

Abstract
Epithelial-mesenchymal transition (EMT) has been considered essential for metastasis, a multistep process including local invasion, intravasation, extravasation, and proliferation at distant sites. However, controversy remains as to whether EMT truly happens and how important it is to metastasis. We studied the involvement of EMT in individual steps of metastasis and found that p12CDK2-AP1, a down-stream effector of TGF-β, induced EMT of hamster cheek pouch carcinoma-1 cells by promoting the expression of Twist2. EMT cells have an increased invasive but decreased metastatic phenotype. When subcutaneously inoculated, both EMT and non-EMT cells established primary tumors, but only EMT cells invaded into the adjacent tissues and blood vessels, however neither cells formed lung metastases. When intravenously inoculated, only non-EMT cells established lung metastases. Moreover, subcutaneous inoculation of a mixture of the two cell types resulted in intravasation of both cell types and formation of lung metastasis from non-EMT cells. Our results allowed us to propose a novel model for the role of EMT in cancer metastasis. We demonstrated that EMT and non-EMT cells cooperate to complete the spontaneous metastasis process. We thus hypothesize that EMT cells are responsible for degrading the surrounding matrix to lead the way of invasion and intravasation. Non-EMT cell then enter the blood stream and reestablish colonies in the secondary sites.
Keywords: Epithelial-mesenchymal transition, Twist2, Invasion, Metastasis, p12CDK2-AP1, E-cadherin
Introduction
EMT is a morphogenetic process in which cells lose their epithelial characteristics such as cell polarity, cell-cell contact and gain mesenchymal properties such as increased motility (1). EMT was originally described during embryogenesis (2) as a developmental process such as gastrulation, renal organogenesis and the formation of neural crest (3). Accumulating evidence has shown that EMT is reactivated in a variety of diseases including cancer progression and metastasis (4). Metastasis is a complex, multi-step process including detachment and migration away from the primary tumor tissues, local invasion of the surrounding matrix, intravasation, survival in the circulation, extravasation, survival and proliferation at the metastatic site (5). It is well documented that during progression to metastatic competence, carcinoma cells change their adhesive properties, activate proteolysis and became motile, allowing them to leave the primary tumor and establish secondary tumor in distant organs (6).
EMT of carcinoma cells is often accompanied by lost expression of E-cadherin, a known tumor suppressor (7). Although irreversible mechanism of E-cadherin inactivation such as LOH of 16q22.1 containing the E-cadherin locus and mutations in its coding region have been demonstrated in human cancer (8, 9), its down-regulation during EMT is believed to be mediated by promoter methylation (10) or by up-regulation of E-box-binding transcription repressors including Twist1, Snail, Slug, ZEB-1, ZEB-2(SIP), E12/E47, CBF-A, FOXC2, Goosecoid, HOXB7, and DeltaEF1. These transcription repressors are often modulated by one of the classic signaling pathways including the WNT, TGF-β, Hedgehog, Notch, and receptor tyrosine kinase that are often disregulated in cancer and have been shown to stimulate EMT (11).
TGF-β is a pluripotent factor that can elicit multiple cellular responses in epithelial cells (12). The role of TGF-β in inducing EMT has been well demonstrated and has been shown to be mediated by its cell surface receptor and its down-stream effector Smad (13). Smad pathway induces the expression of the high mobility group A2 (HMGA2) gene (14), a nuclear factor that is necessary and sufficient for TGF-β-induced EMT by linking the TGF-β signaling pathway with the EMT-inducing transcription factors Snail1, Snail2 and Twist.
On the other hand, the growth suppressor activity of TGF-β and its relationship with EMT and tumor invasion and metastasis is less well studied (15). One established pathway for the growth inhibitory activity of TGF-β is through the induction of p12CDK2-AP1 (p12, Doc-1), a growth suppressor originally isolated from hamster normal oral keratinocytes by subtractive hybridization (16). TGF-β activates Smad 3 and 4 thereby inducing the expression of p12 (17) that, in turn, mediates growth inhibition by interacting with DNA polymerase α/primase (18) and CDK2 (19). Here we report that p12 induces EMT of hamster cheek pouch carcinoma-1 (HCPC-1) cells. EMT cells have increased ability of local invasion but diminished capacity of establishing distant metastasis. Neither non-EMT cells nor EMT cells alone is able to metastasize due to the defect in invasion and in re-growth in distant organs, respectively. However, the mixture of two cell types is able to complete the entire process of spontaneous metastasis. Our results suggest a new model for the role of EMT in tumor metastasis in which the EMT cells lead the way of invasion so that the non-EMT cells can enter the blood stream and re-establish growth in distant organs.
Materials and Methods
Cells
HCPC-1 cells were cultured in DMEM + 10% FBS (16). To distinguish p12 and vector transfectants, they were co-transfected with a green fluorescence protein-expressing plasmid, pEGFPC-1, and a red fluorescence protein-expressing plasmid, pCI-neo-DsRed2, respectively, with a hygromycin-resistent gene, pTk-Hyg. Stable double transfectants were selected with 0.5 mg/ml hygromycine B for 2 weeks. The p12 and vector control transfectants were therefore labeled with green and red fluorescence, respectively. They were used throughout this work except in the MET experiment where p12 transfectants that had not been transfected with pEGFPC-1 were co-transfected with E-cadherin and GFP expression plasmids.
Inducible p12 antisense HaCaT cell line (ip12(−)HaCaT) and vector control cell line (pMTCB6+-HaCaT) were prepared and cultured as described previously (17). p12 antisense expression was induced by 25 μM ZnSO4 for 24 h. To induce EMT of the vector and antisense transfectants of HaCaT cells, a mixture of TGF-β (10 ng/ml) and EGF (30 ng/ml) was added and the cells were incubated in the presence of Zn2+ (25 μM) for 48 h (20).
qRT-PCR assay of E-cadherin
The hamster E-cadherin primer set was designed based on the hamster E-cadherin cDNA sequence. F, 5′-CTGCAGGTCTCATCATGGA-3′; R, 5′- ACCTGTAGACCTCGGCACTG-3′. β-actin was used as an internal control: F, 5′- CACCATGAAGATCAAGATCATTGC-3′; R, 5′- GGCCGGACTCATCGTACTCCTGC-3′.
