, Wynn TA. Global gene expression profiles during acute pathogen-induced pulmonary inflammation reveal divergent roles for Th1 and Th2 responses in tissue repair. J Immunol. 2003;171:3655–3667. doi: 10.4049/jimmunol.171.7.3655. [DOI] [PubMed] [Google Scholar]
- 31.Alabraba EB, Lai V, Boon L, Wigmore SJ, Adams DH, Afford SC. Coculture of human liver macrophages and cholangiocytes leads to CD40-dependent apoptosis and cytokine secretion. Hepatology. 2007 doi: 10.1002/hep.22011. [DOI] [PubMed] [Google Scholar]
- 32.McGaha TL, Bona CA. Role of profibrogenic cytokines secreted by T cells in fibrotic processes in scleroderma. Autoimmun Rev. 2002;1:174–181. doi: 10.1016/s1568-9972(02)00027-7. [DOI] [PubMed] [Google Scholar]
- 33.Henderson NC, Iredale JP. Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci (Lond) 2007;112:265–280. doi: 10.1042/CS20060242. [DOI] [PubMed] [Google Scholar]
- 34.Ide M, Kuwamura M, Kotani T, Sawamoto O, Yamate J. Effects of gadolinium chloride (GdCl(3)) on the appearance of macrophage populations and fibrogenesis in thioacetamide-induced rat hepatic lesions. J Comp Pathol. 2005;133:92–102. doi: 10.1016/j.jcpa.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology. 2005;128:138–146. doi: 10.1053/j.gastro.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 37.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 38.Bonecchi R, Facchetti F, Dusi S, et al. Induction of functional IL-8 receptors by IL-4 and IL-13 in human monocytes. J Immunol. 2000;164:3862–3869. doi: 10.4049/jimmunol.164.7.3862. [DOI] [PubMed] [Google Scholar]
- 39.Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000;204:19–28. doi: 10.1006/cimm.2000.1687. [DOI] [PubMed] [Google Scholar]
- 40.Issa R, Zhou X, Trim N, et al. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. FASEB J. 2003;17:47–49. doi: 10.1096/fj.02-0494fje. [DOI] [PubMed] [Google Scholar]
- 41.Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 42.Friedman SL. Mac the knife? Macrophages- the double-edged sword of hepatic fibrosis. J Clin Invest. 2005;115:29–32. doi: 10.1172/JCI23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cremer I, Dieu-Nosjean MC, Marechal S, et al. Long-lived immature dendritic cells mediated by TRANCE-RANK interaction. Blood. 2002;100:3646–3655. doi: 10.1182/blood-2002-01-0312. [DOI] [PubMed] [Google Scholar]
- 44.Goddard S, Youster J, Morgan E, Adams DH. Interleukin-10 secretion differentiates dendritic cells from human liver and skin. Am J Pathol. 2004;164:511–519. doi: 10.1016/S0002-9440(10)63141-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Randolph GJ, Sanchez-Schmitz G, Liebman RM, Schakel K. The CD16(+) (FcgammaRIII(+)) subset of human monocytes preferentially becomes migratory dendritic cells in a model tissue setting. J Exp Med. 2002;196:517–527. doi: 10.1084/jem.20011608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Neill HC, Ni K, Wilson H. Long-term stroma-dependent cultures are a consistent source of immunostimulatory dendritic cells. Immunol Cell Biol. 1999;77:434–441. doi: 10.1046/j.1440-1711.1999.00851.x. [DOI] [PubMed] [Google Scholar]
- 47.Briard D, Azzarone B, Brouty-Boye D. Importance of stromal determinants in the generation of dendritic and natural killer cells in the human spleen. Clin Exp Immunol. 2005;140:265–273. doi: 10.1111/j.1365-2249.2005.02792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang M, Tang H, Guo Z, et al. Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol. 2004;5:1124–1133. doi: 10.1038/ni1130. [DOI] [PubMed] [Google Scholar]
- 49.Saalbach A, Klein C, Sleeman J, et al. Dermal fibroblasts induce maturation of dendritic cells. J Immunol. 2007;178:4966–4974. doi: 10.4049/jimmunol.178.8.4966. [DOI] [PubMed] [Google Scholar]
- 50.Lai WK, Curbishley SM, Goddard S, et al. Hepatitis C is associated with perturbation of intrahepatic myeloid and plasmacytoid dendritic cell function. J Hepatol. 2007;47:338–347. doi: 10.1016/j.jhep.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 51.Munoz-Fernandez R, Blanco FJ, Frecha C, et al. Follicular dendritic cells are related to bone marrow stromal cell progenitors and to myofibroblasts. J Immunol. 2006;177:280–289. doi: 10.4049/jimmunol.177.1.280. [DOI] [PubMed] [Google Scholar]
