

Abstract
Plasmacytoid dendritic cells (pDCs) are innate sensors that produce IFN-α in response to viral infections. Determining how aging alters the cellular and molecular function of these cells may provide an explanation of increased susceptibility of older people to viral infections. Hence, we examined whether aging critically impairs pDC function during infection with herpes simplex virus type 2 (HSV-2), a viral pathogen that activates TLR9. We found that impaired IFN-α production by aged murine pDCs led to impaired viral clearance with aging. Upon TLR9 activation, aged pDCs displayed defective upregulation of interferon regulatory factor 7 (IRF-7), a key adaptor in the type I IFN pathway, as compared to younger counterparts. Aged pDCs had more oxidative stress, and reducing oxidative stress in aged pDCs partly recovered the age-induced IFN-α defect during TLR9 activation. In sum, aging impairs the type I IFN pathway in pDCs, and this alteration may contribute to the increased susceptibility of older people to certain viral infections.
Keywords: Rodent, viral infection, dendritic cell
Introduction
Clinical studies have demonstrated that aging is associated with an increased susceptibility to pathogens, in particular viruses, implying that aging negatively impacts immunity. Consequently, older people exhibit an increased incidence of morbidity and mortality from infections (1). According to previous work, aging impairs adaptive T cell function (2-5); however, the effect of aging on the innate immune system is less clear. Previous studies reported conflicting results about whether cellular components of the innate system, such as conventional DCs, macrophages, or NK cells are altered with age (4, 6, 7). As innate immunity is the first line of defense against pathogens, understanding how aging modifies this defense is imperative, as this information may be critical for the development of therapies to augment immunity in older people.
Plasmacytoid DCs (pDCs) are key cellular components of the innate immune system that secrete high levels of type I IFNs, such as IFN-α, and IL-12 in response to certain bacteria (8) and viruses (9-12). In contrast to conventional myeloid DCs, which express many TLRs, pDCs express TLR7 and TLR9 (13, 14). The production of type I IFNs and IL-12 occur via separate pathways; the adaptor interferon regulatory factor (IRF)-7 is essential for type I IFN production, whereas IL-12 production is dependent on NF-κB translocation (15). Activation of TLR7 and TLR9 receptors within the endosomes of pDCs is critical for the recognition of single-stranded RNA via TLR7 (16-19) and unmethylated CpG sequences via TLR9 (14, 20-22). The high levels of type 1 IFNs produced by TLR-activated pDCs (21, 23) activate cellular host innate defense mechanisms and initiate adaptive immunity (8, 12, 24, 25). Hence, pDCs are critical for host defense against many viruses.
Prior studies indicate that aging impairs TLR immune responses in macrophages in vitro (26, 27), whereas TLR immune responses are preserved in conventional DCs (28). Regarding pDCs and aging, one clinical study reported an association between increased age and reduced circulating pDC numbers (29). Additionally, decreased IFN-α responses were observed during ex vivo challenge of peripheral blood mononuclear cells (PBMCs) and purified pDCs from people over the age of 65 years when compared to young counterparts (30, 31); however, whether altered pDC function with aging impacts the in vivo response to viral infection is not known. Furthermore, the underlying molecular defects in pDCs that occur with aging have not been examined. One of the leading theories of age-induced cellular damage is the accumulation of damaging reactive oxygen species (ROS) (32, 33), but whether increased ROS levels are important for any age-induced phenotype in pDCs remains unclear.
In this study, we examined the effect of aging on pDC function and any resultant effects on host defense to viral infection. We employed a herpes simplex virus type 2 (HSV-2) infection as an experimental murine model to activate TLR9 in pDCs (21). We demonstrated that aging impaired systemic IFN-α production during viral infection and subsequently resulted in impaired viral clearance, a phenotype that was reversed by the adoptive transfer of young pDCs into aged recipients. Gene and protein analysis demonstrated that aged pDCs exhibited an impaired ability to upregulate IRF-7 and phosphatidylinositol 3-kinase (PI3)-kinase during either TLR9 or IFNαβ receptor activation. These findings were associated with increased oxidative stress during TLR9 activation in aged pDCs as compared to young cells. Importantly, reduction of age-induced oxidative stress led to augmented IFN-α production in TLR9-activated aged pDCs. In conclusion, aging leads to increased oxidative stress and decreased upregulation of IRF-7 within pDCs, and these alterations may be one of the mechanisms by which aging leads to defective host defense to viral infection.
Materials and Methods
Mice
Specific pathogen-free young (2-4 months of age) and aged (18-20 months of age) CBA (H2k) or C57BL/6 (H2b) calorie-restricted (CR) or ad libitum-fed (AL) mice were acquired from the National Institute on Aging (NIA) rodent facility (Baltimore, MD). CR mice were fed 40% of an NIH-31 diet from three months of age. Upon arrival, mice were continued on their respective diets (AL or CR). No animals were used in the study if they had evidence of skin lesions, weight loss, or lymphadenopathy. This study was approved by the Yale University Institutional Animal Care and Use Committee.
HSV-2 and MCMV viral propagation and preparation
HSV-2 (strain 186TKΔKpn) (21), which was generously provided by Dr. Akiko Iwasaki (Yale University, New Haven, CT), was propagated in Vero cells as previously described (21, 34). Virus was purified by centrifugation and was re-suspended in PBS before use. The virus was administered to mice via i.v. (tail vein) injection at 1×107 PFU/mouse. In experiments comparing young versus aged mice, viral dosages were adjusted to body weight. Bone marrow purified pDCs were infected with HSV-2 at 1×103 PFU or PBS as a control, and IFN-α levels in supernatants were measured 24h after infection. HSV-2 viremia in livers and spleens were measured by plaque assay as previously described (21, 34). MCMV Smith strain was generously provided by Anthony Van Den Pol (Yale University, New Haven, CT). The virus was administered to mice via i.p. injection of 2×105 PFU/mouse as previously described (24). MCMV viremia in livers and spleens was measured with a plaque assay as previously described (35).
