

Abstract
Gene silencing through promoter hypermethylation is a growing concept in the development of human cancers. In this study, we examined the contribution of aberrant methylation of promoter regions in methylation-prone tumor suppressors to the pathogenesis of vulvar cancer. Thirteen cell lines from 12 patients with squamous cell carcinoma of the vulva (SCV) were evaluated for aberrant methylation status and gene copy number alterations, concomitantly, using the methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) assay. Of the 22 tumor suppressor genes examined, aberrant methylation was observed for 9 genes: TP73, FHIT, VHL, APC, ESR1, CDKN2B, DAPK1, GSTP1 andI GSF4. The most frequently methylated genes included TP73 in 9 of 13 cell lines, and IGSF4, DAPK1 and FHIT in 3 of 13 cell lines. Methylation specific polymerase chain reaction (MSP) was performed for TP73 and FHIT to confirm aberrant methylation by MS-MLPA. In the context of gene copy number and methylation status, both copies of the TP73 gene were hypermethylated. Loss or decreased mRNA expression of TP73 and IGSF4 by RT-PCR confirmed aberrant methylation. Frequent genetic alterations of loss and gain ofgene copy number included gain of GSTP1 and MEN1, and loss of MFHAS1 and IGSF4 in over 50% of the SCV cell lines. These findings underscore the contribution of both genetic and epigenetic events to the underlying pathogenesis of squamous cell carcinoma of the vulva.
Keywords: Squamous cell carcinoma of vulva (SCV), Epigenetics, Promoter hypermethylation, Methylation, specific Multiplex Ligation, dependent Probe Amplification (MS-MLPA) assay
INTRODUCTION
Chromosome aberrations have served as landmarks in the identification of cancer genes in many tumor types; however, individual genes altered in tumors cannot be deduced solely from the type of chromosome alteration (1). Although the importance of genetic alterations in cancer is well recognized, the appreciation of epigenetic changes is more recent and growing. The term ‘epigenetics’ defines all meiotically and mitotically heritable changes in gene expression that are not coded in the DNA sequence itself (2). Establishment and maintenance of epigenetic control (gene silencing) has several aspects, which include promoter region hypermethylation, methyl-binding proteins, DNA methyltransferases, histone deacetylases and chromatin state. CpG islands, which are stretches of DNA with a GC content greater than 55% (3), located in promoter regions of genes are mainly unmethylated in normal tissues except for imprinted genes (4) and X-chromosome genes in females (5). Methylation of these CpG islands causes stable heritable transcriptional silencing (2). Aberrant methylation of CpG islands is a hallmark of human cancers and is found early during carcinogenesis (2). Genes in cellular pathways which are inactivated by promoter region hypermethylation include MGMT (DNA repair), p16INK4a, p15INK4b (cell cycle), DAPK (apoptosis) and GSTP1 (detoxification) (6–9).
In this study, we focus on aberrant methylation of promoter regions in methylation-prone tumor suppressors in the pathogenesis of vulvar cancer. Vulvar carcinoma accounts for about 4% of all gynecological cancers in the USA (10). It is mainly a disease of elderly women, but has also been reported in some premenopausal women. Squamous cell carcinoma accounts for 90% of vulvar cancers (10). The main risk factors are human papillomavirus infection and chronic vulvar pruritis.
Previous studies from our group(11–13) have reported consistent chromosomal abnormalities in SCV with further characterization of these aberrations at the level of individual genes(1). In this study we examined aberrant promoter methylation of 22 methylation-prone tumor suppressor genes using a high-throughput multi-gene probe panel (41 gene probes, 35 unique genes, including control probes) in 13 SCV cell lines using a modification of the multiplex ligation-dependent probe amplification assay (MLPA) (1, 14–16) termed the methylation specific multiplex ligation-dependent probe amplification (MS-MLPA) assay (7–9, 17, 18).
MATERIALS AND METHODS
Patient histories
The patients’ ages, Tumor–Node–Metastasis (TNM) classification of the disease, the histological grade of the tumor, prior therapy at the time of the tissue biopsy for the establishment of the cell line, and the site of the specimen, are noted in Table 1 (11–13). The phenotypic characterization including karyotypic analysis of the two specimens of vulvar carcinoma from the same patient, UM-SCV-1A and UM-SCV-1B has been previously published (11). UM-SCV-1A was obtained from the primary tumor site and UM-SCV-1B from a malignant pleural effusion.