- 52.Winau F, Hegasy G, Weiskirchen R, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 53.Schnabl B, Purbeck CA, Choi YH, Hagedorn CH, Brenner D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology. 2003;37:653–664. doi: 10.1053/jhep.2003.50097. [DOI] [PubMed] [Google Scholar]
- 54.Marra F, Valente AJ, Pinzani M, Abboud HE. Cultured human liver fat-storing cells produce monocyte chemotactic protein-1. Regulation by proinflammatory cytokines. J Clin Invest. 1993;92:1674–1680. doi: 10.1172/JCI116753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinzani M, Carloni V, Marra F, Riccardi D, Laffi G, Gentilini P. Biosynthesis of platelet-activating factor and its 1O-acyl analogue by liver fat-storing cells. Gastroenterology. 1994;106:1301–1311. doi: 10.1016/0016-5085(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 56.Marra F. Hepatic stellate cells and the regulation of liver inflammation. J Hepatol. 1999;31:1120–1130. doi: 10.1016/s0168-8278(99)80327-4. [DOI] [PubMed] [Google Scholar]
- 57.Eksteen B, Afford SC, Wigmore SJ, Holt AP, Adams DH. Immune-mediated liver injury. Semin Liver Dis. 2007;27:351–366. doi: 10.1055/s-2007-991512. [DOI] [PubMed] [Google Scholar]
- 58.Sakai N, Wada T, Yokoyama H, et al. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci U S A. 2006;103:14098–14103. doi: 10.1073/pnas.0511200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith TJ, Koumas L, Gagnon A, et al. Orbital fibroblast heterogeneity may determine the clinical presentation of thyroid-associated ophthalmopathy. J Clin Endocrinol Metab. 2002;87:385–392. doi: 10.1210/jcem.87.1.8164. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 61.Parsonage G, Falciani F, Burman A, et al. Global gene expression profiles in fibroblasts from synovial, skin and lymphoid tissue reveals distinct cytokine and chemokine expression patterns. Thromb Haemost. 2003;90:688–697. doi: 10.1160/TH03-04-0208. [DOI] [PubMed] [Google Scholar]
- 62.Buckley CD, Amft N, Bradfield PF, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol. 2000;165:3423–3429. doi: 10.4049/jimmunol.165.6.3423. [DOI] [PubMed] [Google Scholar]
- 63.Pilling D, Akbar AN, Girdlestone J, et al. Interferon-beta mediates stromal cell rescue of T cells from apoptosis. Eur J Immunol. 1999;29:1041–1050. doi: 10.1002/(SICI)1521-4141(199903)29:03<1041::AID-IMMU1041>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 64.Scheel-Toellner D, Pilling D, Akbar AN, et al. Inhibition of T cell apoptosis by IFN-beta rapidly reverses nuclear translocation of protein kinase C-delta. Eur J Immunol. 1999;29:2603–2612. doi: 10.1002/(SICI)1521-4141(199908)29:08<2603::AID-IMMU2603>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 65.Azzarone B, Failly-Crepin C, Daya-Grosjean L, Chaponnier C, Gabbiani G. Abnormal behavior of cultured fibroblasts from nodule and nonaffected aponeurosis of Dupuytren’s disease. J Cell Physiol. 1983;117:353–361. doi: 10.1002/jcp.1041170310. [DOI] [PubMed] [Google Scholar]
- 66.Azzarone B, Mareel M, Billard C, Scemama P, Chaponnier C, Macieira-Coelho A. Abnormal properties of skin fibroblasts from patients with breast cancer. Int J Cancer. 1984;33:759–764. doi: 10.1002/ijc.2910330608. [DOI] [PubMed] [Google Scholar]
- 67.Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–367. doi: 10.1186/ar113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 69.Doucet C, Jasmin C, Azzarone B. Unusual interleukin-4 and -13 signaling in human normal and tumor lung fibroblasts. Oncogene. 2000;19:5898–5905. doi: 10.1038/sj.onc.1203933. [DOI] [PubMed] [Google Scholar]