Reagents, in vitro culture, and adoptive transfer of bone marrow cells
The CpG-A sequence used for in vitro and in vivo pDC stimulation (CpG-ODN 5′-CTATTGGAAAACGTTCTTCGGGGCG-3′(36)) was synthesized by MWG Biotech AG (High Point, NC). For in vitro and in vivo administration, various concentrations of the CpG-A sequence were diluted in PBS and then added dropwise to DOTAP methosulfate (1 μg of CpG-A DNA per 5 μg DOTAP; Sigma-Aldrich, St. Louis, MI) to form a liposome:DNA complex. For in vitro experiments, CpG-A (10 μg/ml or indicated dose) was added to 3-5×105 cells in a total volume of 200 μl/well for 18h. For in vivo experiments, CpG-A (5 μg/mg body weight) were administered via i.v. (tail vein) injection. RNA40 was administered at 50 μg/ml for in vitro experiments (Invitrogen, San Diego, CA). In addition, 1 μM N-acetyl cysteine (denoted as NAC) was added to cultures where indicated, as previously described (37, 38). Anti-PDCA-1 antibody (500 μg/mouse, Miltenyi Biotec, Auburn CA) or control antibody was administered via i.p. injection 12h before HSV-2 administration. For adoptive transfer experiments, 5×106 bone marrow cells or 1×104 purified pDCs were transferred via i.v. (tail vein) injection. Murine recombinant IFN-α (PBL Laboratories, Piscataway, NJ) was used in vitro at a dose of 10 units per well.
Primary bone marrow cell isolation and purification
Bone marrow cells were prepared from the femurs and tibias of mice. The pDCs were purified by positive selection by incubating cells with magnetic microbeads that were coupled to a PDCA-1 mAB (Miltenyi Biotec) and then passing the suspension through a MACS LS column according to the manufacturer's instructions (Miltenyi Biotec). We routinely obtained a purity of >90% PDCA-1+CD11clo cells according to flow cytometry analysis. A similar procedure with microbeads coupled to PDCA-1 mAB was used to exclude cells except that depletion columns were employed (Miltenyi Biotec). Flow cytometric analysis demonstrated that the resulting solution contained <0.3% pDCs.
Cytokine analysis
Serum IFN-α (PBL Biomedical Laboratories, Piscataway, NJ) and IL-12p40 (R&D Systems, Minneapolis, MN) levels were assessed by ELISA according to the manufacturer's instructions.
IRF-7 activity ELISA
Nuclear extracts were purified via a nuclear extraction kit (Active Motif, Carlsbad, CA), and IRF-7 activity was measured by ELISA using the TransAM IRF-7 kit, according to the manufacturer's instructions (Active Motif). Results are expressed as absorbance per μg nuclear extract.
RT-PCR
RNA samples were extracted using the Qiagen RNAeasy mini kit (Qiagen, Valencia, CA) and quantified by A260/A280 absorbance readings. Superscript III reverse transcriptase (Invitrogen) was used to create complementary DNA from mRNA according to the manufacturer's instructions. The RT-PCR primer sets were designed with Primer Express software (Applied Biosystems, Foster City, CA). RT-PCR was performed in a final volume of 25 μL containing complementary DNA from 20 ng of reverse-transcribed total RNA, 150 nM forward and reverse primers, and SYBR Green universal PCR master mix (Applied Biosystems). PCR was performed in 96-well plates with the MJ Research detection system (MJ Research Inc, MA). All reactions were performed in triplicate. Analysis of the melting curve and the dilution curve standards was performed to identify primer sets and conditions yielding specific products with 100% amplification efficiency. The primer sequences were as follows: IRF-7: forward primer, acacttcctcatggacctgg; reverse primer, ttcccacttcccattctgag; and PI3-kinase: forward primer, tgtcagatgaggaggctgtg; reverse primer, gggtcaaatcccctttcatt. Relative levels of mRNA were calculated by the comparative cycle threshold method (User Bulletin No. 2, Applied Biosystems). The 18sRNA mRNA levels were used as the invariant control for each sample. Gene expression analysis was performed after 18h of stimulation as this time point exhibited the greatest differences in IFN-α between young and aged pDCs (Fig. 1D).
Figure 1.

IFN-α production is impaired in CpG type A-activated aged pDCs compared to young pDCs in vitro and to systemic administration of CpG type A in vivo. A-B, The pDCs were isolated from the bone marrow of young and aged C57BL/6 mice and incubated for 18h with CpG-ODN at 5 or 10 μg/ml. IFN-α (A) and IL-12p40 (B) production was measured. Control cells were stimulated with either PBS or DOTAP. C, Aged pDCs exhibited defective IFN-α in dose response experiments at 24h post CpG stimulation. IL-12p40 responses were preserved in dose response experiments in aged pDCs as compared to young counterparts (data not shown). D, IFN-α responses by aged pDCs were inferior to young pDCs in time course experiments. *p<0.05 (Student's t-test). The results in A-D are representative of at least four independent experiments with similar findings. N=3 per group for each experiment. E-F, Young and aged C57BL/6 mice were administered type A CpG i.v., and then serum IFN-α (E) and IL-12p40 (F) levels were assessed by ELISA. ***p<0.0001. The results are representative of at least three independent experiments with similar results. N=3 per group for each experiment.
Western blotting
Cells were washed, and cytosolic and nuclear protein fractions were isolated using the NE-PER Nuclear and Cytoplasmic Extraction kit (Pierce Biotechnology, Rockford, IL) according to the manufacturer's protocol. Protein concentrations were determined using the Bio-Rad protein assay reagents (Bio-Rad Laboratories, Hercules, CA) with BSA as the standard. Equal amounts of total cytosolic or nuclear protein (10μg/lane) were separated on 10% bis-acrylamide gels by SDS-PAGE and electrophoretically transferred to nitrocellulose in NuPage buffer (Invitrogen, Carlsbad, CA). Immunodetection was performed using primary anti-IRF7, anti-IKKα mAbs (Santa Cruz Biologicals, Santa Cruz, CA) or rabbit polyclonal anti-mouse Abs to the phosphorylated site of serine 124 of AKT (denoted as phosphorylated AKT) (Abcam, Cambridge, MA) and secondary HRPconjugated anti-goat Ig. The ECL Western Blotting Analysis System was used for detection of the signal. For IRF-7 activation, detectable protein levels were detected at one hour post-CpG-A activation, which peaked after 4h and were maintained up to 18h (data not shown). Eighteen hours after CpG-A activation was chosen for assessment of differences in IRF-7 levels between young and aged pDCs, as this time point demonstrated the largest IFN-α differences between young and aged pDCs (Fig. 1D).
Flow cytometry
Cells were stained with PE-conjugated rat anti-mPDCA-1(Miltenyi Biotec) and FITC-conjugated anti-CD11c (BD Biosciences). In agreement with prior work (39), we confirmed that anti-mPDCA-1 identified CD11clo/int B220hi cells (data not shown). For all staining, incubation was performed for 30 minutes at 4 °C, and a second incubation step with strepavidin PERCP was performed for 15 minutes at 4 °C. Isotype control antibodies were used in every experiment. For staining with dihydroethidium (DHE), which is rapidly oxidized by ROS into its fluorescent derivative (40), cells were incubated for 15 minutes at 37 °C in culture media containing a 1:100 dilution of a 10 μM solution of DHE in DMSO (Invitrogen, Carlsbad, CA). Fluorescence data was acquired with a FACS CALIBUR flow cytometer and analyzed with FlowJo Software (Treestar, Ashland, OR).
