

Abstract
DYT1 is the most common inherited dystonia, a neurological syndrome that causes disabling involuntary muscle contractions. This autosomal dominant disease is caused by a glutamic acid deletion near the carboxy-terminus in the protein torsinA. Cell and animal based studies have shown how the DYT1 mutation causes mutant torsinA to redistribute from the endoplasmic reticulum to the nuclear envelope, acting through a dominant negative effect over the wild type protein. As a result, the wild type:mutant torsinA expression ratio would be important for disease pathogenesis, and events that influence it, such as a differential degradation process for each protein, might modulate DYT1 pathobiology. The DYT1 mutation also triggers the formation of abnormal intermolecular disulfide bonds in torsinA, although the significance of this finding is unclear. How the protein quality control machinery handles torsinA, and whether this process is affected by its abnormal oligomerization remain unknown. Here, we first explored how the disease-linked mutation influences the catabolic process of torsinA, demonstrating that the differences in subcellular localization between both forms of torsinA lead to divergences in their degradation pathways and, whereas torsinA is normally recycled through autophagy, the proteasome is also required for the efficient clearance of the mutated form. Subsequently, we determined that the abnormal disulfide bond-dependent oligomerization of mutant torsinA is not a result of its redistribution to the nuclear envelope, but a direct consequence of the mutation. Finally, we established that the presence of disulfide links in mutant torsinA oligomers interfere with their degradation by the proteasome, thus relying on autophagy as the main pathway for clearance. In conclusion, the abnormal subcellular localization and oligomerization of DYT1-linked torsinA influences its catabolic process, opening the door to the modulation of the wildtype:mutant torsinA ratio through pharmacological manipulation of protein degradation pathways.
Keywords: Dystonia, autophagy, proteasome
Dystonia is a neurological syndrome characterized by disabling twisting involuntary movements (Fahn et al., 1998). While dystonia can be a consequence of different types of brain insults, such as stroke or tumor, several genetic forms of dystonia have been described (Gonzalez-Alegre, 2007). Among those, DYT1 is the most common inherited dystonia. A dominantly inherited disease, DYT1 has limited penetrance with only about a third of mutation carriers developing the clinical syndrome. DYT1 is caused by a GAG deletion in the TOR1A gene that causes the loss of a glutamic acid residue in torsinA (torsinA(ΔE)) (Ozelius et al., 1997). The factors that modify DYT1 penetrance remain unknown, although a genetic polymorphism in the disease causing gene plays a small role (Risch et al., 2007).
TorsinA, a widely expressed AAA protein (ATPases Associated with diverse cellular Activities) (Hanson and Whiteheart, 2005), is an endoplasmic reticulum (ER)-resident glycoprotein (Hewett et al., 2000, Kustedjo et al., 2000). The DYT1 mutation does not alter protein solubility (Kustedjo et al., 2003) but causes torsinA to accumulate in the nuclear envelope (NE) (Gonzalez-Alegre and Paulson, 2004, Goodchild and Dauer, 2004, Naismith et al., 2004). When overexpressed in cultured cells, the accumulation of torsinA(ΔE) in the NE triggers the formation of NE-derived cytoplasmic membranous inclusions or spheroid bodies (Gonzalez-Alegre and Paulson, 2004), probably an artifact of overexpression, but a helpful marker of the abnormal behavior of torsinA(ΔE).
Similar to other AAA proteins, torsinA is predicted to assemble into multimers, deriving energy from ATP hydrolysis to mediate conformational changes on substrate proteins (Breakefield et al., 2001). A dominant negative form of torsinA with a mutation that impairs ATP hydrolysis also localizes to the NE (Goodchild and Dauer, 2004, Naismith et al., 2004). Published reports suggest that the presence of torsinA(ΔE) in multimers “locks” them in the NE, acting through a dominant negative effect over torsinA(wt) and leading to a loss of torsinA function (Goodchild and Dauer, 2004, Naismith et al., 2004, Torres et al., 2004, Gonzalez-Alegre et al., 2005). The number of functional multimers, formed exclusively by torsinA(wt), would depend on the torsinA(wt):torsinA(ΔE) expression ratio. Therefore, factors that modulate this ratio, such as a potential differential degradation process for both forms of torsinA, could influence disease pathogenesis and putatively penetrance.
How neurons handle abnormal proteins is critical in the pathogenesis of many neurological diseases. Whereas the degradation of NE-resident proteins has not been investigated, ER glycoproteins are usually degraded by the proteasome through ERAD (ER-associated degradation) (Meusser et al., 2005, Romisch, 2005) or by the lysosome through macroautophagy (Cuervo, 2004, Kruse et al., 2006) (referred to as autophagy from now on). In ERAD, ER proteins that retain a high mannose tag are selectively retrotranslocated to the cytoplasm for proteasomal degradation. In autophagy, cells recycle macromolecules and organelles such as the ER non-selectively by engulfing them in a double-membrane structure that fuses to the lysosome. As a high-mannose glycoprotein that resides within the secretory pathway, torsinA is a potential a substrate for both systems. However, the different subcellular localization of torsinA(wt) and torsinA(ΔE) (ER versus NE, respectively), could lead to divergences in their catabolic process.
Another differential feature of torsinA(ΔE) is that it forms abnormal intermolecular disulfide links (Gonzalez-Alegre and Paulson, 2004, Torres et al., 2004). Whether this abnormal oligomerization is responsible for the functional defects caused by torsinA(ΔE) is unknown, but a recent report identified torsinA as a redox sensor with critical intramolecular disulfide bonds that regulate its nucleotide binding activity (Zhu et al., 2008), demonstrating a potential role for this abnormal molecular event in DYT1 dysfunction. Furthermore, because reduction of disulfide links is a required step prior to retrotranslocation for proteasomal degradation (Romisch, 2005), this abnormal feature of torsinA(ΔE) could also influence its degradative process and, as explained above, modulate DYT1 pathogenesis.