- 70.Muhlbauer M, Weiss TS, Thasler WE, et al. LPS-mediated NFkappaB activation varies between activated human hepatic stellate cells from different donors. Biochem Biophys Res Commun. 2004;325:191–197. doi: 10.1016/j.bbrc.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 71.Xia Y, Pauza ME, Feng L, Lo D. RelB regulation of chemokine expression modulates local inflammation. Am J Pathol. 1997;151:375–387. [PMC free article] [PubMed] [Google Scholar]
- 72.Oakley F, Mann J, Nailard S, et al. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol. 2005;166:695–708. doi: 10.1016/s0002-9440(10)62291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 74.Nissen LJ, Cao R, Hedlund EM, et al. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest. 2007;117:2766–2777. doi: 10.1172/JCI32479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pinzani M, Rombouts K. Liver fibrosis: from the bench to clinical targets. Dig Liver Dis. 2004;36:231–242. doi: 10.1016/j.dld.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 77.Cassiman D, Libbrecht L, Desmet V, Denef C, Roskams T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol. 2002;36:200–209. doi: 10.1016/s0168-8278(01)00260-4. [DOI] [PubMed] [Google Scholar]
- 78.Rappaport AM, MacPhee PJ, Fisher MM, Phillips MJ. The scarring of the liver acini (Cirrhosis). Tridimensional and microcirculatory considerations. Virchows Arch A Pathol Anat Histopathol. 1983;402:107–137. doi: 10.1007/BF00695054. [DOI] [PubMed] [Google Scholar]
- 79.Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599–1607. doi: 10.5858/2000-124-1599-ROHC. [DOI] [PubMed] [Google Scholar]
- 80.Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416. doi: 10.1055/s-2001-17554. [DOI] [PubMed] [Google Scholar]
- 81.Pinzani M, Marra F, Carloni V. Signal transduction in hepatic stellate cells. Liver. 1998;18:2–13. doi: 10.1111/j.1600-0676.1998.tb00120.x. [DOI] [PubMed] [Google Scholar]
- 82.Gonzalo T, Beljaars L, van de BM, et al. Local inhibition of liver fibrosis by specific delivery of a platelet-derived growth factor kinase inhibitor to hepatic stellate cells. J Pharmacol Exp Ther. 2007;321:856–865. doi: 10.1124/jpet.106.114496. [DOI] [PubMed] [Google Scholar]
- 83.Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–3860. doi: 10.1182/blood.V100.12.3853. [DOI] [PubMed] [Google Scholar]
- 84.Takasaki S, Hano H. Three-dimensional observations of the human hepatic artery (Arterial system in the liver) J Hepatol. 2001;34:455–466. doi: 10.1016/s0168-8278(00)00058-1. [DOI] [PubMed] [Google Scholar]
- 85.Kudo S, Matsuno K, Ezaki T, Ogawa M. A novel migration pathway for rat dendritic cells from the blood: hepatic sinusoids-lymph translocation. J Exp Med. 1997;185:777–784. doi: 10.1084/jem.185.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 87.Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol. 2006;6:244–251. doi: 10.1038/nri1784. [DOI] [PubMed] [Google Scholar]
- 88.Elvevold KH, Nedredal GI, Revhaug A, Smedsrod B. Scavenger properties of cultivated pig liver endothelial cells. Comp Hepatol. 2004;3:4. doi: 10.1186/1476-5926-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong J, Johnston B, Lee SS, et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest. 1997;99:2782–2790. doi: 10.1172/JCI119468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Adams DH, Hubscher SG, Fisher NC, Williams A, Robinson M. Expression of E-selectin and E-selectin ligands in human liver inflammation. Hepatology. 1996;24:533–538. doi: 10.1002/hep.510240311. [DOI] [PubMed] [Google Scholar]
- 91.McNab G, Reeves JL, Salmi M, Hubscher S, Jalkanen S, Adams DH. Vascular adhesion protein 1 mediates binding of T cells to human hepatic endothelium. Gastroenterology. 1996;110:522–528. doi: 10.1053/gast.1996.v110.pm8566600. [DOI] [PubMed] [Google Scholar]
- 92.Lalor PF, Edwards S, McNab G, Salmi M, Jalkanen S, Adams DH. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J Immunol. 2002;169:983–992. doi: 10.4049/jimmunol.169.2.983. [DOI] [PubMed] [Google Scholar]