Statistical analysis
Statistical analysis was performed using GraphPad Prism Software (La Jolla, CA). Consistent results were noted in repeat experiments, allowing pooling of data and subsequent comparisons of means by a two-way Student's t-test. Statistical significance is indicated as p value <0.05.
Results
Aging impairs the ability of pDCs to produce IFN-α in response to in vitro CpG-A activation
Previous work has demonstrated that oligonucleotides containing type-A CpG sequences (CpG-A) induce the production of IFN-α in a TLR9-dependent manner (36). We first examined whether aging altered the above response by isolating pDCs from the bone marrow of young (2-4 months of age) and aged (18-20 months of age) mice and stimulating these cells with CpG-A in vitro. The pDCs isolated from the bone marrow of aged mice produced significantly less IFN-α than the younger mice post CpG-A administration (Fig. 1A). In contrast to the reduced IFN-α response with aging, pDCs from aged and young mice produced similar levels of IL-12p40 following in vitro CpG-A treatment (Fig. 1B). Decreased IFN-α production by aged pDCs remained evident following treatment with different doses of CpG-A and at different time points following CpG-A stimulation (Fig. 1C-D).
The similar IL-12p40 responses induced by CpG-A treatment in aged and young pDCs implied that aged pDCs expressed TLR9 at levels similar to young pDCs, and these levels were confirmed via flow cytometric analysis (data not shown). We also performed cell viability and apoptosis experiments to examine whether CpG-A-induced toxicity was altered with aging. Eighteen hours of activation with CpG-A induced similar levels of cell death and apoptosis in aged and young pDCs (data not shown). Finally, the reduced IFN-α response by aged pDCs was obtained in two different genetic backgrounds, CBA (H2k) and C57BL/6 (H2b) (data not shown), demonstrating that defective IFN-α production with aging was not strain-specific. In sum, these data demonstrate that aging impairs the ability of pDCs to produce IFN-α but not IL-12p40 in response to in vitro TLR9 activation with CpG-A.
Systemic IFN-α levels are impaired in aged hosts in response to in vivo CpG-A administration
To broaden the implications of our in vitro results, we examined the impact of aging on the response of pDCs to CpG-A stimulation in vivo. Hence, young and aged C57BL/6 mice received CpG-A intravenously, and serum IFN-α was measured. Using pDC-depleting mAbs, we demonstrated that administration of CpG-A to young mice increased IFN-α serum levels in a pDC-dependent manner, whereas pDCs were not essential for IL-12p40 production (data not shown). TLR9 specificity of CpG-A treatment was confirmed in TLR9-deficient mice that were unresponsive to such treatment (data not shown). In agreement with our in vitro experiments described above, aged mice exhibited significantly decreased IFN-α levels but maintained IL-12p40 responses as compared to young mice following in vivo CpG-A administration (Fig. 1E-F).
Aged mice manifest decreased IFN-α production in response to HSV-2 infection
We next examined the impact of aging on pDC function in response to viral infection with HSV-2, a pathogen that activates TLR9 (21, 34). Systemic HSV-2 infection in young mice induced IFN-α production and viral clearance in a pDC-dependent fashion (Fig. 2A-B) in agreement with a prior study (34), whereas IL-12p40 production was pDC-independent (data not shown). Subsequently, young and aged C57BL/6 mice were infected with HSV-2, and serum IFN-α production was measured. Again, the specificity of TLR9 for HSV-2 was confirmed by infecting TLR9-deficient mice, which did not produce IFN-α (data not shown). Purified pDCs from aged mice manifested an impaired IFN-α response during in vitro HSV-2 infection as compared to young pDCs (Fig. 2C). Importantly, aged mice exhibited decreased serum IFN-α levels during HSV-2 infection as compared to young infected mice (Fig. 2D). Thus, these results demonstrate that aging impairs the IFN-α response to HSV-2 infection.
Figure 2.

Aged hosts produce less IFN-α during HSV-2 infection as compared with young hosts. A, IFN-α levels during systemic HSV-2 infection were measured in control and anti-PDCA-1 (pDC depleting mAB)-treated young mice. B, Viral load was measured in young mice that were treated with anti-PDCA-1 or control antibody. C, Aged pDCs exhibit an impaired IFN-α response to in vitro HSV-2 infection compared to young pDCs. N = 3/group in at least two independent experiments. D, Young and aged C57BL6 mice were systemically infected with HSV-2 and then serum IFN-α levels were measured. These results are representative of at least four independent experiments (N=3 per group) with similar results. E, Spleen and liver samples were isolated from virally infected mice during HSV-2 infection, and viral load was measured by plaque assay. These results are representative of at least two independent experiments with similar results (N=3 per group).
Next, we examined the effect of aging on viral clearance of HSV-2 by measuring viral titers in spleens and livers isolated from infected young and aged mice. At 96h post-HSV-2 infection, aged hosts demonstrated a significantly increased viral load in comparison to young counterparts (Fig. 2E). These data demonstrate that aged hosts failed to clear HSV-2 infection as effectively as young hosts.
To broaden the implications of our findings, we also employed an alternative viral pathogen, murine cytomegalovirus (MCMV), which relies on pDCs for host defense (41, 42). As this virus has been recently shown to depend on dual signaling of TLR7 and TLR9 (43), we first demonstrated that aged pDCs produced reduced levels of IFN-α in response to RNA40, single stranded RNA that has been shown to stimulate TLR7 (Fig. 3A) (44, 45). We next infected young and aged mice with MCMV and measured IFN-α levels and viral clearance. IFN-α responses and viral clearance were reduced in aged MCMV-infected mice as compared to young mice (Fig. 3B-C). Therefore, in response to a viral pathogen that is known to activate pDCs, IFN-α responses were impaired in aged mice.
Figure 3.

Aged pDCs manifest an impaired IFN-α response to TLR7 activation with RNA40, and aged mice manifest an impaired IFN-α response to MCMV infection and a reduced ability to clear this virus. A, Aged pDCs manifest an impaired IFN-α response during 18h of TLR7 activation with 50 μg/ml RNA40 (N=3/group). Groups were significantly different with a p-value of <0.05. Control cells were treated with PBS. B-C, Aged mice infected with MCMV exhibited reduced IFN-α response (B) and viral load (C) as compared to young mice (N=3/group). Aged and young infected groups were significantly different with p-value of <0.05. Parameters were measured after 36h of viral infection. Control groups received PBS.