Here, we completed experiments to determine whether differences in subcellular localization and disulfide-link dependent oligomerization between torsinA(wt) and torsinA(ΔE) lead to divergences in their catabolic process, which could implicate the protein quality control machinery in DYT1 pathogenesis.
Experimental procedures
Cell culture, transfection, non-reducing lysis and western blot
Cos7 cells were grown, maintained and transiently transfected using LIPOfectamine 2000 (Invitrogen) as described (Gonzalez-Alegre et al., 2003). TorsinA(wt) and torsinA(ΔE) inducible PC6-3 clonal cell lines were grown, differentiated on nerve growth factor, maintained and induced as previously reported (Gonzalez-Alegre and Paulson, 2004). Differentiation with nerve growth factor and 1% HS, 0.5% FBS low serum media were used to avoid cell division and achieve a differentiated neural phenotype. To induce transcription of the transgene we employed doxycycline (DOX) at 1.5μg/mL unless otherwise noted. SH-SY5Y cells were grown in DMEM/F12 with 10% Fetal Bovine Serum, 1.0mM Na Pyruvate and 0.1mM NEAA (all from Gibco). HEK293 cells were grown in DMEM with 10% Fetal Bovine Serum and 1% penicillin/streptomycin (all from Gibco). To obtain protein lysates in reducing conditions, cells were harvested at the indicated times using Laemmli buffer with 200 mM DTT, boiled and sonicated as described (Gonzalez-Alegre and Paulson, 2004). For non-reducing conditions, cells were harvested using Laemmli buffer without DTT, adding 10 mM N-ethylmaleimide (NEM) to avoid aberrant disulfide link formation, boiled and sonicated. An aliquot from each non-reduced lysate was placed into a different Eppendorf, and reduced by addition of 200 mM DTT (Gonzalez-Alegre and Paulson, 2004). Samples were run using SDS-PAGE, transferred to a PVDF membrane and western blot completed as described (Gonzalez-Alegre et al., 2003).
Quantification of western blot signal
To quantify western blot band intensity, signal was determined and normalized to loading control as previously described (Gonzalez-Alegre et al., 2003, Gonzalez-Alegre et al., 2005). In brief, blots were scanned, and the pixel intensity of the bands was quantified using the Scion Image software (Scion, Frederick, MD). TorsinA signal was normalized to the loading control and the result expressed as a percentage of the signal obtained in the control lane.
Plasmid vectors
Expression vectors encoding human torsinA(wt) and torsinA(ΔE) were previously described (Gonzalez-Alegre et al., 2003). Vectors encoding human torsinA carrying the Walker A (K108A) and Walker B (E171Q) mutations were a generous gift of P. Hanson (Washington University) and W. Dauer (Columbia University) (Goodchild and Dauer, 2004, Naismith et al., 2004). Plasmid vectors encoding mouse torsinA carrying an internal myc tag and human torsinA lacking the hydrophobic stretch that follows the signal peptide (torsinA(wt)(Δ24-40)), were a kind gift of Drs. W. Dauer and M. Zolkiewski, respectively.
Antibody generation
TorsinA antibodies were generated by Sigma Genosys by inoculating New Zealand White rabbits with a keyhole limpet hemocyanin-conjugated peptide containing the last 14 amino acids of human torsinA. For affinity purification, immune serum was incubated overnight at 4 °C with the same peptide conjugated to agarose beads, which were then pelleted and washed with TBS (25 mM Tris, pH 7.4, 2.7 mM KCl, 14 mM NaCl). The purified antibody was eluted with four successive bead volumes of glycine, pH 2, and aliquots containing antibody were pooled. TorsinA antibodies (TA913), pre-immune and pre-adsorbed serum were diluted in 5% nonfat dry milk, and antibody specificity was confirmed with Western analysis (supplemental figure 1). Other antibodies employed included FLAG M5 (Sigma), α-tubulin (Sigma), GAPDH (Santa Cruz Biotechnology), DM2A8 antibody against torA (Cell Signaling Technology) and ubiquitin (DakoCytomation).
Indirect immunofluorescence (IF)
Cell were seeded in 4-chamber slides and transfected, or induced by treatment with DOX, and processed as previously described unless otherwise noted (Gonzalez-Alegre et al., 2003, Gonzalez-Alegre and Paulson, 2004, Gonzalez-Alegre et al., 2005). In brief, cells were fixed in 4% paraformaldehyde at room temperature for 15 minutes, incubated sequentially with washes in the indicated primary and corresponding secondary antibodies, nuclei stained with DAPI and slides mounted and visualized with a Zeiss Axioplan fluorescence microscope or an Olympus BX-51 Digital Light Microscope. Digital images were collected on red, green and blue fluorescence channels using an AxiocamHRm (Zeiss) digital camera or a Spot Digital Camera. Secondary antibodies used were Alexa Fluor® 568 (Invitrogen), FITC or Rhodamine conjugated antibodies (Santa Cruz Biotechnology).
Pharmacological treatments
Autophagy and proteasomal inhibition were achieved by adding 10mM 3-MA (Sigma), 10μM lactacystin (Calbiochem) or 10μM epoxomycin (Sigma) to the media at the indicated times. Cycloheximide (Sigma) was used at 10mM with SH-SY5Y and HEK293 cells and 20mM with Cos7 cells.
Results
We first aimed to determine whether our previously reported inducible PC6-3 clonal cell lines (Gonzalez-Alegre and Paulson, 2004) represent an appropriate system to study torsinA degradation, as the same model has been successfully used to study α-synuclein degradation (Webb et al., 2003). To assess torsinA degradation in this system, we induced torsinA transgene transcription by adding DOX to the media for 24h, subsequently washed DOX and tracked torsinA levels by western blot over 7 days. As shown in figure 1A-B, these experiments demonstrated a consistent reduction in torsinA levels of >50% in the 24h after transcription was switched off, illustrating the ability of these cell lines to clear torsinA and their expediency to complete studies on its degradation.
Figure 1. Degradation of overexpressed torsinA in PC6-3 inducible cell lines.