Table 1.
Patient Clinical and Histopathological Data
| Cell line | Age | TNM classification | Grade of the SCC | Prior therapy | Site of the specimen | Survival after initial diagnosis |
|---|---|---|---|---|---|---|
| UM-SCV-1A* | 62 | T3N2M1 | well –poor | none | primary | 2 months |
| UM-SCV-1B* | 62 | T3N2M1 | poor | none | Metastasis | 2 months |
| UM-SCV-2* | 86 | T3N1M0 | poor | surgery | local recurrence | 10 months |
| UM-SCV-3* | 66 | T2N0M0 | mod–well | none | primary | >4, 5 years |
| UM-SCV-4* | 41 | T2N2M0 | well | none | primary | >4 years |
| UM-SCV-6* | 43 | T1N1M0 | mod | surgery | primary | 15 months |
| UM-SCV-7* | 77 | T2N2M0 | well –poor | none | primary | 1 month |
| UT-SCV-1* | 71 | T2N1M0 | well | surgery + RTa | Metastasis | 1 year |
| UT-SCV-2* | 63 | T1N0M0 | mod | surgery + RTa + CTb | local recurrence | >13 years |
| UT-SCV-3* | 73 | T3N2M0 | mod | none | primary | 3 months |
| UT-SCV-4† | 31 | T2N0M0 | mod | none | primary | 10 years |
| UT-SCV-5† | 46 | T3N2M1 | well | none | pleural effusion | 5 weeks |
| UT-SCV-6† | 72 | T3N2N0 | well | none | primary | 8 weeks |
RT indicates radiotherapy.
CT indicates chemotherapy.
Raitanen M, et. al. Characterization of 10 vulvar carcinoma cell lines by karyotyping, comparative genomic hybridization and flow cytometry. Gynecol Oncol 2004;93(1):155–63.
personal communication Dr. Seija Grenman, Turku Central University, Turku, Finland
This study performed MS-MLPA on previously cytogenetically characterized cell lines UM-SCV-1A, -1B, -2, -3, -4, -6, -7, UT-SCV-1, -2, -3 and three additional cell lines, UT-SCV-4, -5, and -6, for a total of 13 cell lines.
DNA Extraction
DNA, extracted using the QIAamp Kit (Qiagen Inc, Chatsworth, CA), was obtained from passaged cell lines that originated from fresh tumor samples as described previously(11, 19, 20). The number of passages for the cell lines were as follows: UM-SCV-1A – passage(P) 22, UM-SCV-1B – P8, UM-SCV-2 – P22, UM-SCV- 3 – P26, UM-SCV- 4 – P19, UM-SCV-6 – P17, UM-SCV-7 – P13, UT-SCV-1 – P26, UT-SCV-2 and -3 – P27, UT-SCV-4 – P12, UT-SCV-5 – P9, and UT-SCV-6 – P12.
Isolation of total RNA from cultured cells
Cells for tissue culture for isolation of mRNA for gene expression studies using Real Time PCR (RT-PCR) were available only for cell lines UT-SCV-2, -3, -4 and -6. Cultured cells from these cell lines were grown in a monolayer, rinsed with 1x PBS, trypsinized and collected as a cell pellet. The latter was washed 2 times with PBS, spun, and the supernatant completely removed to obtain cell pellets for immediate RNA extraction. RNA was extracted using the QIAamp RNA mini protocol for isolation of total RNA from cultured cells (Valencia, CA USA).
Reverse Transcriptase (RT-PCR)
Total RNA (1μg) for synthesis was done using the SuperScript First-strand Synthesis System for RT-PCR (Invitrogen Life Technologies, Carlsbad, CA) according to the manufacturer’s protocol. Briefly, RNA, Oligo (dt) primer and dNTP mix were denatured at 65°C for 5 minutes and immediately chilled on ice. This was followed by an annealing step with addition of buffer, DTT, RNaseout inhibitor, and MgCl2 (without SuperScriptII {SSII}) and incubated at 42°C for 2min, followed by the addition of SSII. cDNA synthesis was allowed to continue at 42°C for 50 min. The reaction was terminated at 70°C for 15 minutes and immediately placed on ice. Removal of residual RNA was performed by the addition of 1μl of RNase H incubated at 37°C for 20 min. PCR was performed using 2 μl of RNA free cDNA.