RT-PCR analysis of E-cadherin repressors
The mRNA levels of E-cadherin repressors including E12/E47, Snail, Slug, Twist1, Twist2, ZEB-1, and ZEB-2 were analyzed by RT-PCR with the primer sets shown bellow. E12/E47: F, 5′-TCAGGACGGTTCTGGATGAGACAG-3′; R, 5′-TTTTCTGCTTGGTCACTGGGGC-3′. Snail: F, 5′-CGAGCCATAGAACTAAAGCCAACC-3′; R, 5′-ATGTGTCCAGTAACCACCCTGCTG-3′. Slug: F, 5′-AACTTCAAACATTCCTGGTGCG-3′; R, 5′-TGGAGGGGCATTGTATTACTGC-3′. Twist1: F, 5′-ACGCTGCCCTCGGACAAG-3′; R, 5′-CCCTCCATCCTCCAGACGG-3′. Twist2: F, 5′-GGCAAGAAGGGCAGTCCG-3′; R, 5′-TTGTCAGAGGGAAGTGTGGGG-3′. ZEB-1: F, 5′-TGGGGCATCTCACACTTTTGTTC-3′; R, 5′-TTCATCACCTGGGTCCGTAATAAC-3′. ZEB-2: F, 5′-AAGTGTGTGTGCTCTGAAGCCC-3′, R, 5′-CTGCTGAGT GTGTTTGTTTGGATG-3′.
Twist2 siRNA
Twist2 specific siRNA (sense, 5′- ACAGUAAGAAGUCGAGCGAAG AUGG-3′; antisense, 5′-CCAUCUUCGCUCGACUUCUUACUGUAG-3′) was designed using a DsiRNA Web Design Tool from Integrated DNA Technologies, Inc. The p12 transfectants of HCPC-1 cells (∼90% confluence) were transfected with 1 nM siRNA duplex together with 0.8 μg pEGFP-C1 plasmid using Lipofectamine 2000. After 48 h, the cells were fixed with ethanol and the transfectants were identified by green fluorescence.
Tumor invasion and metastasis assays
Animal experiments were approved by IACUC of Harvard Medical School. For spontaneous metastasis assay, tumor cells were inoculated subcutaneously. Vector and p12 transfectants, or a 1:1 mixture of the two cell types, 1 × 106 each, were inoculated subcutaneously into the back of BALB/c athymic mice. For direct lung metastasis assay, tumor cells were injected intravenously. Vector or p12 transfectants, or the E-cadherin re-expressed p12 transfectants, 1 × 106 cells, were injected into the tail vein directly. Blood samples were collected weekly from the eye corner and analyzed for the existence of tumor cells by PCR analysis of GFP and DsRed sequences. Four weeks after tumor cell inoculation, the animals were sacrificed; primary tumors and the lung tissues were removed and processed for histology, immunofluorescence (IF), and immunohistochemistry (IHC) examinations. A total of 0.5-1.0 ml of blood samples were collected from the hearts when animals were sacrificed. These blood samples were cultured for 2-3 weeks in vitro in an attempt to recover tumor cells from circulation. They were also analyzed for GFP and DsRed by nested PCR. GFP, First primer set: F, 5′-ATCTGCACCACCGGCAAGC-3′; R, 5′-TCGAACTTCACCTCGGCGC-3′. Nested primer set, F, 5′-ACCCTCGTGACCACCCTGAC-3′; R, 5′-CCGTCGTCCTTGAAGAAGATGG-3′. DsRed, first primer set: F, 5′-CTTCGCCTGGGACATCCTG-3′; R, 5′-TGGGTCACGGTCGCCACGCC-3′. Nested primer set: F, 5′-CCTGTCCCCCCAG TTCCAG -3′; R, 5′-CCCACTTGAAGCCCTCGG-3′.
Fluorescence, IF, IHC, and Western blotting
For fluorescent detection of GFP and DsRed that were expressed in Vector and p12 transfectants, respectively, the culture dishes were monitored under a fluorescent microscope directly. For IF of E-cadheirn, vimentin, and Twist2, cells were fixed with 4% paraformaldehyde for 30 min, permeablized with 0.2% Triton X-100 in PBS for 5 min, washed with PBS, and incubated with a mouse anti-E-cadherin mAb (BD Transduction Lab. #610404), a goat anti-vimentin pAb (Santa Cruz, #sc-7939), and a mouse anti-human Twist2 mAb (abcam, #ab57997) at RT for 1 h. After washing with PBS, Alexa 488-labeled goat anti-rabbit IgG, Alexa 555-labeled donkey anti-goat IgG, and Alexa 555-labeled goat anti-mouse IgG were used to visualize E-cadherin, vimentin, and Twist2, respectively. For double IF of blood vessels and tumor cells in the primary tumors derived from vector transfectants, a rabbit anti-mouse CD31 pAb (abcam, #ab28364) and a mouse anti-DsRed mAb (Clontech, #632392) were used and were visualized with Alexa 488-labeld goat anti-rabbit IgG and Alexa 555-labeld rabbit-anti mouse IgG, respectively. For double IF of blood vessels and tumor cells in the primary tumors derived from p12 transfectants, a goat anti-mouse CD31 pAb and a rabbit anti-GFP pAb (Santa Cruz, # sc-8334) were used and were visualized with a Alexa 555-labeld goat anti-mouse IgG and Alexa 488-labeld goat-anti rabbit IgG, respectively. For IHC staining, HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG were used as the secondary antibodies. For Western blotting, 150 μg protein was loaded on each lane and blotted with antibodies against desmoplakin (abcam, #ab16434), N-cadherin (SantaCruz, #sc-7939), E-Cadherin, vimentin, Twist, and p12 (pAb86) (17), and β-actin (Santa Cruz, #sc-1616-R).