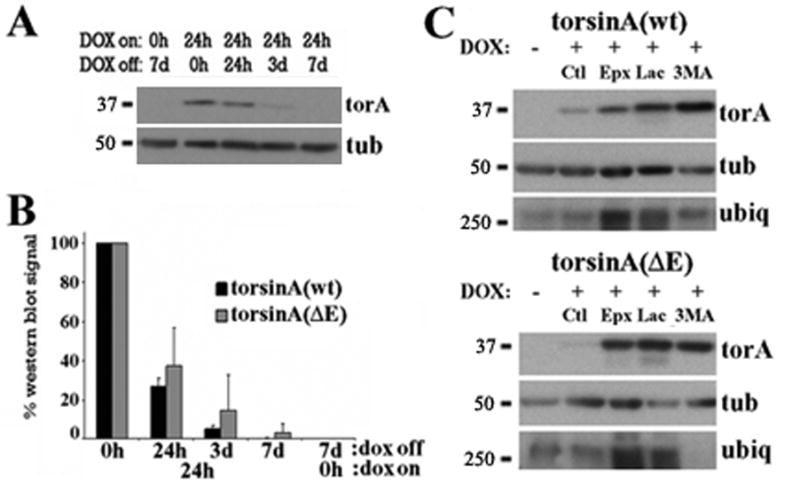
(A) Representative western blot of PC6-3 torsinA(wt) inducible cell lines that were induced with DOX for 24 h, DOX was then removed from the media and torsinA levels assessed by western blot at the indicated times. (B) Quantification of western blot signal normalized to loading control from three experiments as in (A) for torsinA(wt) and torsinA(ΔE). (C) PC6-3 cells were induced to express torsinA for 24h, DOX was removed from the media and treated as indicated for another 24h followed by western blot analysis. Increased ubiquitin immunoreactivity was only found after proteasomal inhibition. Ct: control; Epx: epoxomycin; 3MA: 3-methyladenine; Lac: lactacystin; torA: torsinA; tub: α-tubulin; ubiq: ubiquitin.
To identify appropriate incubation times for pharmacological inhibition of catabolic pathways, cells were treated for 6, 18, 24 or 48 hours with 10μM lactacystin or 10μM epoxomycin (proteasome) or 10mM 3MA (autophagy). Treatment with 3MA was well tolerated for 48h whereas, in contrast to previous reports (Webb et al., 2003), proteasome inhibition prolonged more than 24h induced significant cell death (not shown). These findings, together with the results shown in figure 1, made 24h an appropriate time point for subsequent studies. To identify the degradation process responsible for the clearance of torsinA, transcriptional induction by DOX as above was followed by a 24h wash and then 24h catabolic inhibition. Assessment of torsinA levels by western blotting suggested that, when highly overexpressed, both forms of torsinA are a substrate for both the proteasome and autophagy, perhaps with more proteasomal involvement for the disease-linked protein compared to the wild type protein (figure 1C). The increased ubiquitin immunoreactivity found after lactacystin and epoxomycin treatments, but not with 3MA, demonstrated the specificity of the drugs used.
When assessing torsinA levels by IF, we detected increased torsinA(wt) immunoreactivity within its normal ER distribution after inhibiting either catabolic pathway (not shown). Interestingly, in torsinA(ΔE)-expressing cells the number of spheroid bodies (membranous cytoplasmic inclusions that derive from its accumulation in the NE (Gonzalez-Alegre and Paulson, 2004)) was increased only after inhibition of autophagy, while increased NE immunostaining, but not spheroid bodies, was seen after pharmacological impairment of the proteasome (figure 2). Overall, these findings suggest that NE-located torsinA is degraded by the proteasome, but when torsinA(ΔE) accumulates in spheroid bodies it requires autophagy for its clearance. It should be noted here again that spheroid bodies are thought to originate from the abnormal accumulation of torsinA(ΔE) in the NE only when highly overexpressed, but are probably not present at normal levels of expression in the disease state and, therefore, represent an experimental artifact.
Figure 2. Spheroid bodies are degraded through autophagy.
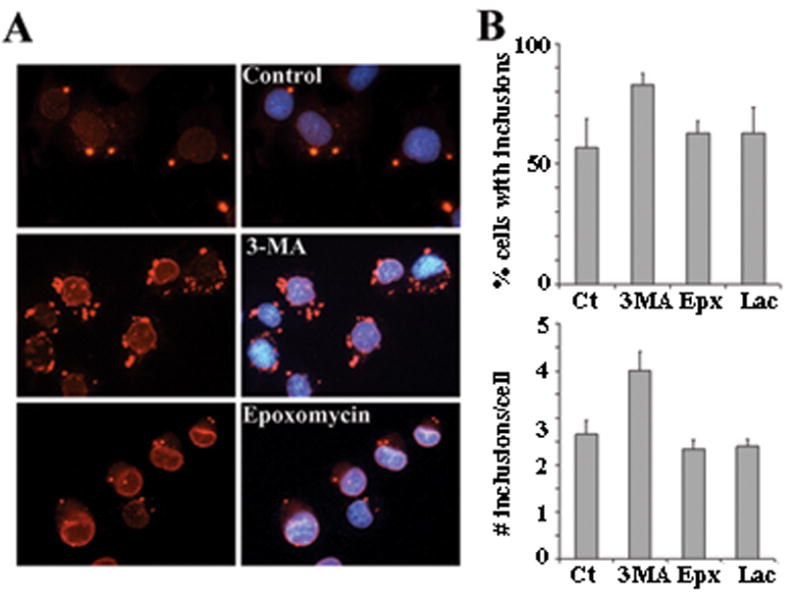
(A) IF analysis of PC6-3 cells inducibly expressing torsinA(ΔE) and treated as in figure 1C with the indicated drugs. TorsinA is seen in red and nuclear DAPI staining in blue. (B) Quantification of percentage of cells with spheroid bodies and average number of spheroid bodies per cell in randomly selected fields from three independent experiments under the indicated treatment conditions. Ct: control; Epx: epoxomycin; 3MA: 3-methyladenine; Lac: lactacystin.