Real-time Reverse Transcription Polymerase Chain Reaction
Expression of TP73, IGSF4 and DAPK1 was analyzed using a real-time quantification method (LightCycler) according to the manufacturer’s recommendations (Roche Diagnostics, Indianapolis, IN). PCR primers were as follows: TP73: 5′-GGC TGC GAC GGC TGC AGA G-3′ (forward) and 5′-GCT CAG CAG ATT GAA CTG GGC CAT G-3′ (reverse), giving a product of 256 base pairs; IGSF4: 5′-GGC TTC TGC TGT TGC TC-3′ (forward) and 5′-CCC TGA AAT AAA TGG TCT GC-3′ (reverse), producing a product of 183 base pairs, and for DAPK1: 5′-TGA CAG TTT ATC ATG ACC GTG TTC AG-3′ (forward) and 5′-GTG CTG GAT CTC CTT CAG GAT-3′ (reverse), for a product size of 231 base pairs (Table 2).
Table 2.
RT-PCR Primer Sequences for SCV
| Gene | Forward Primer | Reverse Primer | Product Size |
|---|---|---|---|
| TP73 | 5′-GGCTGCGACGGCTGCAGAG-3′ | 5′-GCTCAGCAGATTGAACTGGGCCATG-3′ | 256 |
| IGSF4 | Exon 1–2: 5′-GGCTTCTGCTGTTGCTC-3′ | Exon 1–2: 5′-CCCTGAAATAAATGGTCTGC-3′ | 183 |
| DAPK1 | Exon 1–2: 5′-TGACAGTTTATCATGACCGTGTTCAG-3′ | Exon 1–2: 5′-GTGCTGGATCTCCTTCAGGAT-3′ | 231 |
Because the precise amount of total RNA added to each reaction and its quality are difficult to determine, expression of TP73, IGSF4 and DAPK1 were normalized with an endogenous standard β-actin. Standard curves were used to determine the concentration of TP73, IGSF4, DAPK1 and β-actin gene products. Quantification of TP73, IGSF4, and DAPK1 expression were obtained by direct comparison with β-actin dilution standards amplified in parallel reactions in the same run (Table 3). After real-time data acquisition, the parameter Ct (threshold cycle/crossing point) was calculated by determining the point at which the fluorescence exceeds an arbitrary threshold limit, which is set to cross the fluorescent signal of all standards in the exponential phase. The target load in the normal and the 4 cell lines, UT-SCV-2, -3, -4 and -6 were quantified by measuring Ct and using a standard curve to determine the starting target message quantity.
Table 3.
RT-PCR results for TP73, IGSF4, and DAPK1
| TP73 Gene expression | |||||
|---|---|---|---|---|---|
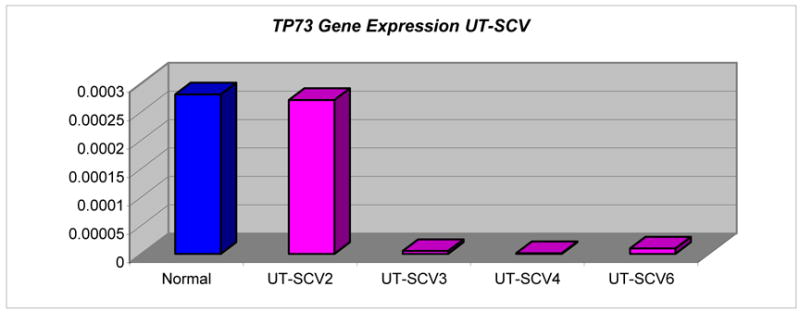