Results
p12 m ediates TGF-β-induced EMT in HaCaT cells
We have previously shown that TGF-β induces expression of p12 that mediates the growth inhibitory activity of TGF-β (17). TGF-β is a known inducer of EMT (21, 22), we therefore first examined the effect of p12 on TGF-β-induced EMT of HaCaT cells. Zn-inducible p12 antisense transfectants (ip12(−)HaCaT) that have been shown to resist TGF-β-induced growth inhibition (17) and the corresponding vector control transfectants (pMTCB6+-HaCaT) were incubated with a mixture of TGF-β (10 ng/ml) and EGF (30 ng/ml), a condition known to induced EMT of HaCaT cells (20). As shown in Fig. 1, the morphology of the pMTCB6+-HaCaT vector control transfectants changed from epithelial to fibroblastic (Fig. 1A, top panels) in the presence of TGF-β and EGF, but that of the ip12(−)HaCaT p12 antisense transfectants remained epithelial (Fig. 1A, bottom panels). Western blotting shows that the p12 protein level in the vector control transfectants was induced by TGF-β and EGF and was decreased in the antisense transfectants both in the absence and presence of TGF-β and EGF (Fig. 1B, bottom panel). Consistently, treatment with TGF-β and EGF decreased the protein level of E-Cadherin in the vector control transfectants but not in the p12 antisense transfectants (Fig. 1B, top panel). These results indicate that p12 is required for TGF-β-induced morphology change and E-Cadherin expression, two prominent markers of EMT.
Figure 1.

p12 antisense inhibits TGF-β-induced EMT on HaCaT cells. A, Phase-contrast images of p12 antisense transfectants of HaCaT cells. Zn-inducible p12 antisense transfectants (ip12(−)HaCaT) and vector control transfectants (pMTCB6+-HaCaT) were cultured in DMEM + 5% FBS in the presence of 25 μM ZnSO4, and treated with or without a mixture of TGF-β (10 ng/ml) and EGF (30 ng/ml) for 48 h. B, Western blotting analysis of the E-cadherin and p12 protein levels in vector and p12 antisense transfectants with β-actin as a loading control.
p12 induces EMT of HCPC-1 cells
We then used HCPC-1 cells to study the effect of p12 on EMT and on tumor invasion and metastasis as p12 was originally identified from hamster oral keratinocytes. HCPC-1 cells were co-transfected with pcDNA3 control vector and pCI-neo-DsRed2 encoding a red fluorescent protein, or with a pcDNA3-p12 vector encoding the p12 protein and pEGFPC1 encoding a green fluorescent protein. Therefore, the vector- and p12-transfected cells are labeled with red and green fluorescence, respectively. HCPC-1-vector transfectants remained the epithelial characteristics such as polygonal structures and cell-cell contacts (Fig. 2A, left). The morphology of p12 transfectants exhibited spindle-like fibroblastic structures without tight cell-cell contacts (Fig. 2A, middle). The vector- and p12-transfected cells clearly segregated from each other in co-culture (Fig. 2A, right). The polygonal vector transfectants were surrounded by the p12 transfectants as shown by red (Fig. 2B, left) and green (Fig. 1B, middle) fluorescence and the merge of the two colors (Fig. 2B, right).
Figure 2.
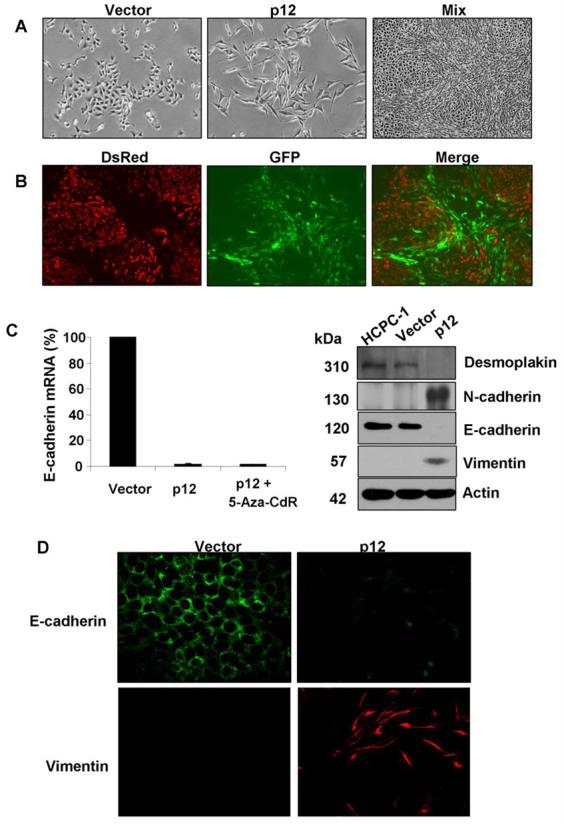
p12 induces EMT in HCPC-1 cells. A, Phase-contrast images of vector and p12 transfectants. p12 expression altered cell morphology from polygonal, epithelial structure (left) to spindle-like, fibroblastoid (middle) structure. Mixed culture of p12 and vector transfectants segregated from each other (right). B, Fluorescent images of the mixed culture of vector and p12 transfectants shown in A, right panel. Red and green fluorescence generated from DsRed and GFP, respectively, represented vector and p12 transfectants. C, qRT-PCR analysis of E-cadherin mRNA levels in vector and p12 transfectants and in p12 transfectants treated with 5-Aza-cdR (left). Western blotting analysis of the desmoplakin, N-cadherin, E-cadherin and vimentin protein levels in HCPC-1 parental cells and in vector and p12 transfectants with β-actin as a loading control (right). D, IF of E-cadherin (green) and vimentin (red) in vector and p12 transfectants.