Because the normal degradation process of ER proteins can saturate with high levels of expression leading to recruitment of alternative pathways (Kruse et al., 2006, Pandey et al., 2007) and, as shown above, the degradation of spheroid bodies could differ from the normal degradation process for NE-localized torsinA(ΔE), we designed experiments with lower transgene expression levels. Taking advantage of the inducibility of the PC6-3 system and the human-specific torsinA antibody (TA913) we have developed (supplementary figure 1), we induced transcription for 12h with DOX at different dilutions, assessing transgene levels with the TA913 antibody (human specific) and its relative expression to endogenous torsinA with DM2A8 antibody (recognizes both human and rat torsinA). Incubation with 0.06ng/mL DOX (1:25,000 dilution from the original 1.5μg/mL) resulted in transgene expression similar to endogenous torsinA (figure 3A). Immunostaining of torsinA(ΔE) cells at that level of induction demonstrated only rare spheroid body formation (figure 3B), reducing this potential confounding factor. We then repeated the 24h catabolic inhibition experiments with these near physiological torsinA induction levels (0.06ng/mL DOX) and consistently found that torsinA(wt) is preferentially degraded through autophagy while both autophagy and the proteasome contribute similarly to the clearance of torsinA(ΔE) (figure 3C and 3D). To confirm the role of autophagy on torsinA(wt) turnover, we employed human cell lines (SH-SY5Y and HEK293) to determine whether we could track the degradation of endogenous torsinA after translational inhibition with cycloheximide as the low steady state levels of torsinA was difficult to track without inhibition of new protein synthesis. These cell lines were incubated with cycloheximide for 6, 12, 18, 24 and 48h, and torsinA levels assessed by western blot and compared to tubulin. We found a detectable steady reduction in torsinA levels, beginning at 12h (see figure 4) and most obvious at 48h (not shown). We picked the 12h time point to perform experiments coupling cycloheximide treatment with inhibition of both proteolytic pathways, demonstrating that torsinA degradation was reversed by inhibition of autophagy and not the proteasome (figure 4).
Figure 3. Differential degradation of torsinA(wt) and torsinA(ΔE) at near physiological expression levels.
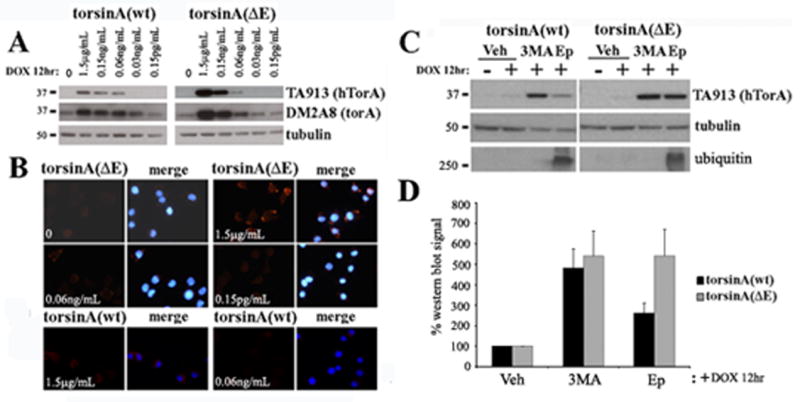
(A) PC6-3 cells were induced with DOX for 12h at the indicated dilutions of the concentration used in the previous experiments shown in figures 1 and 2, and expression levels assessed by western blot analysis. TA913 antibody recognizes only the transgene (human torsinA), while DM2A8 equally detects both the transgene and endogenous rat torsinA. (B) IF analysis after induction at different concentrations to assess spheroid body formation. TorsinA is seen in red and nuclear DAPI staining in blue. (C) Induction at the 1/25,000 dilution for 12 h, followed by a 24 h treatment with the indicated proteolytic inhibitors and western blot analysis of transgene levels. (D) Quantification of 4 experiments as in (C). Vh: vehicle; Epx: epoxomycin; 3MA: 3-methyladenine; hTorA: human torsinA; torA: torsinA.
Figure 4. Endogenous torsinA(wt) is preferentially degraded through autophagy.
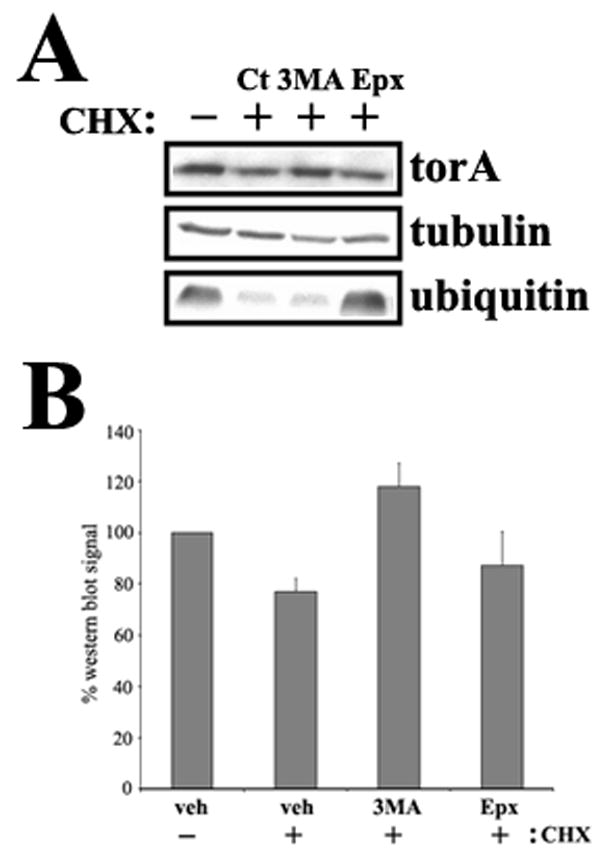
(A) HEK293 cells were incubated with cycloheximide and other indicated drugs for 12 hours and torsinA levels assessed by western blot. Proteasome inhibition is demonstrated by ubiquitin levels (B) Quantification of 3 experiments as in (A). CHX: cycloheximide; Ct: control; Epx: epoxomycin; 3MA: 3-methyladenine.