- 93.Suniara RK, Jenkinson EJ, Owen JJ. An essential role for thymic mesenchyme in early T cell development. J Exp Med. 2000;191:1051–1056. doi: 10.1084/jem.191.6.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marra F, Romanelli RG, Giannini C, et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29:140–148. doi: 10.1002/hep.510290107. [DOI] [PubMed] [Google Scholar]
- 95.Hellerbrand Wang SC, Tsukamoto H, Brenner DA, Rippe RA. Expression of intracellular adhesion molecule 1 by activated hepatic stellate cells. Hepatology. 1996;24:670–676. doi: 10.1002/hep.510240333. [DOI] [PubMed] [Google Scholar]
- 96.Lalor PF, Shields P, Grant A, Adams DH. Recruitment of lymphocytes to the human liver. Immunol Cell Biol. 2002;80:52–64. doi: 10.1046/j.1440-1711.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 97.Curbishley SM, Eksteen B, Gladue RP, Lalor P, Adams DH. CXCR3 Activation Promotes Lymphocyte Transendothelial Migration across Human Hepatic Endothelium under Fluid Flow. Am J Pathol. 2005;167:887–899. doi: 10.1016/S0002-9440(10)62060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Heydtmann M, Lalor PF, Eksteen JA, Hubscher SG, Briskin M, Adams DH. CXC chemokine ligand 16 promotes integrin-mediated adhesion of liver-infiltrating lymphocytes to cholangiocytes and hepatocytes within the inflamed human liver. J Immunol. 2005;174:1055–1062. doi: 10.4049/jimmunol.174.2.1055. [DOI] [PubMed] [Google Scholar]
- 99.Heydtmann M, Hardie D, Shields PL, et al. Detailed analysis of intrahepatic CD8 T cells in the normal and hepatitis C-infected liver reveals differences in specific populations of memory cells with distinct homing phenotypes. J Immunol. 2006;177:729–738. doi: 10.4049/jimmunol.177.1.729. [DOI] [PubMed] [Google Scholar]
- 100.Efsen E, Grappone C, Defranco RM, et al. Up-regulated expression of fractalkine and its receptor CX3CR1 during liver injury in humans. J Hepatol. 2002;37:39–47. doi: 10.1016/s0168-8278(02)00065-x. [DOI] [PubMed] [Google Scholar]
- 101.Isse K, Harada K, Zen Y, et al. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41:506–516. doi: 10.1002/hep.20582. [DOI] [PubMed] [Google Scholar]
- 102.Roth SJ, Carr MW, Springer TA. C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-gamma inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur J Immunol. 1995;25:3482–3488. doi: 10.1002/eji.1830251241. [DOI] [PubMed] [Google Scholar]
- 103.Taub DD, Proost P, Murphy WJ, et al. Monocyte chemotactic protein-1 -2 and -3 are chemotactic for human T lymphocytes. J Clin Invest. 1995;95:1370–1376. doi: 10.1172/JCI117788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Czaja MJGAXJSPaJY. Monocyte Chemoattractant Protein-1 expression occurs in toxic rat liver injury and human liver disease. Journal of Leukocyte Biology. 1994;55:120–126. doi: 10.1002/jlb.55.1.120. 55, 120–126. [DOI] [PubMed] [Google Scholar]
- 105.Safadi R, Ohta M, Alvarez CE, et al. Immune stimulation of hepatic fibrogenesis by CD8 cells and attenuation by transgenic interleukin-10 from hepatocytes. Gastroenterology. 2004;127:870–882. doi: 10.1053/j.gastro.2004.04.062. [DOI] [PubMed] [Google Scholar]
- 106.Nelson DR, Lauwers GY, Lau JY, Davis GL. Interleukin 10 treatment reduces fibrosis in patients with chronic hepatitis C: a pilot trial of interferon nonresponders. Gastroenterology. 2000;118:655–660. doi: 10.1016/s0016-5085(00)70134-x. [DOI] [PubMed] [Google Scholar]
- 107.Tiggelman AM, Boers W, Linthorst C, Brand HS, Sala M, Chamuleau RA. Interleukin-6 production by human liver (myo)fibroblasts in culture. Evidence for a regulatory role of LPS, IL-1 beta and TNF alpha. J Hepatol. 1995;23:295–306. [PubMed] [Google Scholar]
- 108.Adams DH, Hathaway M, Shaw J, Burnett D, Elias E, Strain AJ. Transforming growth factor-beta induces human T lymphocyte migration in vitro. J Immunol. 1991;147:609–612. [PubMed] [Google Scholar]
- 109.Hathaway M, Burnett D, Elias E, Adams DH. Secretion of soluble chemotactic factors, including interleukin-6: a mechanism for the recruitment of CD8-positive T lymphocytes to human liver allografts during rejection. Hepatology. 1993;18:511–518. [PubMed] [Google Scholar]