The morphological change from polygonal to fibroblastoid structure upon p12 transfection prompted us to examine the expression levels of desmoplakin and E-cadherin, epithelial markers, and N-cadherin and vimentin, mesenchymal markers. Real-time quantitative RT-PCR analysis showed that E-cadherin expression in HCPC-1-p12 was repressed by 98% as compared to that of HCPC-1-Vector (Fig. 2C, left). Treatment with 5′-Aza-2′-deoxycytidine (5-Aza-CdR), a demethylating agent, did not recover E-cadherin expression in HCPC-1-p12, indicating that aberrant methylation of the CpG islands within the E-cadherin promoter is not the cause of E-cadherin suppression. Vimentin mRNA is not detectable in the Vector transfectants but appeared in the p12 transfectants (data not shown). The loss of E-cadherin expression, and the gain of vimentin expression in p12 transfectants were confirmed by Western blotting analysis (Fig. 2C, right) and IF staining (Fig. 2D). Loss of desmoplakin expression and increased N-cadherion expression, two additional epithelial and mesenchymal markers, confirmed that these cells underwent EMT (Fig. 2C, right). Taken together, these results indicated that p12 overexpression in HCPC-1 cells induced EMT.
p12-induced EMT is mediated by Twist2
We next examined the effect of p12 on the expression of E-cadherin repressors known to play a role in EMT. RT-PCR analysis shows that Snail and Slug were undetectable in the parental HCPC-1 cells, and in the Vector and p12 transfectants (Fig. 3A), whereas E12/E47, Twist1, ZEB-1, and ZEB-2 were constitutively expressed in HCPC-1 cells and was not changed after p12 transfection (Fig. 3A). Twist2 mRNA was detectable in the parental HCPC-1 cells and in the Vector transfectants but was significantly increased in p12 transfectants (Fig. 3A). Sequence analysis confirmed that the PCR amplicon was hamster Twist2 with a 96.2% homologue to the mouse Twist2. Increase of Twist2 protein in the p12 transfectants was confirmed by Western blotting (Fig. 3B). IF showed that Twist2 was barely detectable in the vector transfectants (Fig. 3C) but was strongly stained in p12 transfectants. Merge with DAPI staining shows that Twist was detectable in the nucleus of p12-transfected cells (indicated by arrows). To confirm that Twist2 is responsible for p12-induced EMT, siRNA was used to knockdown Twist2 expression in p12 transfectants. Transient transfection with synthetic double-stranded siRNA was used because we failed to establish stable Twist-2 siRNA transfectants. A Twist2-specifc siRNA and a scrambled non-specific control siRNA were co-transfected with a plasmid encoding GFP into p12-transfected HCPC-1 cells that had not been already transfected with GFP so that siRNA transfected-cells could be easily identified with green fluorescence. Due to the low transfection efficiency, only a low percentage of the cells were transfected, as shown by GFP expression. Characterization of MET by Western blotting analysis of EMT markers were not feasible, but it is clear that Twist2-specific siRNA-transfected cells converted back to polygonal structure (Fig. 3D, left). However, the control siRNA-transfected cells remained the fibroblastoid morphology (Fig. 3D, right).
Figure 3.

Dco-1 induces the expression of Twist2 that mediates EMT. A, RT-PCR analysis of known E-cadherin repressors. Twist2 expression was induced by p12. B, Western blotting analysis of Twist2 protein in vector and p12 transfectants. C, IF of Twist 2 protein in vector and p12 transfectants. Cell nuclei were stained with DAPI. Twists were detected both in the cytoplasm and in the nucleus (arrows). D, Knocking down Twist 2 reverted cell morphology to epithelial like structures (arrows). Twist2-specific synthetic siRNA and a plasmid encoding GFP were co-transfected into p12 transfectants (GFP free). Twist-2 siRNA-transfected cells are thus labeled with GFP expression.
EMT enhances local invasion of xenograft HCPC-1 tumors
Enhanced migration is a hall marker for mesenchymal cells. An in vitro cell migration assay showed that p12-transfected cells migrated away from the wounded age (Fig. 4A, right) as compared to the vector-transfected cells that barely migrated (Fig. 4A, left). The average distance of vector and p12 transfectants that migrated from the wounded edge was 100 ± 90 and 550 ± 150 μm, respectively, in 24 h (Fig. 4B). Therefore, p12-induced EMT cells have acquired a migratory phenotype.
Figure 4.
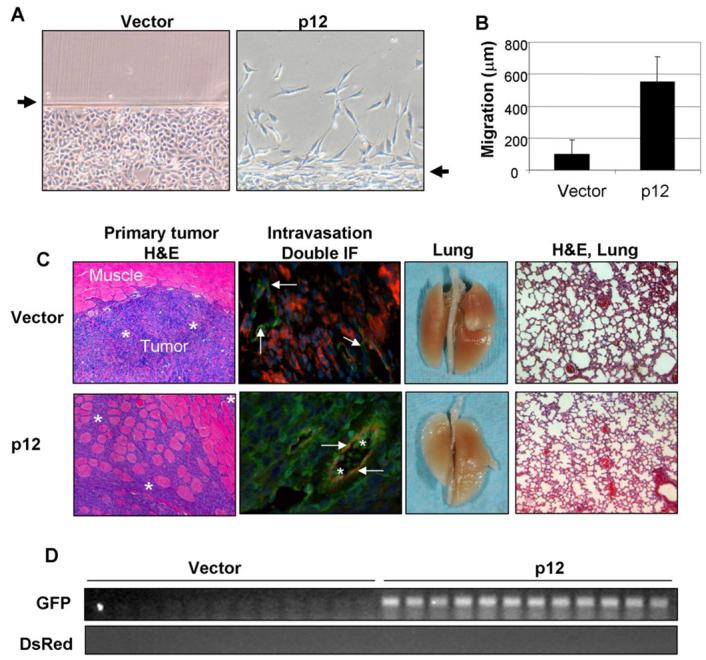
p12-induced EMT enhances cell motility and local tumor invasion. A, Migration of vector and p12 transfectants from wounded edges in an in vitro cell migration assay. B, Average distances of cell migration in 24 h. A total of 100 cells were measured under a phase contrast microscope. C, H&E staining showed local invasion of the primary tumors derived from p12 transfectants (EMT) but not from vector transfectants (non-EMT). Tumor cells were indicated by stars. Double IF of tumor cells (indicated by stars) and blood vessels (indicated by arrows) showed intravasation of tumor cells only in the tumor tissues derived from p12 transfectants. Macro photos of the lungs and H&E examinations did not detect metastasis in the lung in all the animals in both groups (n=12). D, PCR analysis of GFP and DsRed genomic DNA collected from blood samples of the tumor-bearing mice.