Aged mice display similar numbers of pDCs in the spleen, bone marrow, and blood compared to younger mice
Since prior clinical reports suggested that aging may lead to a reduction in pDC numbers (29), we next assessed whether aged mice had altered absolute numbers of pDCs in the spleen, bone marrow, and peripheral blood as this decrease may account for the reduced IFN-α during viral infection in aged mice. However, we did not find evidence that aging led to reduced numbers of pDCs within these compartments (Fig. 4). (Aged mice did not exhibit reduced spleen or bone marrow cellularity as compared to young mice, data not shown). Thus, in agreement with our in vitro data, the diminished IFN-α response to in vivo viral infection with aging is likely due to a defect in pDC IFN-α production on a per cell basis.
Figure 4.
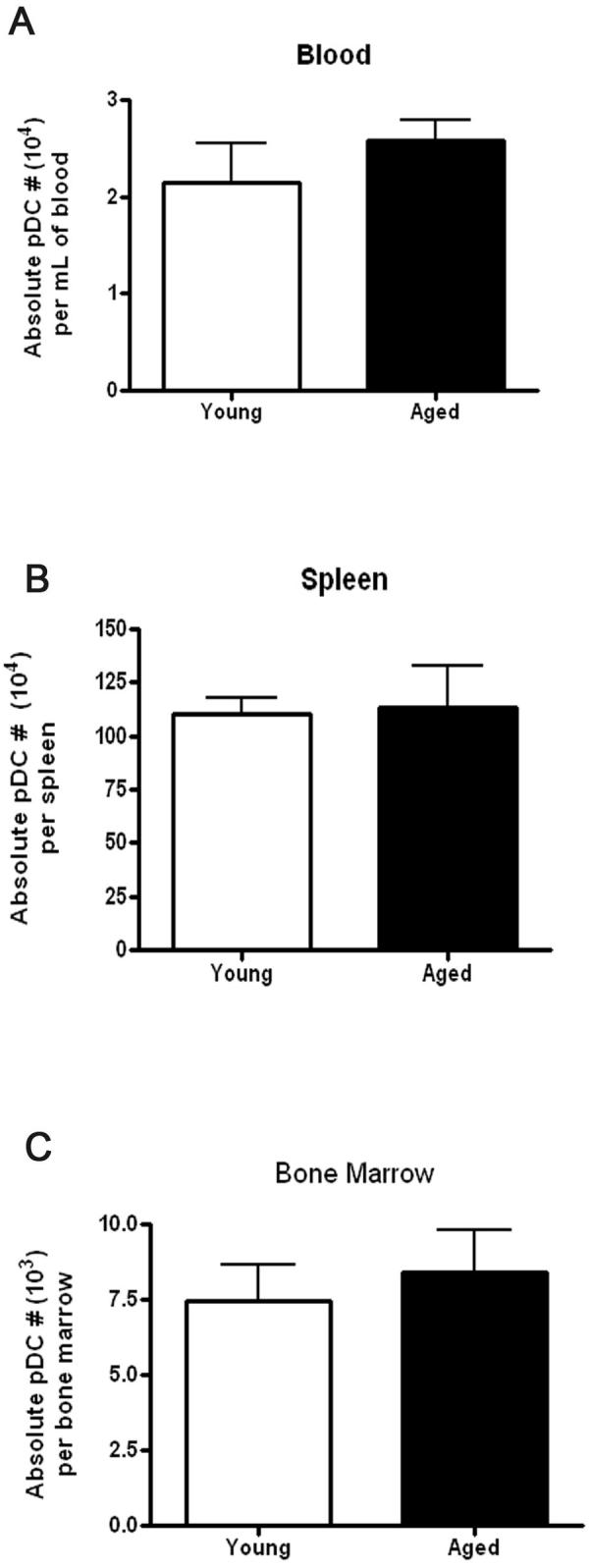
Aged mice maintain similar numbers of pDC within the blood, spleen and bone marrow as compared to young mice. The pDCs from blood, bone marrow, and spleen from young and aged mice were enumerated using flow cytometry (PDCA-1+ staining; N>6/group).
Adoptive transfer of young pDCs into aged mice augments IFN-α during HSV-2 viral infection and increases viral clearance
We next examined whether the reduced IFN-α response and impaired clearance of HSV-2 in aged hosts could be recovered by adoptive transfer of young cells. In preliminary experiments, we established the efficacy of augmenting IFN-α responses by adoptively transferring young wild-type bone marrow cells into TLR9-deficient hosts. Specifically, intravenous administration of CpG-A 72h post-transfer of bone marrow cells resulted in detectable serum IFN-α levels in the TLR9-deficient mice that received transfer of wild-type cells when compared to control TLR9-deficient mice that received transfer of TLR9-deficient cells (data not shown). Hence, aged mice received either young whole bone marrow cells or young bone marrow cells that were depleted of pDCs. The aged hosts that received young whole bone marrow cells produced a superior IFN-α response and a reduction in viral load when compared to aged hosts that received young pDC-depleted bone marrow cells (Fig. 5A-B). We next examined whether the presence of young pDCs transferred to aged hosts were sufficient to elevate IFN-α responses and lead to increased viral clearance. Indeed, transfer of young but not aged pDCs to aged hosts led to elevated IFN-α responses and reduced viral load during HSV-2 infection (Fig. 5C-D). As young pDCs were functional in an aged environment, our results indicate that aging does not induce factors that suppress pDC responses to HSV-2 infection.
Figure 5.

Transfer of young whole bone marrow cells into aged hosts augments IFN-α levels and viral clearance in HSV-2-infected aged hosts in a pDC-dependent fashion. Aged C57BL/6 mice were adoptively transferred i.v. with 5×106 whole or pDC-depleted bone marrow cells isolated from young C57BL/6 mice. A, Mice were infected with HSV-2 at 72h post-transfer, and serum IFN-α levels were measured by ELISA. B, Liver and spleen samples were isolated at 96h post-HSV-2 infection, and viremia was measured. *p<0.05 (Student's t-test). C, Aged mice were adoptively transferred with either 1×104 young or aged pDCs from syngeneic donors 72 hours prior to infection with HSV-2. IFN-α responses were measured at 24h post-infection. Non-infected (denoted as control) and non-adoptively transferred infected controls (denoted as HSV-2) are shown. D, Viral load was measured in the liver and spleen from aged mice that were adoptively transferred with aged or young pDCs (N=3/group). Results are representative of data from at least two independent experiments with similar results.
IRF-7 upregulation is impaired with aging either in response to TLR9 activation or IFN-α stimulation
TLR9-mediated IFN-α induction in pDCs is dependent on the adaptor protein MyD88 and subsequent assembly of a signaling complex that upregulates IRF-7 (46, 47). Indeed, IRF-7 is essential for IFN-α production during infection with HSV-1, a virus that also activates TLR9 (46). IRF-7 upregulation also occurs in response to stimulation via the IFNα/β receptor. Importantly, after TLR9 activation, type I IFN production in pDCs is sustained by a positive feedback loop in which IFN-α activates the IFNα/β receptor to maintain IRF-7 expression (46).