- 110.Delaney B, Koh WS, Yang KH, Strom SC, Kaminski NE. Hepatocyte growth factor enhances B-cell activity. Life Sci. 1993;53:89–93. doi: 10.1016/0024-3205(93)90654-l. [DOI] [PubMed] [Google Scholar]
- 111.Adams DH, Harvath L, Bottaro DP, et al. Hepatocyte growth factor and macrophage inflammatory protein 1 beta: structurally distinct cytokines that induce rapid cytoskeletal changes and subset-preferential migration in T cells. Proc Natl Acad Sci U S A. 1994;91:7144–7148. doi: 10.1073/pnas.91.15.7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7:344–353. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 113.Hjelmstrom P, Fjell J, Nakagawa T, Sacca R, Cuff CA, Ruddle NH. Lymphoid tissue homing chemokines are expressed in chronic inflammation. Am J Pathol. 2000 Apr;156(4):1133–8. doi: 10.1016/S0002-9440(10)64981-4. 156:1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bonacchi A, Petrai I, Defranco RM, et al. The chemokine CCL21 modulates lymphocyte recruitment and fibrosis in chronic hepatitis C. Gastroenterology. 2003;125:1060–1076. doi: 10.1016/s0016-5085(03)01194-6. [DOI] [PubMed] [Google Scholar]
- 115.Grant AJ, Goddard S, Ahmed-Choudhury J, et al. Hepatic expression of secondary lymphoid chemokine (CCL21) promotes the development of portal-associated lymphoid tissue in chronic inflammatory liver disease. Am J Pathol. 2002;160:1445–55. doi: 10.1016/S0002-9440(10)62570-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 117.Bajenoff M, Egen JG, Koo LY, et al. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mempel TR, Henrickson SE, von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427:154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 119.Melhem A, Muhanna N, Bishara A, et al. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 120.Neubauer K, Eichhorst ST, Wilfling T, Buchenau M, Xia L, Ramadori G. Sinusoidal intercellular adhesion molecule-1 up-regulation precedes the accumulation of leukocyte function antigen-1-positive cells and tissue necrosis in a model of carbontetrachloride-induced acute rat liver injury. Lab Invest. 1998;78:185–194. [PubMed] [Google Scholar]
- 121.Kordes C, Sawitza I, Muller-Marbach A, et al. CD133+ hepatic stellate cells are progenitor cells. Biochem Biophys Res Commun. 2007;352:410–417. doi: 10.1016/j.bbrc.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 122.McGettrick HM, Lord JM, Wang KQ, Rainger GE, Buckley CD, Nash GB. Chemokine- and adhesion-dependent survival of neutrophils after transmigration through cytokine-stimulated endothelium. J Leukoc Biol. 2006;79:779–788. doi: 10.1189/jlb.0605350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ross EA, Douglas MR, Wong SH, et al. Interaction between integrin alpha9beta1 and vascular cell adhesion molecule-1 (VCAM-1) inhibits neutrophil apoptosis. Blood. 2006;107:1178–1183. doi: 10.1182/blood-2005-07-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Filer A, Parsonage G, Smith E, et al. Differential survival of leukocyte subsets mediated by synovial, bone marrow, and skin fibroblasts: site-specific versus activation-dependent survival of T cells and neutrophils. Arthritis Rheum. 2006;54:2096–2108. doi: 10.1002/art.21930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tough DF, Borrow P, Sprent J. Induction of bystander T-cell proliferation by viruses and type- i Interferon in-vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 126.Abrignani S. Bystander activation by cytokines of intrahepatic T cells in chronic viral hepatitis. Semin Liver Dis. 1997;17:319–322. doi: 10.1055/s-2007-1007208. [DOI] [PubMed] [Google Scholar]
- 127.Mitchell T, Kappler J, Marrack P. Bystander virus infection prolongs activated T cell survival. J Immunol. 1999;162:4527–4535. [PubMed] [Google Scholar]
- 128.Inaba M, Kurasawa K, Mamura M, Kumano K, Saito Y, Iwamoto I. Primed T cells are more resistant to Fas-mediated activation-induced cell death than naive T cells. J Immunol. 1999;163:1315–1320. [PubMed] [Google Scholar]