Next, we injected vector and p12 transfectants into a subcutaneous region of BALB/c athymic mice to test the ability of these cells to establish xenograft tumors, to invade into surrounding tissues and to metastasize to the lung. All mice from both groups (n=12) formed tumor burdens in the subcutaneous region 4 weeks after injection. The average tumor volumes were 864 and 888 cm3, respectively, for the vector and p12 transfectants. However, H&E staining revealed that vector transfectants formed a clear border between the primary tumor tissue and the adjacent muscle tissues without any evidence of invasion, whereas p12 transfectants aggressively invaded into the adjacent muscle tissue (Fig. 4C, tumor cells were indicated by stars). Double IF staining with an anti-von Williebrand factor (vWF, green fluorescence) and an anti-DsRed IgG (red fluorescence) did not detect any tumor cells in the blood vessels (indicated by arrows) located within the tumor mass derived from vector-transfected HCPC-1 cells (Fig. 4C, 2nd panel, top). However, tumor cells (indicated by stars) were detected in the blood vessels (indicated by arrows) located in the tumor mass derived from p12-transfected cells (Fig. 4C, 2nd panel, bottom) as shown by double IF with an anti-vWF (red fluorescence) and an anti-GFP IgG (green fluorescence). In both cases, no metastasis was formed in the lung, as shown by visual and microscopic examinations after H&E staining (Fig. 4C, 3rd and 4th panels). Culture of the blood samples collected from both groups of animals failed to establish colonies in the soft agar. However, PCR analysis for DsRed and GFP DNA sequence as an indicator of the existence of vector and p12 transfectants, respectively, showed that p12-transfected cells (GFP-labeled) but not vector transfectants (DsRed-labeled) were detected in the blood stream of all mice (Fig. 4D). These results indicated that non-EMT cells failed to intravasate, whereas EMT cells have enhanced ability to invade into the surrounding tissues and intravasate into the blood vessels but failed to establish metastasis.
EMT decreases lung metastasis derived from intravenous HCPC-1 cells
To more directly examine the effect of EMT on the metastasis, we injected vector and p12 transfectants into the tail vein of athymic mice to bypass the initial steps of metastasis process. Numerous metastatic lesions were formed from intravenously injected vector transfectants after 4 weeks in all animals (n=6) (Fig. 5A, top panel). However, intravenously injected p12 transfectants failed to form any metastases in the lung (Fig. 5A, middle panel). No lung metastases were detected by H&E staining even when the numbers of injected cells were 5 times higher and after a prolonged period (12 weeks). These results indicated that p12-induced EMT is accompanied with a decreased ability to establish metastatic growth in the lung although they have an enhanced migratory and local invasive phenotype.
Figure 5.
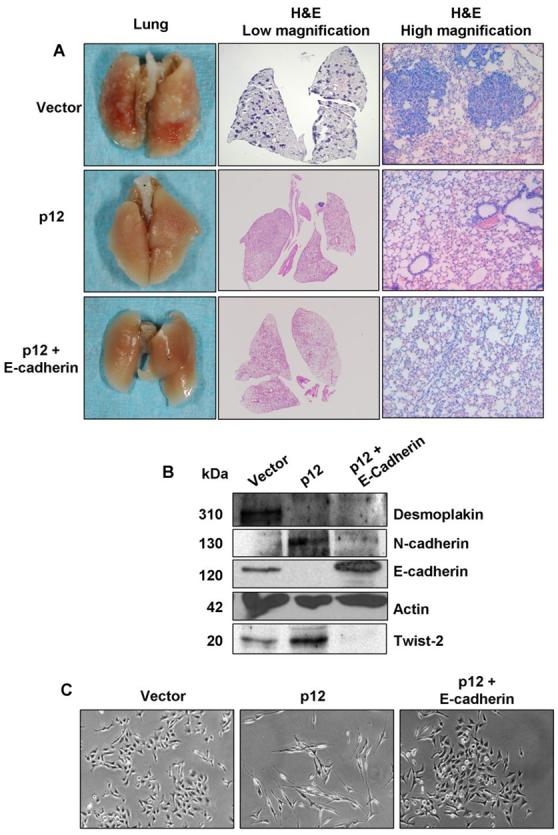
Ability of non-EMT, EMT, and MET cells to establish lung metastasis from intravenous inoculation. A, All the animals (n=6) developed lung metastases from intravenous inoculation of vector transfectants (non-EMT cells) as shown by macro photos and H&E staining at both low and high magnifications. None of the animals developed lung metastasis from intravenous inoculation of p12 transfectants (EMT cells) and E-cadherin re-expressed p12 transfectants (MET cells). B, Western blotting analysis of E-cadherin confirmed E-cadherin re-expression. C, E-cadherin re-expression reverted the fibroblastoid structures of p12 transfectants to epithelial structures, suggesting an MET process.
It has been proposed that regain of epithelial phenotype by de-repression of E-cadherin is necessary for metastatic growth of carcinomas (4). To test whether re-expression of E-cadherin in p12-induced EMT cells will allow the cells to form metastasis, we transfected an E-cadherin expression plasmid into p12 transfectants and tested their ability to form lung metastasis from intravenous injection. E-cadherin reexpression was confirmed by Western blotting analysis (Fig. 5B). The morphology of E-cadherin re-expressed cells have a polygonal structure (Fig. 5C, right) that is clearly different from the fibroblastoid structure of the original p12-transfected cells (Fig. 5C, middle) but is similar to that of the original vector transfectants (Fig. 5C, left). Therefore, from the cell morphology viewpoint, mesenchymal-epithelial transition (MET) occurred by re-expressing E-cadherin. However, E-cadherin re-expressed MET cells failed to establish lung metastasis when they were directly injected into the tail vein of athymic mice (Fig. 5A, bottom panel). We therefore examined the protein levels of desmoplakin, N-cadherin as well as Twist-2 in E-cadherin re-expressed cells. Although the expression of N-cadherin and Twist-2 decreased by in E-cadherin re-expressed cells, the expression of desmoplakin did not change (Fig. 5B). The possibility that the failure to establish lung metastases from i.v.-injected cells is due to a lack of desmoplakin could not be excluded. The ability of E-cadherin re-expressed MET cells to form subcutaneous tumors in athymic mice was not significantly different from that of vector-transfected non-EMT cells and p12-transfected EMT cells (data not shown).