Because it has been shown that the presence of abnormal oligomers, such as those caused by intermolecular disulfide links, can modify the degradation process of ER proteins (Fujita et al., 2007), we examined the origin and fate of the abnormal intermolecular disulfide link-dependent torsinA(ΔE) oligomers we previously described (Gonzalez-Alegre and Paulson, 2004, Torres et al., 2004) (for clarity, we will refer to multimers when addressing the putative torsinA/ATP/ substrate complex, and oligomers when describing the abnormal disulfide-linked species). Non-reducing, NEM-containing lysates obtained from PC6-3 cells inducibly expressing torsinA(ΔE), with or without a c-terminal FLAG tag, resulted in a stable higher molecular weight species of ∼75 kDa detected by western blot analysis that disappeared in reducing conditions (figure 5A-B). Although the intensity of this band varied between experiments it was consistently found with torsinA(ΔE) and only rarely and weakly with torsinA(wt), demonstrating a clear biochemical difference resulting from the DYT1 mutation. Through this abnormal molecular event, torsinA(ΔE) could dimerize with other torsin proteins. We assessed for the presence of torsinB, another member of the torsin gene family that resides within the ER, but did not detect torsinB signal at the ∼75 kDa torsinA(ΔE) band (figure 5C).
Figure 5. Disulfide link-dependent oligomerization of torsinA(ΔE) is not a consequence of its abnormal redistribution to the NE.
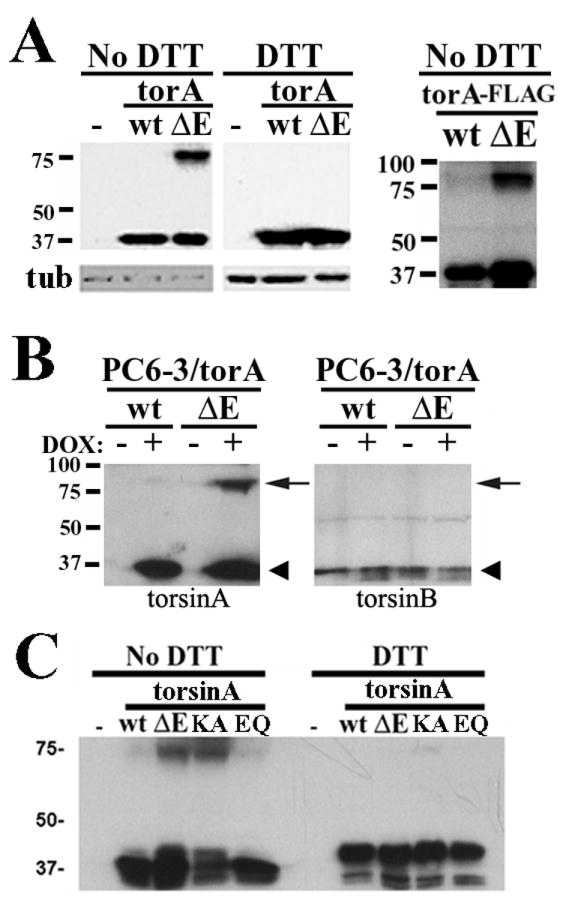
(A) Reducing and non-reducing lysates of Cos7 cells transiently expressing torsinA(wt) or torsinA(ΔE) were electrophoresed, showing an ∼75 kDa torsinA immunoreactive band in non-reducing (no DTT) conditions for torsinA(ΔE). A FLAG epitope tag at the c-terminus of torsinA did not affect the presence of this abnormal species. (B) Blotting the same membrane sequentially for torsinB and torsinA did not detect any torsinB signal at the ∼75 kDa torsinA immunoreactive band (arrow). Monomeric torsinA and torsinB have a similar molecular weight (arrowhead). (C) Expression of different forms of torsinA in Cos7 cells followed by western blot analysis as in (A). TorA: torsinA; KA: torsinA(K108A); EQ: torsinA(E171Q).
We next asked whether oligomerization depends on torsinA localization to the NE or is a direct consequence of the mutation. To answer that question, we performed experiments with previously described functional mutant forms of torsinA, including torsinA(K108A) (unable to bind ATP, localizes to the ER) and torsinA(E171Q) (binds but does not hydrolyze ATP, localizes to the NE) (Goodchild and Dauer, 2004, Naismith et al., 2004). Interestingly, ER-located torsinA(K108A) displayed the highest propensity to oligomerize (figure 5D), while torsinA(E171Q), which localizes to the NE, did oligomerize more than torsinA(wt), but not as much as torsinA(ΔE), suggesting that NE-localization is not the cause of this abnormal molecular event.
We then explored if the presence of intermolecular disulfide links modify the degradation process for torsinA(ΔE) oligomers. Employing non-reducing lysates in the inducible PC6-3 cell lines with low concentration of DOX as in figure 3, pharmacological inhibition of catabolic pathways showed that, in contrast to the proteasomal degradation of monomers, abnormal oligomers are degraded preferentially through autophagy (figure 6A). Similar results were obtained with the oligomers detected with torsinA(K108A) and torsinA(E171Q), suggesting this is not a consequence from their different subcellular localization but from the oligomerization itself (figure 6B).
Figure 6. TorsinA(ΔE) oligomers are preferentially cleared by autophagy.