| Sample | Ct β-actin | Ct TP73 | Ct Δ(delta) | 2−(Ct Δ) | |
| Normal | 18.7 | 30.5 | 11.8 | 0.000280444 |
|
| UT-SCV2 | 12.49 | 24.34 | 11.85 | 0.000270891 | |
| UT-SCV3 | 10.17 | 27.7 | 17.53 | 5.28377E-06 | |
| UT-SCV4 | 11.9 | 31.66 | 19.76 | 1.12628E-06 | |
| UT-SCV6 | 12.85 | 29.48 | 16.63 | 9.85987E-06 | |
| IGSF4 Gene expression | |||||
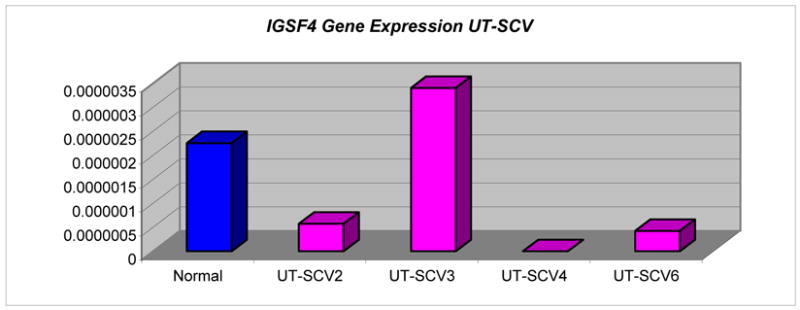
| Sample | Ct β-actin | Ct IGSF4 | Ct Δ(delta) | 2−(Ct Δ) |
|
| Normal | 16.18 | 34.94 | 18.76 | 2.25256E-06 | |
| UT-SCV2 | 13.1 | 33.84 | 20.74 | 5.71002E-07 | |
| UT-SCV3 | 11.85 | 24.09 | 18.17 | 3.40244E-06 | |
| UT-SCV4 | 12.63 | 43.62 | 30.99 | 4.689E-10 | |
| UT-SCV6 | 13.87 | 35.04 | 21.17 | 4.23833E-07 | |
| DAPK1 Gene expression | |||||
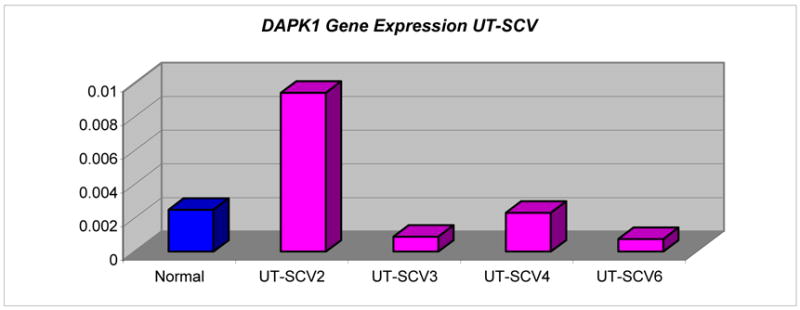
| Sample | Ct β-actin | Ct DAPK1 | Ct Δ(delta) | 2−(Ct Δ) |
|
| Normal | 16.3 | 24.95 | 8.65 | 0.002489376 | |
| UT-SCV2 | 13.28 | 20.01 | 6.73 | 0.009420374 | |
| UT-SCV3 | 11.87 | 22 | 10.13 | 0.000892414 | |
| UT-SCV4 | 12.81 | 21.57 | 8.76 | 0.002306626 | |
| UT-SCV6 | 14.17 | 24.54 | 10.37 | 0.000755647 | |
For accurate quantification of cDNA targets TP73, IGSF4, and DAPK1, the amplification efficiency of the target should be similar to that of the internal standard. To obtain this, the slope of the standard curve was converted to amplification efficiency E by the following algorithm: E=10 −1/slope. To calculate TP73, IGSF4, or DAPK1 expression levels, initially the difference in Ct between TP73, IGSF4, or DAPK1 and β-actin, termed CtΔ (delta), was obtained (Table 3). Expression levels of the target were plotted as exponent values 2−ΔCt (Table 3).
The Methylation Specific Multiplex Ligation Dependent Probe Amplification (MS-MLPA) Assay
The Methylation Specific Multiplex Ligation Dependent Probe Amplification assay allows for the relative quantification of approximately 41 different DNA sequences in a single reaction requiring only 20 ng of human DNA (21). The standard use of the technique to observe quantitative changes in copy number has been outlined in other studies (1, 14, 15, 22). Modification of the MLPA to detect aberrant methylation (MS-MLPA) has been detailed elsewhere (7, 9, 17, 21).