Subcutaneous injection of a mixture of EMT and non-EMT cells results in the formation of lung metastasis
Since p12-induced EMT cells have enhanced invasive properties (Fig. 4) and non-EMT cells are able to form metastasis when injected into the tail vein (Fig. 5), we next examined whether a mixture of the two cell types would be able to complete the entire process of spontaneous metastasis from the primary subcutaneous region to the lung. A 1:1 mixture of the vector and p12 transfectants, 1×106 cells each per mouse, was inoculated subcutaneously onto athymic mice. Primary subcutaneous tumors formed at the injection sites in all mice (n=6) after 4 weeks. H&E staining shows that the edges of the primary tumors have an invasive appearance (Fig. 6A, left), reminiscent to that seen in the tumors derived from p12 transfectants alone (Fig. 4C). Double IF stating for GFP and DsRed showed that the invasive fronts were composed of layers of mainly EMT cells (Green, p12 transfectants) followed by non-EMT cells (red, vector transfectants) (Fig. 6A, middle). The center of the primary tumors is composed of entirely non-EMT cells (red) (Fig. 6A, right). Both DsRed and GFP DNA were detected by PCR analysis of the blood samples collected from these animals (Fig. 6B, right lane), indicating that both EMT and non-EMT cells were able to enter the blood stream under this experimental setting. Since we have shown in Figure 4D that only EMT cells were able to intravasate, a conceivable interpretation of these results would be that EMT cells invaded into the surrounding tissues and intravasated so that the obstacles were cleared for non-EMT cells to migrate and to enter the circulation.
Figure 6.
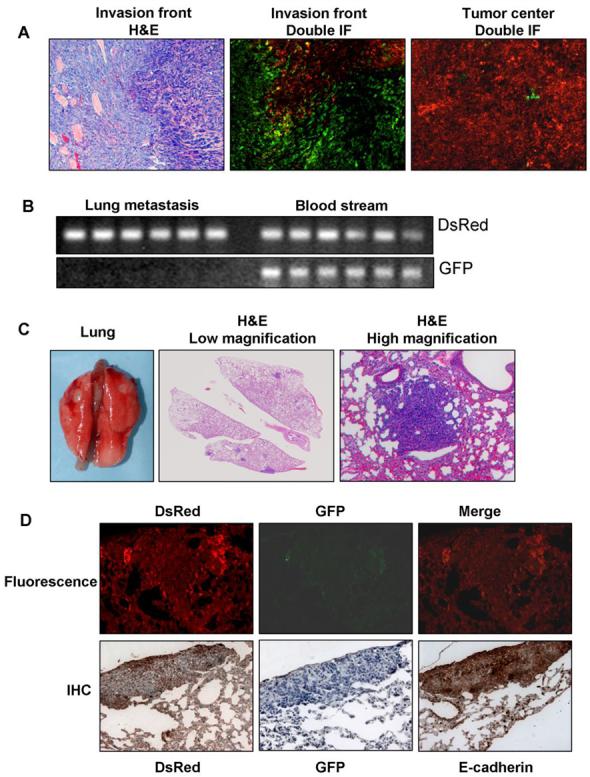
A mixture of EMT and non-EMT cells completed the entire spontaneous metastasis process. A, Primary tumor tissues. H&E staining of the tumor edge showed invasive phenotype. Double IF of GFP and DsRed showed that the invasion front is composed of mainly p12 transfectants (green) but the tumor center consists of mainly vector transfectants (red). B, PCR detection of DsRed and GFP DNA from the blood samples and lung metastases. Both GFP and DsRed were detected in the blood stream but only DsRed was detectable in the metastatic foci in the lung. C, Lung metastases shown by macro photos and H&E staining. D, Fluorescent images and IHC staining of DsRed, GFP, and E-cadherin in the lung metastases. GFP was not detectable but both DsRed and E-cadherin are strongly expressed, indicating they were originated from vector transfectants (non-EMT cells).
Visible lung metastases formed in 3 of the 6 animals (Fig. 6C). When the lung tissues were examined microscopically after H&E staining, all 6 animals have lung metastases. Direct fluorescent detection of DsRed and GFP showed that all the metastatic tumors emitted red but not green fluorescence (Fig. 6D, top panels) indicating that they were derived from DsRed-positive cells. IHC of the serial sections of the metastatic tumors with an anti-DsRed and an anti-GFP IgG was carried out and showed that the metastatic tumors expressed DsRed but not GFP (Fig. 6D, bottom panels), confirming that the metastases were originated from the non-EMT cells. IHC with an anti-E-cadherin IgG demonstrated strong E-cadherin expression in the metastatic tumor cells (Fig. 6D, bottom right), consistent with them being non-EMT cells. PCR analysis showed that DsRed but not GFP was detectable from the DNA extracted from the metastatic tumor tissues in the lung (Fig. 6B, left lane). However, both DsRed and GFP DNA were detected in the blood stream of all the animals in both groups. These results further demonstrated that the metastatic tumor cells are the original non-EMT cells rather than the EMT cells or the MET version of the EMT cells.
Discussion
Numerous experimental data support the notion that EMT occurs and plays an important role in tumor progression and metastasis (1). It is undisputed that carcinoma cells need to change their adhesive properties and become motile in order to leave the primary tumors and to invade into the surrounding tissues. However, it is still controversial as to whether transformation of a non-invasive tumor into a metastatic tumor is truly an EMT (23, 24). The main argument for the lack of a role of EMT in cancer is that metastases appear histologically similar to the primary tumor from which they are derived (25). To reconcile this apparent contradiction, it has been proposed that reversion to an epithelial morphology, an MET process, occurs in metastatic foci by re-expression of E-cadherin (4, 23). Dynamic expression of E-cadherin in cancer progression has been documented (10, 26). However, direct experimental data supporting MET in cancer metastasis are scarce (27).