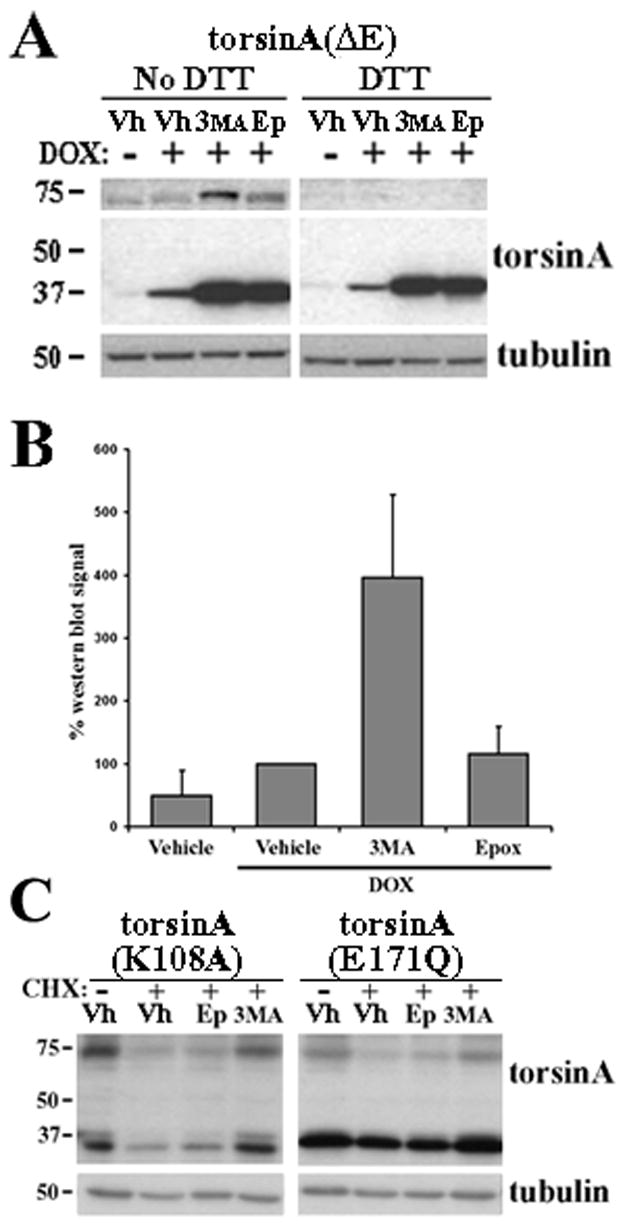
(A) PC6-3 torsinA(ΔE) cells were induced with low concentration DOX for 12h and treated with the different catabolic inhibitors as in figure 3C, non-reducing lysates were obtained and western blot completed to assess torsinA expression under different treatment conditions. The lower panel of the gel is a slightly shorter exposure than the upper panel to avoid overexposure. A faint, non-specific band also seen in the uninduced lane that migrates slightly faster than this oligomer is seen as a consequence of the longer exposure time required to detect the oligomer at lower/near physiological levels of expression. (B) Quantification of oligomer expression in 3 experiments as in (A). (C) Cos7 cells were transfected with plasmid vectors encoding torsinA(K108A) or torsinA(E171Q). Cycloheximide and proteolytic inhibitors were added 18 h after transfection as indicated, non-reducing lysates obtained and western blot completed. CHX: cycloheximide; Vh: vehicle; Epx: epoxomycin; 3MA: 3-methyladenine.
Discussion
In this work, we have identified how the DYT1 mutation influences the degradation process and oligomerization of torsinA, offering potential strategies to modify disease pathogenesis. First, we identified differences in the degradation process for torsinA caused by the DYT1 mutation. Second, we determined that disulfide link-dependent oligomerization of disease-linked torsinA is not a result of its localization to the NE, but likely a direct consequence of the mutation itself. Third, the presence of intermolecular disulfide links modulates the catabolic process of torsinA(ΔE) oligomers. Overall, these results suggest that differences in subcellular localization and oligomerization of torsinA influence its degradation process and may play an important role in disease pathogenesis. Pharmacological manipulation of protein degradation pathways could, therefore, be used to modify the pathobiology underlying DYT1.
Our findings show that the turnover of torsinA(wt) is mostly dependent on autophagy, in contrast to torsinA(ΔE) which depends to a higher degree on selective degradation by the proteasome. These results confirm those of a study published in a different cell system while this manuscript was being prepared (Giles et al., 2008). Using a different experimental design, the authors demonstrated that torsinA is a very stable protein and, similar to our findings, is degraded by autophagy whereas torsinA(ΔE) has a significantly shorter half life as it is efficiently targeted to the proteasome for degradation. However, the authors did not address the presence of disulfide links or spheroid bodies and their influence on this process.
Recent reports demonstrated the presence of constitutive autophagy in mammalian neurons (Hara et al., 2006, Komatsu et al., 2006). This non-selective process that participates in ER recycling would be responsible for eliminating torsinA in normal conditions. However, physiological (i.e., starvation) or pharmacological (i.e., lithium) interventions that activate or inhibit autophagy could have an effect on the half life of torsinA(wt), with perhaps less effect on torsinA(ΔE) levels. As a result, this could modify the torsinA(wt):torsinA(ΔE) expression ratio, thought to be important for disease pathogenesis (Breakefield et al., 2001). These potential “environmental” modifiers of torsinA clearance could have an impact on disease pathogenesis and perhaps influence the reduced penetrance of the DYT1 mutation. Furthermore, genetic variations in components of the quality control machinery involved in this process could also represent genetic modifiers of DYT1 pathogenesis and penetrance. Ongoing interventional and genetic studies in animal models of DYT1 will help elucidate some of these questions.
Why is torsinA(ΔE) recognized by ERAD, a more selective process than autophagy, as an abnormal protein? In vitro studies suggest the DYT1 mutation does not cause torsinA to misfold (Kustedjo et al., 2003). In ERAD, ER proteins that retain a high mannose tag, acting as a “timer” for their progression through the secretory pathway, are identified as abnormal, retrotranslocated to the cytosol, ubiquitinated and sent to the proteasome for degradation. However, torsinA(wt) is a stable high mannose ER resident protein that does not normally exit the ER and is preferentially degraded by autophagy. This suggests that retaining the high mannose tag is not sufficient to target torsinA(ΔE) for proteasomal degradation. One possibility is that, due to its redistribution to the NE, this form of torsinA would be less accessible to the autophagic machinery and require the actions of the proteasome for its clearance, as suggested by our experiments with immunostaining. Alternatively, as it occurs for individual subunits of multimeric surface receptors that fail to assemble in the secretory pathway, defects in the normal incorporation of torsinA(ΔE) into multimeric complexes could be the basis for their rapid retrotranslocation for proteasomal degradation.