The probe design in MS-MLPA is similar to ordinary MLPA probes. For 26/41 probes, the recognition sequence detected by the MLPA probe is contained within a restriction site for the methyl-sensitive enzyme, HhaI (Figure 1). The 41 gene probe panel (Table 4) interrogates 35 unique genes implicated in cancer for losses and gains in a separate reaction in the absence of the methyl-sensitive enzyme HhaI. Because there are two probes each for VHL, CDKN2A, BRCA1 and BRCA2, and 3 probes for MLH1, a normal control DNA sample will generate 41 individual peaks in the absence of HhaI (Figures 2 & 3). A concurrently run reaction with the 41 gene probe set in the presence of HhaI is designed to detect aberrant promoter hypermethylation by taking advantage of a HhaI site in the promoter region of 22 of the 35 unique genes (note that one of the two BRCA1 probes is designed to recognize a region outside the HhaI recognition site, Table 4). Fifteen of the 41 gene probes are designed outside a Hha1 site and serve as undigested controls (Figure 2–5). Upon digestion of the sample DNA with HhaI, probes that recognize the unmethylated regions will not generate a signal because these sequences have become cut by HhaI and cannot bind to the probe (Figure 1). Conversely, a MLPA probe will bind to an intact methylated site, spared by HhaI, and generate an amplification signal (Figures 1, 4 & 5). To detect both aberrant methylation and changes in copy number, each sample requires 2 MLPA reactions. Details of the assay including its interpretation are described elsewhere (21). Aberrant methylation is identified as the appearance of a signal peak that is otherwise absent in normal DNA samples (Figures 2–5). To quantify whether one, both, or more copies of a specific gene locus becomes aberrantly hypermethylated, a previously described mathematical algorithm was employed (17).
Figure 1.
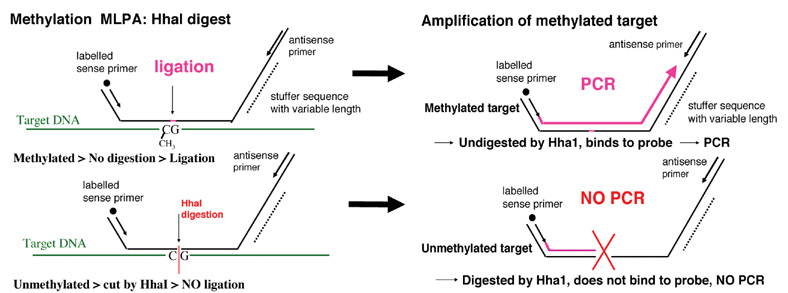
(17). Methylation Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA).
Table 4.
MS-MLPA Probe Panel
| # | PCR Product size | Gene probe | Chrom Loc | # | PCR Product size | Gene probe | Chrom Loc | # | PCR Product size | Gene probe | Chrom Loc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M-238 | TP73 | 01p36 | 481 | MFHAS1 | 08p23.1 | 310 | RB1 | 13q14.3 | ||
| 472 | NRAS | 01p13.2 | 12 | M-160 | CDKN2A | 09p21 | 202 | MLH3 | 14q24.3 | ||
| 364 | MSH6 | 02p11 | 13 | M-427 | CDKN2A | 09p21 | 283 | TSC2 | 16p13.3 | ||
| 2 | M-220 | VHL | 03p26 | 14 | M-211 | CDKN2B | 09p21 | 21 | M-400 | ASC | 16p12 |
| 3 | M-274 | VHL | 03p26 | 15 | M-346 | DAPK1 | 09q34.1 | 22 | M-247 | CDH13 | 16q24.2 |
| 4 | M-265 | FANCD2 | 03p26 | 136 | CREM | 10p12.1 | 23 | M-355 | HIC1 | 17p13.3 | |
| 5 | M-166 | MLH1 | 03p21.3 | 16 | M-193 | MEN1 | 11q13 | 337 | BRCA1 | 17q21 | |
| 6 | M-292 | MLH1 | 03p21.3 | 17 | M-454 | GSTP1 | 11q13 | 24 | M-436 | BRCA1 | 17q21 |
| 7 | M-463 | MLH1 | 03p21.3 | 18 | M-319 | IGSF4 | 11q23 | 256 | BCL2 | 18q21.3 | |
| 8 | M-328 | RASSF1 | 03p21.3 | 175 | TNFRSF1A | 12p13 | 391 | KLK3 | 19q13 | ||
| 9 | M-409 | FHIT | 03p14.2 | 445 | TNFRSF7 | 12p13 | 25 | M-184 | KLK10 | 19q13.3 | |
| 10 | M-148 | APC | 05q21 | 19 | M-382 | CDKN1B | 12q13.1 | 229 | NF2 | 22q12 | |
| 154 | IL4 | 05q31.1 | 20 | M-301 | BRCA2 | 13q12 | 26 | M-142 | TIMP3 | 22q12.3 | |
| 11 | M-373 | ESR1 | 06q25.1 | 418 | BRCA2 | 13q12.3 |
 probes with Hha I sites in CpG rich promoter regions
probes with Hha I sites in CpG rich promoter regions
 genes represented by more than one gene probe
genes represented by more than one gene probe
Figure 2.