For individual EMT cells to complete the entire metastasis process including detaching from primary site, degradation of the surrounding matrix, migration and invasion through the basement membrane, intravasation and survival in the circulation, extravasation, attach and proliferation at the secondary site, the cells would need an incredible plasticity to accomplish these complex tasks. On the other hand, cancers are known to consist of a highly heterogeneous population of cells that display a remarkable range of phenotypes (28). Therefore, investigation of an alternative pathway for the involvement of EMT in cancer metastasis is warranted. We hypothesized that EMT is necessary but not sufficient for metastasis.
To test this hypothesis, we took the advantage of the unique properties of p12 in regulating SSC growth and studied the effect of p12 overexpression on EMT of HCPC-1 cells and on their progression and metastasis. p12 is a growth suppressor originally isolated from hamster normal oral keratinocytes by subtractive cloning (16). Ectopic expression of p12 in HCPC-1 induced morphological alteration from polygonal to fibroblastoid structure, but was accompanied by an elongation of doubling time and loss of anchorage-independent growth in soft agar (16). We have now demonstrated that p12 overexpression in HCPC-1 cells induced EMT as shown by a complete loss of E-cadherin expression and the gain of vimentin expression (Fig. 2). This is consistent with our previous report that p12 is a down-stream effector of TGF-β signaling cascade (17). TGF-β represses E-cadherin expression (29) and is an important mediator of EMT (22).
E-cadherin represents the best-characterized molecular marker in epithelial cells. E-cadherin expression is mainly regulated by promoter hypermethylation (30) or by activation and upregulation of transcription repressors (31) that bind to the three E-box elements in the promoter region (32). p12-induced E-cadherin suppression could not be reverted by treatment of 5′-Aza-CdR (Fig. 2C), a universal demethylating agent, indicating that aberrant methylation is not the underlying mechanism. Instead, we found that Twist2 was upregulated after p12 expression in HCPC-1 cells (Fig. 3A). Twist2 is a bHLH transcription factor (33) that forms a heterodimer complex with E12/E47 protein. E12/E47 is known to bind to the E-box motives in the E-cadherin promoter and represses E-cadherin expression (34). Thus, an E-cadherin repressor activity of Twist2 can be envisioned. However, direct evidence for Twist2 to repress E-cadherin repression was lacking. We found that E12/E47 gene is constitutively expressed in HCPC-1 cells (Fig. 3A), so induction of Twist2 gene expression by p12 allows the formation of the Twist2/E12 complex thereby repressing E-cadherin expression. We also confirmed that p12-induced EMT was mediated by Twist2 by showing the reversion of EMT cells after Twist2 siRNA treatment (Fig. 3D). Therefore, our data showed that Twist2 is a new member to the growing list of the EMT-inducing E-cadherin repressors that have now included Twist1 (35), Snail (36), Slug (37), SIP (38), E12/E47 (34), CBF-A (39), FOXC2 (40), Goosecoid (41), HOXB7 (42), and DeltaEF1 (43).
p12 transfectants clearly have enhanced motility and ability of local invasion when inoculated subcutaneously on athymic mice (Fig. 4), consistent with an EMT phenotype observed in numerous reports (1, 6, 25, 32). However, p12-induced EMT cells have a reduced capacity to form metastasis when they were directly inoculated into the blood stream (Fig. 5). In contrast, non-EMT cells formed overt lung metastases when inoculated into the blood stream of athymic mice (Fig. 5). These results are in agreement with p12 being a growth suppressor (16) but are not consistent with the hypothesis that EMT is able to mediate the entire metastasis process (35). Re-expression of E-cadherin in p12 transfectants elicited MET of the EMT cells but was unable to promote metastasis from intravenously inoculated cells (Fig. 5), suggesting that either there are uncharacterized factors in our experimental system that are important or MET is not an integral part for metastasis.
The failure for subcutaneously injected non-EMT cells to metastasize is because they were unable to invade locally and to intravasate. However, the reason for p12-induced EMT cells, inoculated either subcutaneously or intravenously, not to form metastasis is unclear at present. It could be due to the intrinsic property of p12 as a growth suppressor whose over-expression has been shown to decrease anchorage-independent growth (16) and may thus reduce the survival of the tumor cells in circulation. However, it is clear that p12 is a new inducer of EMT. It is also consistent with our previous observation that distribution of p12 protein in normal human oral mucosal tissue is limited to the basal cell layer and to the mesenchymal cells in the adjacent connective tissues (44). The expression pattern of p12 in normal human oral tissues suggest that p12 may segregate the stem cell-like basal cells from those that are entering a differentiation program by regulating E-cadherin expression, and thus may play a role in maintaining the mesenchymal phenotype. p12 was initially thought as a tumor suppressor but it is currently considered as a growth suppressor because no mutations have yet been found in human cancers. The apparent contradiction for a growth suppressor to induce EMT, a process associated with tumor malignancy, can be reconciled by the facts that p12-induced EMT actually suppresses distant metastasis. In this regard, p12 is not alone. For example, Snail, a known E-cadherin repressor and an EMT inducer (45), was found to attenuate cell proliferation by inducing cyclin-dependent kinase inhibitor p21 (WAF/CIP1) (46). Also, a low proliferation rate in the invasive front of human colorectal adenocarcinomas has been reported (47).
Most interestingly, we found that subcutaneous inoculation of a mixture of EMT and non-EMT cells were able to form spontaneous lung metastases (Fig. 6). Under this circumstance, both EMT and non-EMT cells, represented by GFP and DsRed expression, respectively, were detectable in blood stream but only non-EMT cells were detected in the lung metastases (Fig. 6). When individual non-EMT and EMT cells were inoculated subcutaneously, only EMT cells were detectable in the blood stream but no lung metastasis formed in both cases (Fig. 4). However, when they were inoculated intravenously, only non-EMT cells were able to from lung metastases (Fig. 5). Taken together, our results suggested that EMT and non-EMT cells cooperate to complete the spontaneous metastasis process. We thus hypothesize that EMT cells are responsible for degrading the surrounding matrix to lead the way of intravasation. Non-EMT cells follow the EMT cells to enter the blood stream where both cell types survive. But only non-EMT cells are able to reestablish colonies in the secondary sites.
Acknowledgments
Grant support: NIH grant CA10044 (T. Tsuji), CA10524 (G.-f. Hu), and the Alexander and Margaret Stewart Trust (G.-f. Hu).