We extended our studies to analyze the influence of abnormal disulfide link-dependent oligomerization on the degradation process for torsinA(ΔE). Employing functional mutant forms of torsinA that localize to the ER or NE, we demonstrated that oligomerization is neither a consequence of the NE localization of torsinA nor causes torsinA to localize to the NE, but is likely a direct consequence of the glutamic acid deletion independent of its localization. It is tempting to speculate that the DYT1 mutation causes a conformational change in torsinA that favors the formation of an intermolecular disulfide link when torsinA subunits are assembled into multimers. A recent report demonstrated the presence of intramolecular disulfide links in torsinA in vitro that are coupled to the nucleotide binding capacity of this protein (Zhu et al., 2008). The formation of an abnormal intermolecular bond could “sequester” the cysteines implicated in the normal intramolecular linking process and, as a result, interfere with the completion of the normal ATPase cycle leading to the dominant negative effect of torsinA(ΔE) over torsinA(wt) in DYT1 dystonia.
The ER is a highly oxidizing environment where aberrant bonds can form when favored by structural protein changes. TorsinA(ΔE) has six evolutionarily conserved cysteines, two predicted to lie close to an α-helix in torsinA that could be disrupted by the DYT1 mutation (Kock et al., 2006) and perhaps trigger the formation of this abnormal bond between adjacent torsinA subunits when hexamers are assembled. These abnormal oligomers, and not monomeric torsinA(ΔE), could perhaps constitute the pathogenic species leading to neuronal dysfunction in DYT1 dystonia, and the cellular ability to eliminate them being critical to prevent this pathogenic event. Our findings suggest that the presence of disulfide links, which commonly need to be reduced before retrotranslocation to the cytosol for degradation (Romisch, 2005), lead to the requirement of autophagy for the clearance of oligomers, as opposed to the implication of the proteasome for the degradation of the monomeric form. The fact that torA(K108A) oligomers, a torsinA form that localizes to the ER, are also cleared by autophagy suggests the molecular basis for specific degradation of oligomers by autophagy is not the subcellular localization but the oligomerization itself. In a similar finding, soluble dysferlin, another disease-linked protein, was found to be degraded by the proteasome through ERAD. However, aggregates of mutated dysferlin within the ER were cleared by autophagy (Fujita et al., 2007). Therefore, abnormal aggregation or oligomerization of mutated proteins within the secretory pathway can modify their catabolic process.
In summary, our results suggest that, whether torsinA(ΔE) oligomers or monomers are responsible for neuronal dysfunction in DYT1, the quality control system could play a major role in DYT1 pathogenesis. By eliminating monomeric torsinA(ΔE), the proteasome would potentially diminish the formation of abnormal oligomers that, once formed, would be cleared by autophagy, thus modifying the torsinA(wt):torsinA(ΔE) ratio (figure 7). Consequently, genetic or environmental impairment of the ability of ERAD to clear monomeric torsinA(ΔE) or autophagy to dispose of the oligomeric form, could modulate DTY1 pathogenesis and penetrance. A therapeutic strategy aimed at enhancing the torsinA(wt):torsinA(ΔE) ratio is being developed through RNA interference-mediated, allele-specific silencing of torsinA(ΔE) as a potential therapy for DYT1 (Gonzalez-Alegre et al., 2003, Gonzalez-Alegre et al., 2005). Pharmacological manipulation of both proteolytic pathways is feasible (Nalepa et al., 2006, Rubinsztein et al., 2007) and could be employed to achieve this same goal.
Figure 7. Schematic representation of torsinA degradation pathways.
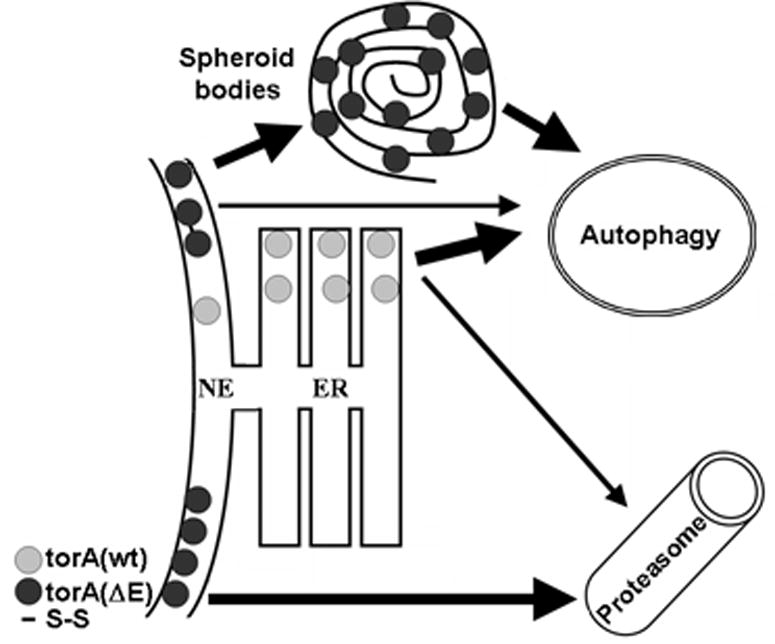
The turnover of torsinA(wt), preferentially located within the ER, is mostly dependent on the actions of autophagy. On the other hand, the NE localization of torsinA(ΔE) targets it for proteasomal degradation when existing as monomers. However, the presence of intermolecular disulfide links in torsinA(ΔE) oligomers would interfere with the retrotranslocation step, requiring autophagy for their clearance. Similarly, torsinA(ΔE) accumulated in spheroid bodies would be degraded through autophagy.