Normal (control), UT-SCV-3 and UT-SCV-4 MS-MLPA assay results of MS-MLPA probe mix with and without HhaI enzyme. Note methylation of both copies of TP73 in UT-SCV-3 and 4 and methylation of both copies of IGSF4 and FHIT in UT-SCV-4.
Figure 3.

Normal (control), UM-SCV-2 and UT-SCV-6 MS-MLPA assay results of MS-MLPA probe mix with and without HhaI enzyme. Note methylation of both copies of TP73 in UM-SCV-2 and UT-SCV-6. Methylation of one copy of DAPK1 and gain of GSTP1 copy number in UM-SCV-2.
Figure 5.

MS-MLPA assay results showing 15 peaks in the normal (control) with aberrant methylation peaks for TP73, IGSF4 and FHIT in UT-SCV-4 and TP73 in UT-SCV-6.
Figure 4.

MS-MLPA assay results showing 15 peaks in the normal (control) with an aberrant methylation peak for CDKN2B in UT-SCV-2 and TP73 in UT-SCV-3.
Bisulfite Modification and Methylation-Specific Polymerase (MSP) Chain Reaction Assay
MSP was performed for cell lines with sufficient amounts of DNA. Genomic DNA (100ng) from SCV cell line DNA and control universal methylated DNA (Chamicon International, Inc) and control unmethylated DNA (normal genomic DNA) were modified using the EZ DNA methylation kit (Zymo Research, Orange, CA) during which methylated DNA is protected and unmethylated cytosine is converted to uracil (9).
The modified DNA served as a template using primers specific for either the methylated or modified unmethylated TP73 and FHIT sequences. TP73 methylation specific primers (MS) were sense: 5′-TCGGGGGGCGGTTCGGTTTCGC-3′, anti-sense: 5′-CTCTAACCGCTCCAACCTCTCGCGAACG-3′ (21). Unmethylated DNA specific primers (UMS) were sense: 5′-GGGGGTGGTTTGGTTTTGTGGGT-3′, antisense: 5′-CTAAACTCTAACCACTCCAACCTCTCACAAACA-3′ (21). FHIT methylation specific primers (MS) were sense: 5′-TTGGGGCGCGGGTTTGGGTTTTTA CGC-3′, anti-sense: 5′-CGTAAACGACGCCGACCCCACTA-3′ (23). Unmethylated DNA specific primers (UMS) were sense: 5′-TTGGGGTGTGGGTTTGGGTTTTTATG-3′, antisense: 5′-CATAAACAACACCAACCCCACTA-3′ (23).
MSP amplification for TP73 was performed using 3ul of bisulfite modified DNA in a PCR mix containing 1X PCR buffer, 2mM MgCl2 and 2U Amp gold Taq DNA polymerase, 0.4uM primer followed by 38 cycles at 95°C 50 seconds, 62°C 50 seconds, 72°C 1min. PCR generated a 193bp methylated product and a 195bp unmethylated product (Figure 6).
Figure 6.

Methylation Specific PCR (MSP) confirmation of aberrant methylation detected by MS-MLPA for TP73.