Thus, we examined whether aging altered signaling in adaptors downstream of either the TLR9 receptor, the IFNα/β receptor, or IRF-7, an adaptor common to both pathways. The fact that IL-12p40 production was preserved in aged pDCs (Fig. 1B) indicated that adaptors downstream of TLR9 that were independent of the IFNα/β receptor were likely intact during aging. Indeed, upon TLR9 activation via CpG-A, aged pDCs were able to upregulate MyD88, TRAF-6, IRAK2, IRAK4, IRAKM, and IKKα similarly to young pDCs (data not shown and Fig. 6B IKKα). We next determined whether IFNα/β receptor expression was reduced in aged pDCs at rest and following TLR activation. The expression of this receptor was preserved in aged pDCs under these conditions, according to flow cytometric analysis and RT-PCR (data not shown). Furthermore, gene expression analysis indicated that aged pDCs exhibit similar expression of signal adaptors downstream of the IFNα/β receptor, including Stat 1, Stat 2, Jak 1, Tyk2, and IKKε, as compared to young cells at rest and during activation with recombinant IFN-α (data not shown). Thus, no evidence indicating that aging impaired the basal expression and upregulation of adaptors downstream of either the TLR9 or the IFNα/β receptor but upstream of IRF-7 was found.
Figure 6.

Aged pDCs manifest an impaired ability to upregulate IRF-7 or PI3-kinase during either TLR9 or IFN-α receptor stimulation. A, Aged or young pDCs were stimulated with CpG-A, IFN-α, or both, and then the upregulation of IRF-7 mRNA levels from each experimental group were measured, normalized to 18sRNA, and compared to young controls. B, The same groups as in (A) except that IRF-7 or Iκκα protein levels were measured by western blot. Results are representative of data from at least two independent experiments with similar results (N=3/group). C, The experimental groups are the same as in (A), except that PI3-kinase levels were measured by real-time PCR. D, IRF-7 nuclear translocation was analyzed by western blotting. Young and aged nuclear protein fractions were electrophoresed on two separate SDS-PAGE gels that were run concurrently under the same conditions. Results are representative of data from at least two independent experiments with similar results (N=3/group). E, Nuclear IRF-7 activity was measured in young and aged pDCs by ELISA.
As a result, we next examined whether aging impairs IRF-7 upregulation in response to either TLR9 or IFN-α activation. Neither IFN-α treatment nor TLR9 activation via CpG-A induced IRF-7 gene or protein upregulation in aged pDCs, whereas either condition induced IRF-7 upregulation in young pDCs (Fig. 6A-B). Furthermore, combined treatment with IFN-α and CpG-A also failed to upregulate IRF-7 in aged pDCs, whereas this combination effectively induced IRF-7 upregulation in young pDCs (Fig. 6A-B). These data demonstrate that aging impairs IRF-7 upregulation in response to either TLR9 or IFN-αβ receptor activation.
Recent evidence has indicated that PI3-kinase is critical for the nuclear translocation of IRF-7 by pDCs (48). Furthermore, a recent study found that aging impairs PI3-kinase signaling in human myeloid DCs (49). In our study, PI3-kinase upregulation was impaired in aged pDCs relative to young pDCs during activation with either CpG-A or IFN-α (Fig. 6C). In response to IFN-α treatment, aged pDCs downregulated PI3-kinase, whereas IFN-α was upregulated in young pDCs (Fig. 6C). Young pDCs were able to translocate IRF-7 to the nucleus during either CpG-A or IFN-α treatment, whereas aged pDCs failed to do so (Fig. 6D). This result was expected given the fact that aged pDCs also failed to upregulate cytosolic IRF-7 during these experimental conditions. Furthermore, nuclear IRF-7 transcriptional activity was reduced in aged pDCs after CpG-A activation as compared that in young pDCs (Fig. 6E). In sum, aging impairs PI3-kinase upregulation and IRF-7 upregulation during TLR9 or IFNαβ receptor activation.
Aging increases reactive oxygen species (ROS) levels in pDCs, and reducing ROS in aged pDCs augments IFN-α production in response to TLR9 activation
One of the leading theories of age-induced cellular damage is the accumulation of damaging ROS (32, 33). Therefore, we assessed whether aged pDCs exhibit elevated levels of ROS as compared to young pDCs. Indeed, aged pDCs displayed increased amounts of ROS at rest and during TLR9 activation as compared to young pDCs (Fig. 7A-B). To determine whether increased ROS in aged pDCs influenced the type I IFN response during TLR9 activation, aged pDCs were pre-treated with the anti-oxidant NAC, which has been shown to reduce ROS (37, 38, 50). NAC treatment reduced ROS and increased IFN-α production by TLR9-activated aged pDCs as compared to aged pDCs activated with TLR9 in the absence of NAC (Fig. 7C-D). Young pDCs that were pre-treated with NAC also exhibited an increased IFN-α response compared to control treated cells (Fig. 7D). These data indicate that reducing ROS in aged pDCs augments IFN-α production during TLR9 activation.
Figure 7.

Aging leads to increased oxidative stress in pDCs at rest and during CpG-A activation. A-B, Aged pDCs exhibit evidence of increased oxidative stress (measured by DHE fluorescence) at rest (A) and during CpG-A (B) stimulation. Grey line represents young cells, and the black line indicates aged cells. Data represents at least two independent experiments with similar results (N=3/group). C, Pre-treating aged pDCs with NAC (black line) reduces the ROS profile during CpG-A activation compared to aged pDCs pre-treated with PBS (grey line). Data represents at least two independent experiments with similar results (N=3/group). D, NAC treatment augments IFN-α responses in aged pDCs during CpG-A stimulation. *p<0.05 Student's t-test. Young and aged pDCS were pre-treated for 1 h at 37 °C with or without 1 μM NAC diluted in normal cell culture media. Cells were cultured with 10 μg of CpG-A at 37 °C for 18h. NAC control refers to cells that were pre-treated with NAC but not activated with CpGA. Control cells that were neither pre-treated with NAC nor activated with CpG-A are denoted as control. Data represents at least two independent experiments with similar results (N=3/group).