Supplementary Material
Characterization of the TA913 antibody. This rabbit polyclonal antibody (TA913) detects human torsinA but not mouse or rat torsinA, or c-terminus tagged human torsinA. (A) Western blot analysis with preimmune and torsinA antiserum showing specificity of the antibody in torsinA(wt) inducible cells. (B) Western blot of human (lanes 3-8) and mouse brain lysates (lane 9) showing human specificity. Lanes 1-2 are PC6-3 controls. The amount of protein loaded of PC6-3 lysates was 1/50 of the rest to avoid overexposure. Therefore, the loading control is not detected. (C) TA913 detects endogenous torsinA in human and primate cell lines. (D) IF of inducible cell lines shows torsinA(wt) in the ER, whereas torsinA(ΔE) localizes to the NE and spheroid bodies. TorsinA is seen in red and DAPI nuclear staining in blue. (E) TA913 recognizes untagged human, but not overexpressed mouse torsinA (mtorA) or c-terminus FLAG-tagged human torsinA.
Acknowledgments
We thank all members of the Gonzalez-Alegre laboratory and Dr. Kevin Glenn for their helpful comments. This work was supported by NIH/NINDS K02 NS058450 (PGA) and the Neuroscience Training Program NIH T32 NS007421 (KG).
List of abbreviations
- 3MA
3-methyladenine
- AAA
ATPases Associated with diverse cellular Activities
- ATP
adenosine triphosphate
- DOX
doxycycline
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- IF
indirect immunofluorescence
- NGF
nerve growth factor
- NE
nuclear envelope
- NEM
N-ethylmaleimide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Breakefield XO, Kamm C, Hanson PI. TorsinA: movement at many levels. Neuron. 2001;31:9–12. doi: 10.1016/s0896-6273(01)00350-6. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Fahn S, Bressman SB, Marsden CD. Classification of dystonia. Adv Neurol. 1998;78:1–10. [PubMed] [Google Scholar]
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- Giles LM, Chen J, Li L, Chin LS. Dystonia-associated mutations cause premature degradation of torsinA protein and cell type-specific mislocalization to the nuclear envelope. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alegre P. The inherited dystonias. Semin Neurol. 2007;27:151–158. doi: 10.1055/s-2007-971170. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Bode N, Davidson BL, Paulson HL. Silencing primary dystonia: lentiviral-mediated RNA interference therapy for DYT1 dystonia. J Neurosci. 2005;25:10502–10509. doi: 10.1523/JNEUROSCI.3016-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Miller VM, Davidson BL, Paulson HL. Toward therapy for DYT1 dystonia: allele-specific silencing of mutant. TorsinA Ann Neurol. 2003;53:781–787. doi: 10.1002/ana.10548. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24:2593–2601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT. Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci U S A. 2004;101:847–852. doi: 10.1073/pnas.0304375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO. Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Genet. 2000;9:1403–1413. doi: 10.1093/hmg/9.9.1403. [DOI] [PubMed] [Google Scholar]
- Kock N, Naismith TV, Boston HE, Ozelius LJ, Corey DP, Breakefield XO, Hanson PI. Effects of genetic variations in the dystonia protein torsinA: identification of polymorphism at residue 216 as protein modifier. Hum Mol Genet. 2006;15:1355–1364. doi: 10.1093/hmg/ddl055. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kruse KB, Brodsky JL, McCracken AA. Autophagy: an ER protein quality control process. Autophagy. 2006;2:135–137. doi: 10.4161/auto.2.2.2388. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Bracey MH, Cravatt BF. Torsin A and its torsion dystonia-associated mutant forms are lumenal glycoproteins that exhibit distinct subcellular localizations. J Biol Chem. 2000;275:27933–27939. doi: 10.1074/jbc.M910025199. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Deechongkit S, Kelly JW, Cravatt BF. Recombinant expression, purification, and comparative characterization of torsinA and its torsion dystonia-associated variant Delta E-torsinA. Biochemistry. 2003;42:15333–15341. doi: 10.1021/bi0349569. [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- Naismith TV, Heuser JE, Breakefield XO, Hanson PI. TorsinA in the nuclear envelope. Proc Natl Acad Sci U S A. 2004;101:7612–7617. doi: 10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat Rev Drug Discov. 2006;5:596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Risch NJ, Bressman SB, Senthil G, Ozelius LJ. Intragenic Cis and Trans modification of genetic susceptibility in DYT1 torsion dystonia. Am J Hum Genet. 2007;80:1188–1193. doi: 10.1086/518427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romisch K. Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol. 2005;21:435–456. doi: 10.1146/annurev.cellbio.21.012704.133250. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- Torres GE, Sweeney AL, Beaulieu JM, Shashidharan P, Caron MG. Effect of torsinA on membrane proteins reveals a loss of function and a dominant-negative phenotype of the dystonia-associated DeltaE-torsinA mutant. Proc Natl Acad Sci U S A. 2004;101:15650–15655. doi: 10.1073/pnas.0308088101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Zhu L, Wrabl JO, Hayashi AP, Rose LS, Thomas PJ. The Torsin-family AAA+ Protein OOC-5 Contains a Critical Disulfide Adjacent to Sensor-II that Couples Redox State to Nucleotide Binding. Mol Biol Cell. 2008 doi: 10.1091/mbc.E08-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of the TA913 antibody. This rabbit polyclonal antibody (TA913) detects human torsinA but not mouse or rat torsinA, or c-terminus tagged human torsinA. (A) Western blot analysis with preimmune and torsinA antiserum showing specificity of the antibody in torsinA(wt) inducible cells. (B) Western blot of human (lanes 3-8) and mouse brain lysates (lane 9) showing human specificity. Lanes 1-2 are PC6-3 controls. The amount of protein loaded of PC6-3 lysates was 1/50 of the rest to avoid overexposure. Therefore, the loading control is not detected. (C) TA913 detects endogenous torsinA in human and primate cell lines. (D) IF of inducible cell lines shows torsinA(wt) in the ER, whereas torsinA(ΔE) localizes to the NE and spheroid bodies. TorsinA is seen in red and DAPI nuclear staining in blue. (E) TA913 recognizes untagged human, but not overexpressed mouse torsinA (mtorA) or c-terminus FLAG-tagged human torsinA.