Figure 6A: Lanes 1 & 2: universal methylated and unmethylated controls; Lanes 3-12 span UM cell lines 1a, 2-4 and 6. Note presence of methylated product in UM-SCV-1a, -2, -3 and -6. Note absence of methylated product in UM-SCV-4; Lanes 13 & 14: negative control.
Figure 6B: Lanes 1 & 2: universal methylated and unmethylated controls; Lanes 3-12 span UT cell lines 1-4 and 6. Note presence of methylated product in UT-SCV-2, -3, -4, and -6. Note absence of methylated product in UT-SCV-1; Lanes 13 & 14: negative control.
MSP amplification for FHIT was performed using 3ul of bisulfite modified DNA in a PCR mix containing 1X PCR buffer, 2mM MgCl2 and 2U Amp gold Taq DNA polymerase, 0.4uM primer followed by 38 cycles at 95°C 50 seconds, 62°C 50 seconds, 72°C 1min. PCR generated a 74bp methylated and unmethylated product (Figure 7).
Figure 7.

Methylation Specific PCR (MSP) confirmation of aberrant methylation detected by MS-MLPA for FHIT. Lanes 1 & 2: universal methylated and unmethylated controls; Lanes 3-12 span UT cell lines 1-4 and 6. Note presence of methylated product in UT-SCV-2 and UT-SCV-4. Note absence of methylated product in UT-SCV-1, -3 and -6; Lanes 13 & 14: negative control.
The resultant PCR for TP73 and FHIT were separated on 2% agarose gel, stained with ethidium bromide and visualized under UV illumination (Figures 6 & 7).
RESULTS
Aberrant methylation was observed for 9 genes, APC (5q21), CDKN2B (9p21), VHL (3p26), TP73 (1p36.3), IGSF4 (11q23.2), DAPK1 (9q34.1), ESR1 (6q25.1), FHIT (3p14.2) and GSTP1 (11q13) in 11 of 13 SCV cell lines. The most frequently methylated genes were TP73 in 9 of 13 cell lines, detected by MS-MLPA in 6/13 and MSP in 3/13, followed by IGSF4, DAPK1 and FHIT in 3 of 13 cell lines (Table 5). UM-SCV-3 showed aberrant methylation for 6 of the 9 genes (APC, VHL, TP73, IGSF4, DAPK1 and ESR1), all of which had both copies methylated. In UM-SCV-7, hypermethylation was observed for both copies of APC and FHIT, the sole copy of IGSF4 and one of two copies of ESR1. UT-SCV-2 and UT-SCV-4 showed hypermethylation of 3 of the 9 aberrantly methylated genes: CDKN2B, FHIT and GSTP1 in UT-SCV-2 and TP73, IGSF4 and FHIT in UT-SCV-4 (Table 5). Promoter hypermethylation of VHL, CDKN2B, and GSTP1 was infrequent, occurring in only 1 of 13 cell lines (UM-SCV-3 and UT-SCV-2). Aberrant methylation was not observed in UM-SCV-1B, UM-SCV-6, UT-SCV-1 and UT-SCV-5.
Table 5.