We next employed calorie restriction (CR) as a model to reduce oxidative stress during the lifespan of mice. CR has been shown to decrease ROS generation and increase ROS clearance pathways, and this model has been established to reduce age-induced oxidative stress in yeast, C. elegans, Drosophila, and rodents (33). Furthermore, CR leads to increased lifespan in all of these organisms. Hence, we compared TLR9 responses in pDCs purified from aged CR, and AL-fed aged and AL-fed young mice. First, we demonstrated that pDCs from aged CR mice manifested reduced oxidative stress at rest and during TLR9 stimulation compared to pDCs from aged AL-fed controls (Fig. 8A-B). The pDCs purified from aged CR mice produced more IFN-α than pDCs from aged ALfed mice in response to in vitro CpG-A stimulation or HSV-2 infection (Fig. 8C-D); however, the responses from pDCs from aged CR mice remained inferior to the responses of pDCs from AL-fed young mice (Fig. 8C-D). These results demonstrate that CR reduced age-induced ROS in pDCs and partially recovered the defective IFN-α response to TLR9 activation in aged pDCs.
Figure 8.

CR partly recovers the age-induced defective type I IFN signaling response in pDC. A-B, The pDCs harvested from CR aged mice exhibit reduced oxidative stress (grey line) at rest and during 18h of activation with CpG-A as compared to pDCs harvested from AL fed aged controls (black line). Young AL fed controls are shown (dashed line). Data represent at least four independent experiments with similar results (N=3/group). C-D, The pDCs harvested from CR aged mice displayed augmented IFN-α responses during in vitro activation with 18h CpG-A or in vivo infection with HSV-2 as compared to pDCs from AL-fed aged and young controls. Data represent at least two independent experiments with similar results (N=3/group). E, The pDCs from CR aged mice exhibit increased IRF-7 gene expression during either CpG-A or IFN-α stimulation as compared to pDCs from AL-fed aged mice. Gene expression from pDCs harvested from young AL-fed controls is shown. Data represent at least three independent experiments with similar results (N=3/group). F, The pDCs from CR aged mice exhibit increased IRF-7 activity, expressed as absorbance per μg nuclear extract, during CpG-A activation compared to pDCs from age-matched AL-fed mice. The pDCs from young AL-fed mice are shown. G, The pDCs from CR aged mice exhibit increased PI3-kinase upregulation during either CpG-A or IFN-α stimulation as compared to pDCs from ALfed aged mice. Gene expression from pDCs harvested from young AL-fed controls is shown. Data represents at least three independent experiments with similar results (N=3/group). H, The pDCs from CR aged mice exhibit increased phosphorylation of AKT during CpG-A activation compared to pDCs from age-matched AL-fed mice. The pDCs from young AL-fed mice are shown.
Next, we examined whether pDCs purified from aged CR mice manifested altered PI3-kinase and IRF-7 upregulation as compared to pDCs harvested from aged AL-fed mice following either CpG-A or IFNαβ receptor activation. The pDCs from aged CR mice exhibited superior IRF-7 upregulation and IRF-7 activity compared to pDCs from aged AL-fed mice during TLR9 activation with CpG-A, although the response was not quite as robust as pDCs from young AL-fed controls (Fig. 8E-F). Furthermore, PI3-kinase upregulation was elevated in pDCs from aged CR mice relative to pDCs from aged AL-fed mice under these conditions. In fact, PI3-kinase upregulation by pDCS from aged CR mice was similar to pDCs from young AL-fed mice (Fig. 8G). Additionally, pDCs from aged CR mice displayed superior phosphorylation of AKT, which signals downstream of PI3-kinase, during CpG-A activation as compared to pDCs from aged AL-fed mice and exhibited a similar response to that of pDCs from young ALfed mice (Fig 8H). In sum, our results indicate that the age-induced impairment in the upregulation of P13-kinase and IRF-7 in pDCs during TLR9 activation is, at least partly, recovered by CR.
Discussion
In our study, we first examined the impact of aging on pDC function and whether any age-induced defects within pDCs impaired host defense to viral infection. Using CpG-A or HSV-2 virus, which both activate pDCs via TLR9, we found that aged pDCs exhibit an impaired ability to produce IFN-α, but not IL-12p40, as compared to pDCs from young animals. During HSV-2 infection, defective pDC function was responsible for an impaired ability of aged mice to clear the virus. Thus, our study provides evidence that defective type I IFN responses by aging pDCs impairs host defense to viral infection in these experimental models.
Gene and protein analyses were employed to uncover the age-related defects within pDCs during TLR9 stimulation. Aged pDCs displayed an impaired ability to upregulate IRF-7 and PI3-kinase during either TLR9 or IFNα/β receptor activation. We did not find evidence that adaptors upstream of IRF-7 that were unique to either the TLR9 or IFNαβ receptor pathways were altered by aging under our experimental conditions. Therefore, our study has demonstrated that aging impairs the upregulation of PI3-kinase and IRF-7 within pDCs during TLR9 or IFNα/β receptor activation.
We also noted that aged pDCs exhibited increased oxidative stress both at rest and during TLR9 activation compared to young pDCs (Fig. 7). This result is in agreement with prior work indicating that aging leads to accumulation of damaging ROS, which augment oxidative stress and subsequently impair cellular functions (33).Reducing ROS by pre-treating aged pDCs with NAC increased TLR9-induced IFN-α responses by these cells. Furthermore, pDCs from aged CR mice showed a partly recovered phenotype. In fact, these pDCs had reduced ROS compared to pDCs from AL-fed age-matched controls (Fig. 8), and these pDCs demonstrated elevated IFN-α levels and IRF-7 upregulation and activity during TLR9 activation as compared to pDCs from aged AL-fed controls (Fig. 8). Prior work indicates that CR may mediate its effects by reducing ROS generation or increasing the clearance of ROS, in part, by improving mitochondrial function (33). Our data support these findings as pDCs from aged CR mice exhibited reduced ROS as compared to pDCs from aged AL-fed controls (Fig. 8A-B). Thus, age-induced elevations in ROS levels may lead to IRF-7 protein degradation; however, the possibility that CR may be mediating its effects independently of ROS reduction cannot be excluded by this study. In addition, perhaps other age-induced cellular perturbations besides elevated ROS levels may be important for the impaired type I IFN response by aged pDCs, since NAC treatment reduced ROS levels in aged pDCs but did not recover the response to the same level noted in non-NAC-treated, CpG-A-activated young pDCs (Fig. 7D). Future studies will be required to determine the mechanism of susceptibility of the type I IFN signaling pathway to the damaging effects of ROS generation during aging. Overall, our study suggests that increased oxidative stress during aging leads to impaired type I IFN function in pDCs in response TLR9 activation.
IFN-α produced by pDCs is essential for host defense against certain viruses (8, 11). Importantly, reduced IFN-α levels result in increased mortality during viral infections (51, 52). Furthermore, the early release of IFN-α during viral infection leads to direct inhibition of viral replication in addition to activation of NK cells, cytotoxic T lymphocytes, and macrophages to eliminate virally infected cells (51-53). Here, we demonstrated that adoptive transfer of young pDCs into aged hosts augmented IFN-α responses and improved viral clearance during HSV-2 infection. These results suggest that augmenting the IFN-α response in aged hosts may be a potential therapeutic approach to protect aged hosts from viral infections.