- 129.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Holt AP, Stamataki Z, Adams DH. Attenuated liver fibrosis in the absence of B cells. Hepatology. 2006;43:868–871. doi: 10.1002/hep.21155. [DOI] [PubMed] [Google Scholar]
- 131.Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115:3072–3082. doi: 10.1172/JCI24798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chiaramonte MG, Schopf LR, Neben TY, Cheever AW, Donaldson DD, Wynn TA. IL-13 is a key regulatory cytokine for Th2 cell-mediated pulmonary granuloma formation and IgE responses induced by Schistosoma mansoni eggs. J Immunol. 1999;162:920–930. [PubMed] [Google Scholar]
- 134.Hoffmann KF, McCarty TC, Segal DH, et al. Disease fingerprinting with cDNA microarrays reveals distinct gene expression profiles in lethal type 1 and type 2 cytokine-mediated inflammatory reactions. FASEB J. 2001;15:2545–2547. doi: 10.1096/fj.01-0306fje. [DOI] [PubMed] [Google Scholar]
- 135.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–1089. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 136.Parekh VV, Prasad DV, Banerjee PP, Joshi BN, Kumar A, Mishra GC. B cells activated by lipopolysaccharide, but not by anti-Ig and anti-CD40 antibody, induce anergy in CD8+ T cells: role of TGF-beta 1. J Immunol. 2003;170:5897–5911. doi: 10.4049/jimmunol.170.12.5897. [DOI] [PubMed] [Google Scholar]
- 137.Mosmann T. Complexity or coherence? Cytokine secretion by B cells. Nat Immunol. 2000;1:465–466. doi: 10.1038/82707. [DOI] [PubMed] [Google Scholar]
- 138.Harris DP, Haynes L, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1:475–482. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 139.Harris DP, Goodrich S, Mohrs K, Mohrs M, Lund FE. Cutting edge: the development of IL-4-producing B cells (B effector 2 cells) is controlled by IL-4, IL-4 receptor alpha, and Th2 cells. J Immunol. 2005;175:7103–7107. doi: 10.4049/jimmunol.175.11.7103. [DOI] [PubMed] [Google Scholar]
- 140.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha(2) receptor is involved in induction of TGF-beta(1) production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 141.Afford SC, Randhawa S, Eliopoulos AG, Hubscher SG, Young LS, Adams DH. CD40 activation induces apoptosis in cultured human hepatocytes via induction of cell surface fas ligand expression and amplifies fas-mediated hepatocyte death during allograft rejection. J Exp Med. 1999;189:441–446. doi: 10.1084/jem.189.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ferru I, Roye O, Delacre M, Auriault C, Wolowczuk I. Infection of B-cell-deficient mice by the parasite Schistosoma mansoni: demonstration of the participation of B cells in granuloma modulation. Scand J Immunol. 1998;48:233–240. doi: 10.1046/j.1365-3083.1998.00376.x. [DOI] [PubMed] [Google Scholar]
- 143.de LC, Galli G, Aldrighetti L, et al. Production of profibrotic cytokines by invariant NKT cells characterizes cirrhosis progression in chronic viral hepatitis. J Immunol. 2004;173:1417–1425. doi: 10.4049/jimmunol.173.2.1417. [DOI] [PubMed] [Google Scholar]
- 144.Safadi R, Zigmond E, Pappo O, Shalev Z, Ilan Y. Amelioration of hepatic fibrosis via beta-glucosylceramide-mediated immune modulation is associated with altered CD8 and NKT lymphocyte distribution. Int Immunol. 2007;19:1021–1029. doi: 10.1093/intimm/dxm069. [DOI] [PubMed] [Google Scholar]
- 145.Kim JH, Kim HY, Kim S, Chung JH, Park WS, Chung DH. Natural killer T (NKT) cells attenuate bleomycin-induced pulmonary fibrosis by producing interferon-gamma. Am J Pathol. 2005;167:1231–1241. doi: 10.1016/s0002-9440(10)61211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chuang YH, Lian ZX, Yang GX, et al. Natural killer T cells exacerbate liver injury in a transforming growth factor beta receptor II dominant-negative mouse model of primary biliary cirrhosis. Hepatology. 2007 doi: 10.1002/hep.22052. [DOI] [PubMed] [Google Scholar]
- 147.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–2777. doi: 10.1084/jem.20051759. [DOI] [PMC free article] [PubMed] [Google Scholar]