References
- 1.Berx G, Raspe E, Christofori G, Thiery JP, Sleeman JP. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis. 2007;24:587–97. doi: 10.1007/s10585-007-9114-6. [DOI] [PubMed] [Google Scholar]
- 2.Shook D, Keller R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev. 2003;120:1351–83. doi: 10.1016/j.mod.2003.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Duband JL, Monier F, Delannet M, Newgreen D. Epithelium-mesenchyme transition during neural crest development. Acta Anat. 1995;154:63–78. doi: 10.1159/000147752. [DOI] [PubMed] [Google Scholar]
- 4.Hugo H, Ackland ML, Blick T, et al. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–83. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 5.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 6.Thompson EW, Newgreen DF, Tarin D. Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res. 2005;65:5991–5. doi: 10.1158/0008-5472.CAN-05-0616. [DOI] [PubMed] [Google Scholar]
- 7.Semb H, Christofori G. The tumor-suppressor function of E-cadherin. Am J Hum Genet. 1998;63:1588–93. doi: 10.1086/302173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12:226–37. doi: 10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 9.Kouvaraki M, Gorgoulis VG, Rassidakis GZ, et al. Alterations of the 16q22.1 and 16q24.3 chromosomal loci in sporadic invasive breast carcinomas: correlation with proliferative activity, ploidy and hormonal status of the tumors. Anticancer Res. 2001;21:991–9. [PubMed] [Google Scholar]
- 10.Lombaerts M, van Wezel T, Philippo K, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer. 2006;94:661–71. doi: 10.1038/sj.bjc.6602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 12.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–3. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. Embo J. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thuault S, Valcourt U, Petersen M, et al. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–83. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 16.Todd R, McBride J, Tsuji T, et al. Deleted in oral cancer-1 (doc-1), a novel oral tumor suppressor gene. Faseb J. 1995;9:1362–70. doi: 10.1096/fasebj.9.13.7557027. [DOI] [PubMed] [Google Scholar]
- 17.Hu MG, Hu GF, Kim Y, et al. Role of p12(CDK2-AP1) in transforming growth factor-beta1-mediated growth suppression. Cancer Res. 2004;64:490–9. doi: 10.1158/0008-5472.can-03-2284. [DOI] [PubMed] [Google Scholar]
- 18.Matsuo K, Shintani S, Tsuji T, et al. p12(DOC-1), a growth suppressor, associates with DNA polymerase alpha/primase. Faseb J. 2000;14:1318–24. doi: 10.1096/fj.14.10.1318. [DOI] [PubMed] [Google Scholar]
- 19.Shintani S, Ohyama H, Zhang X, et al. p12(DOC-1) is a novel cyclin-dependent kinase 2-associated protein. Mol Cell Biol. 2000;20:6300–7. doi: 10.1128/mcb.20.17.6300-6307.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilkins-Port CE, Higgins PJ. Regulation of extracellular matrix remodeling following transforming growth factor-beta1/epidermal growth factor-stimulated epithelial-mesenchymal transition in human premalignant keratinocytes. Cells Tissues Organs. 2007;185:116–22. doi: 10.1159/000101312. [DOI] [PubMed] [Google Scholar]
- 21.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–36. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 23.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 24.Tarin D, Thompson EW, Newgreen DF. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- 25.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–81. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bukholm IK, Nesland JM, Borresen-Dale AL. Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients. J Pathol. 2000;190:15–9. doi: 10.1002/(SICI)1096-9896(200001)190:1<15::AID-PATH489>3.0.CO;2-L. [seecomments] [DOI] [PubMed] [Google Scholar]
- 27.Chaffer CL, Brennan JP, Slavin JL, et al. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271–8. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 28.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 29.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–8. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 30.Graff JR, Herman JG, Lapidus RG, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–9. [PubMed] [Google Scholar]
- 31.Liu YN, Lee WW, Wang CY, et al. Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene. 2005;24:8277–90. doi: 10.1038/sj.onc.1208991. [DOI] [PubMed] [Google Scholar]
- 32.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Cserjesi P, Olson EN. Dermo-1: a novel twist-related bHLH protein expressed in the developing dermis. Dev Biol. 1995;172:280–92. doi: 10.1006/dbio.1995.0023. [DOI] [PubMed] [Google Scholar]
- 34.Perez-Moreno MA, Locascio A, Rodrigo I, et al. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276:27424–31. doi: 10.1074/jbc.M100827200. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 36.Cano A, Perez-Moreno MA, Rodrigo I, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 37.Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62:1613–8. [PubMed] [Google Scholar]
- 38.Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–78. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 39.Venkov CD, Link AJ, Jennings JL, et al. A proximal activator of transcription in epithelial-mesenchymal transition. J Clin Invest. 2007;117:482–91. doi: 10.1172/JCI29544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mani SA, Yang J, Brooks M, et al. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A. 2007;104:10069–74. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartwell KA, Muir B, Reinhardt F, et al. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proc Natl Acad Sci U S A. 2006;103:18969–74. doi: 10.1073/pnas.0608636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu X, Chen H, Parker B, et al. HOXB7, a homeodomain protein, is overexpressed in breast cancer and confers epithelial-mesenchymal transition. Cancer Res. 2006;66:9527–34. doi: 10.1158/0008-5472.CAN-05-4470. [DOI] [PubMed] [Google Scholar]
- 43.Eger A, Aigner K, Sonderegger S, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375–85. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- 44.Tsuji T, Duh FM, Latif F, et al. Cloning, mapping, expression, function, and mutation analyses of the human ortholog of the hamster putative tumor suppressor gene Doc-1. J Biol Chem. 1998;273:6704–9. doi: 10.1074/jbc.273.12.6704. [DOI] [PubMed] [Google Scholar]
- 45.Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 46.Vega S, Morales AV, Ocana OH, et al. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–43. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung A, Schrauder M, Oswald U, et al. The invasion front of human colorectal adenocarcinomas shows co-localization of nuclear beta-catenin, cyclin D1, and p16INK4A and is a region of low proliferation. Am J Pathol. 2001;159:1613–7. doi: 10.1016/s0002-9440(10)63007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]