Aged pDCs exhibited impaired responses to TLR7 activation in vitro (Fig. 3). We found that during infection with MCMV, a virus that requires pDCs for host defense (41) and has recently been shown to activate both TLR7 and TLR9 (43), aging also impaired IFN-α responses and viral clearance (Fig. 3). Additionally, a prior report demonstrated that aged mice produced reduced type I IFN response after stimulation with poly I:C, a substance that activates TLR3 (54), although this study did not focus on pDCs. Overall, our study indicates that aging impairs both TLR7 and TLR9 responses in pDCs.
A prior clinical report indicated that pDCs purified from the peripheral blood of older people yielded impaired IFN-α responses as compared to cells from younger control subjects (31). This result agrees with our study and suggests that the findings of our report may be translatable to humans. Thus, confirmation of our results in human cells will be critical.
Our study clearly demonstrates that aged pDCs exhibit a defective response to TLR9 activation. Other studies have indicated that aging also impairs the function of other cellular components of the immune system, including T cells, B cells, macrophages, and NK cells. In our study, however, viral clearance was augmented in aged hosts by the adoptive transfer of young bone marrow cells or young pDCs but not with pDC-depleted young bone marrow. These data imply that pDCs are both necessary and sufficient for the adoptive transfer to alter the phenotype in our experimental model. Recently, innate cytosolic receptors of viral infection, such as the RNA helicase system, have been identified (55). Future investigation will be required to determine whether aging alters this pathway of innate viral recognition and to integrate the many alterations that occur in the immune response with aging. Such avenues of investigation will have important implications for the design of potential vaccines to augment immune function with aging.
In conclusion, our study indicates that impaired IFN-α responses within pDCs lead to defective host defense to viral infections. Importantly, aging impaired the upregulation of IRF-7, a key signal adaptor in the type I IFN signaling pathway, in response to TLR9 activation. This fundamental information helps to clarify the complex interaction between aging and host immunity and also provides a potential explanation for the increased susceptibility to viral infections observed in older people.
Acknowledgments
This work was supported in part by NIH grants AG028082 and AG026772 to DRG. HWSD is supported by NIH grant T32AG019134. WEW is supported by NIH grant 5T32HL007778.
Non-standard abbreviations
- AL
ad libitum
- CR
calorie restriction
- DHE
dihydroethidium
- HSV-2
herpes simplex virus-2
- IRF
interferon regulatory factor
- NAC
N-acetyl cysteine
- pDC
plasmacytoid DC
- PI3-kinase
phosphoinositide 3-kinase
- ROS
reactive oxygen species
References
- 1.Pawelec G, Barnett Y, Forsey R, Frasca D, Globerson A, McLeod J, Caruso C, Franceschi C, Fulop T, Gupta S, Mariani E, Mocchegiani E, Solana R. T cells and aging, January 2002 update. Front Biosci. 2002;7:d1056–1183. doi: 10.2741/a831. [DOI] [PubMed] [Google Scholar]
- 2.Linton P-J, Dorschkind K. Age related changes in lymphocyte development and function. Nat Immunol. 2004;5:133–139. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 3.Yung R. Changes in immune function with age. Rheum. Dis. Clin. N. Am. 2000;26:455–473. doi: 10.1016/s0889-857x(05)70151-4. [DOI] [PubMed] [Google Scholar]
- 4.Weng NP. Aging of the Immune System: How Much Can the Adaptive Immune System Adapt? Immunity. 2006;24:495. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haynes L, SL S. Why Aging T cells Fail: Implications for vaccination. Immunity. 2006;24:663–666. doi: 10.1016/j.immuni.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sambhara S, Kurichh A, Miranda R, James O, Underdown B, Klein M, Tartaglia J, Burt D. Severe Impairment of Primary but Not Memory Responses to Influenza Viral Antigens in Aged Mice: Costimulation in Vivo Partially Reverses Impaired Primary Immune Responses. Cellular Immunology. 2001;210:1. doi: 10.1006/cimm.2001.1799. [DOI] [PubMed] [Google Scholar]
- 7.Solana R, Pawelec G, Tarazona R. Aging and Innate Immunity. Immunity. 2006;24:491. doi: 10.1016/j.immuni.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 9.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O'Garra A, Biron C, Briere F, Trinchieri G. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 10.Asselin-Paturel C, Trinchieri G. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005;202:461–465. doi: 10.1084/jem.20051395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 12.Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 13.Wagner H. The immunobiology of the TLR9 subfamily. Trends Immunol. 2004;25:381–386. doi: 10.1016/j.it.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 15.Pichlmair A, Reis e Sousa C. Innate Recognition of Viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Shao Y, Bennett TA, Shankar RA, Wightman PD, Reddy LG. The functional effects of physical interactions among Toll-like receptors 7, 8, and 9. J Biol Chem. 2006;281:37427–37434. doi: 10.1074/jbc.M605311200. [DOI] [PubMed] [Google Scholar]
- 17.Wang JP, Asher DR, Chan M, Kurt-Jones EA, Finberg RW. Cutting Edge: Antibody-mediated TLR7-dependent recognition of viral RNA. J Immunol. 2007;178:3363–3367. doi: 10.4049/jimmunol.178.6.3363. [DOI] [PubMed] [Google Scholar]
- 18.Heil F, Ahmad-Nejad P, Hemmi H, Hochrein H, Ampenberger F, Gellert T, Dietrich H, Lipford G, Takeda K, Akira S, Wagner H, Bauer S. The Toll-like receptor 7 (TLR7)-specific stimulus loxoribine uncovers a strong relationship within the TLR7, 8 and 9 subfamily. Eur J Immunol. 2003;33:2987–2997. doi: 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
- 19.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 20.Latz E, Visintin A, Espevik T, Golenbock DT. Mechanisms of TLR9 activation. J Endotoxin Res. 2004;10:406–412. doi: 10.1179/096805104225006525. [DOI] [PubMed] [Google Scholar]
- 21.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like Receptor 9-mediated Recognition of Herpes Simplex Virus-2 by Plasmacytoid Dendritic Cells. J. Exp. Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J Immunol. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 23.Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, Lee K-D, Coffman RL, Barrat FJ. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J. Exp. Med. 2006;203:1999–2008. doi: 10.1084/jem.20060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EEM, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. MyD88-Dependent and -Independent Murine Cytomegalovirus Sensing for IFN-{alpha} Release and Initiation of Immune Responses In Vivo. J Immunol. 2005;175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 25.Montoya CJ, Jie HB, Al-Harthi L, Mulder C, Patino PJ, Rugeles MT, Krieg AM, Landay AL, Wilson SB. Activation of plasmacytoid dendritic cells with TLR9 agonists initiates invariant NKT cell-mediated crosstalk with myeloid dendritic cells. J Immunol. 2006;177:1028–1039. doi: 10.4049/jimmunol.177.2.1028. [DOI] [PubMed] [Google Scholar]
- 26.Boehmer ED, Meehan MJ, Cutro BT, Kovacs EJ. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Age Develop. 2005;126:1305. doi: 10.1016/j.mad.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Plowden J, Renshaw-Hoelscher M, Gangappa S, Engleman C, Katz JM, Sambhara S. Impaired antigen-induced CD8+ T cell clonal expansion in aging is due to defects in antigen presenting cell function. Cell. Immunol. 2004;229:86–92. doi: 10.1016/j.cellimm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Tesar BM, Walker WE, Unternaehrer J, Joshi NS, Chandele A, Haynes L, Kaech S, Goldstein DR. Murine myeloid dendritic cell-dependent toll-like receptor immunity is preserved with aging. Aging Cell. 2006;5:473–486. doi: 10.1111/j.1474-9726.2006.00245.x. [DOI] [PubMed] [Google Scholar]
- 29.Teig N, Moses D, Gieseler S, Schauer U. Age-related changes in human blood dendritic cell subpopulations. Scand J Immunol. 2002;55:453–457. doi: 10.1046/j.1365-3083.2002.01068.x. [DOI] [PubMed] [Google Scholar]
- 30.Abb J, Abb H, Deinhardt F. Age-related decline of human interferon alpha and interferon gamma production. Blut. 1984;48:285–289. doi: 10.1007/BF00320399. [DOI] [PubMed] [Google Scholar]
- 31.Shodell M, Siegal F. Circulating, Interferon-producing plasmacytoid dendritic cells decline during human ageing. Scand J. Immunol. 2002;56:518–521. doi: 10.1046/j.1365-3083.2002.01148.x. [DOI] [PubMed] [Google Scholar]
- 32.Wallace DC. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Ann Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech Ageing Dev. 2004;125:811. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 34.Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting Edge: Plasmacytoid Dendritic Cells Provide Innate Immune Protection against Mucosal Viral Infection In Situ. J Immunol. 2006;177:7510–7514. doi: 10.4049/jimmunol.177.11.7510. [DOI] [PubMed] [Google Scholar]
- 35.Jonjic S, Krmpotic A, Arapovic J, UH K. Dissection of the antiviral NK cell response by MCMV mutants. Methods Mol Biol. 2008;415:127–149. doi: 10.1007/978-1-59745-570-1_8. [DOI] [PubMed] [Google Scholar]
- 36.Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O'Garra A, Vicari A, Trinchieri G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J. Exp. Med. 2005;201:1157–1167. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homandberg GA, Hui F, Wen C. Fibronectin fragment mediated cartilage chondrolysis. I. Suppression by anti-oxidants. Biochim Biophys Acta. 1996;1317:134–142. doi: 10.1016/s0925-4439(96)00046-4. [DOI] [PubMed] [Google Scholar]
- 38.Gressier B, Cabanis A, Lebegue S, Brunet C, Dine T, Luyckx M, Cazin M, Cazin JC. Decrease of hypochlorous acid and hydroxyl radical generated by stimulated human neutrophils: comparison in vitro of some thiol-containing drugs. Methods Find Exp Clin Pharmacol. 1994;16:9–13. [PubMed] [Google Scholar]
- 39.Wendland M, Czeloth N, Mach N, Malissen B, Kremmer E, Pabst O, Forrster R. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc Natl Acad Sci U S A. 2007;104:6347–6352. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fink B, Laude K, McCann L, Doughan A, Harrison DG, Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am J Physiol Cell Physiol. 2004;287:C895–902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- 41.Krug A, French AR, Barchet W, Fischer JAA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-Dependent Recognition of MCMV by IPC and DC Generates Coordinated Cytokine Responses that Activate Antiviral NK Cell Function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M. Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. 2008;180:5799–5803. doi: 10.4049/jimmunol.180.9.5799. [DOI] [PubMed] [Google Scholar]
- 44.Crozat K, Beutler B. TLR7: A new sensor of viral infection. Proc Natl Acad Sci U S A. 2004;101:6835–6836. doi: 10.1073/pnas.0401347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 46.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 47.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Role of a transductionaltranscriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guiducci C, Ghirelli C, Marloie-Provost M-A, Matray T, Coffman RL, Liu Y-J, Barrat FJ, Soumelis V. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J. Exp. Med. 2008;205:315–322. doi: 10.1084/jem.20070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agrawal A, Agrawal S, Cao J-N, Su H, Osann K, Gupta S. Altered Innate Immune Functioning of Dendritic Cells in Elderly Humans: A Role of Phosphoinositide 3-Kinase-Signaling Pathway. J Immunol. 2007;178:6912–6922. doi: 10.4049/jimmunol.178.11.6912. [DOI] [PubMed] [Google Scholar]
- 50.Luo J, Tsuji T, Yasuda H, Sun Y, Fujigaki Y, Hishida A. The molecular mechanisms of the attenuation of cisplatin-induced acute renal failure by N-acetylcysteine in rats. Nephrol. Dial. Transplant. 2008;23:2198–2205. doi: 10.1093/ndt/gfn090. [DOI] [PubMed] [Google Scholar]
- 51.Kadowaki N, Antonenko S, Lau JY-N, Liu Y-J. Natural Interferon {alpha}/{beta}-producing Cells Link Innate and Adaptive Immunity. J. Exp. Med. 2000;192:219–226. doi: 10.1084/jem.192.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadowaki N, Liu Y-J. Natural type I interferon-producing cells as a link between innate and adaptive immunity. Hum Immunol. 2002;63:1126–1132. doi: 10.1016/s0198-8859(02)00751-6. [DOI] [PubMed] [Google Scholar]
- 53.Bogdan C. The function of type I interferons in antimicrobial immunity. Curr Opin Immunol. 2000;12:419–424. doi: 10.1016/s0952-7915(00)00111-4. [DOI] [PubMed] [Google Scholar]
- 54.Jiang J, Gross D, Nogusa S, Elbaum P, Murasko DM. Depletion of T Cells by Type I Interferon: Differences between Young and Aged Mice. J Immunol. 2005;175:1820–1826. doi: 10.4049/jimmunol.175.3.1820. [DOI] [PubMed] [Google Scholar]
- 55.Stetson DB, Medzhitov R. Antiviral defense: interferons and beyond. J. Exp. Med. 2006;203:1837–1841. doi: 10.1084/jem.20061377. [DOI] [PMC free article] [PubMed] [Google Scholar]