SCV Aberrant gene methylation and copy number
| APC | CDKN2B | VHL | TP73 | IGSF4 | DAPK1 | ESR1 | FHIT | GSTP1 | |
|---|---|---|---|---|---|---|---|---|---|
| UM-SCV-1A | 2 | 2 | 2 | *2 | 1 | n=2; m=1 | 2 | 2 | 2 |
| UM-SCV-1B | 2 | 2 | 2 | 2 | 1 | 2 | 3 | 3 | 4 |
| UM-SCV-2 | 2 | 2 | 2 | n=2; m=2 | 1 | n=2; m=1 | 2 | 2 | 7 |
| UM-SCV-3 | n=2; m=2 | 2 | n=2; m=2 | n=2; m=2 | n=2; m=2 | n=2; m=2 | n=2; m=2 | 2 | 3 |
| UM-SCV-4 | 2 | 0 | 2 | n=2; m=2 | 3 | 2 | 2 | 3 | 3 |
| UM-SCV-6 | 2 | 2 | 2 | *2 | 1 | 2 | 3 | 2 | 4 |
| UM-SCV-7 | n=2; m=2 | 3 | 2 | 2 | n=1; m=1 | 3 | n=2; m=1 | N=2; m=2 | 4 |
| UT-SCV-1 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 3 |
| UT-SCV-2 | 2 | n=3; m=1 | 2 | *2 | 2 | 3 | 3 | N=2; m=2 | n=3; m=1 |
| UT-SCV-3 | 2 | 2 | 2 | n=2; m=2 | 1 | 2 | 3 | 2 | 2 |
| UT-SCV-4 | 2 | 3 | 2 | n=2; m=2 | n=2; m=2 | 2 | 2 | N=2; m=2 | 3 |
| UT-SCV-5 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 |
| UT-SCV-6 | 2 | 1 | 2 | n=2; m=2 | 1 | 3 | 2 | 1 | 3 |
n = copy number without Hha1; m = methylated copy number after Hha1 digestion detected by MS-MLPA
= unmethylated by MS-MLPA but methylated by MSP
Gene copy number in the context of methylation status, indicated both copies of TP73, FHIT, IGSF4, APC, VHL, DAPK1, and ESR1 were methylated in 6, 3, 2, 2, 1, 1, and 1 cell lines, respectively (Table 5). For IGSF4, in UM-SCV-7, the sole copy was methylated. For DAPK1 and ESR1, one of two copies was methylated in UM-SCV-1A and UM-SCV-2, and UM-SCV-7, respectively (Table 5). Gain of GSTP1 and MEN1 was most frequent occurring in 11 of 13 cell lines. Aberrant methylation of GSTP1, observed in only UT- SCV-2 indicated hypermethylation of 1 of 3 copies. In 6 of 13 cell lines, the sole IGSF4 copy was unmethylated. Homozygous loss of CDKN2B was noted in UM-SCV-4, loss of one copy in UT-SCV-6, gain of copies in UM-SCV-7 and UT-SCV-4, and methylation of 1 of 3 copies in UT-SCV-2. Five of 13 cell lines with gain of ESR1 did not show methylation. FHIT and DAPK1 showed gain in 3 of 13 cell lines, none of which were methylated (Table 5). Common losses consisted of MFHAS1, IGSF4, VHL, BCL2, MLH1 and FANCD2 in 9, 7, 6, 6, 5 and 5 of 13 cell lines, respectively.
Absence or decreased mRNA expression of TP73 was confirmed by RT-PCR in UT-SCV-3, -4 and -6, supporting aberrant methylation of TP73 by MS-MLPA. UT-SCV-2 with two unmethylated copies of TP73 had normal expression of its mRNA transcript (Tables 3 & 6).
Table 6.
Gene expression by RT-PCR and Copy Number in UT-SCV Cell lines
| Sample | TP73 | IGSF4 | DAPK1 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Expression | Copy | Methylated | Expression | Copy | Methylated | Expression | Copy | Methylated | |
| UT-SCV-2 | Normal | 2 | 0 | L | 2 | 0 | H | 3 | 0 |
| UT-SCV-3 | L | 2 | 2 | H | 1 | 0 | L | 2 | 0 |
| UT-SCV-4 | L | 2 | 2 | absent | 2 | 2 | normal | 2 | 0 |
| UT-SCV-6 | L | 2 | 2 | L | 1 | 0 | L | 3 | 0 |
Absence of expression of IGSF4 in UT-SCV-4 was concordant with promoter hypermethylation of both copies. Decreased expression of unmethylated IGSF4 in UT-SCV-6 was consistent with loss of 1 gene copy. Unmethylated IGSF4 expression was variable and discordant with gene copy number in UT-SCV-2 (2 copies) and UT-SCV-3 (1 copy) (Table 3). Expression levels of unmethylated DAPK1 and gene copy number were concordant for UT-SCV-2 (3 copies) and UT-SCV-4 (2 copies), but varied in UT-SCV-3 and UT-SCV-6 (Tables 3 & 6).
MSP was performed for TP73 and FHIT. MSP of TP73 confirmed aberrant methylation detected by MS-MLPA for UM-SCV-2, UM-SCV-3, UT-SCV-3, -4 and -6. In addition to these cell lines, MSP indicated aberrant methylation of TP73 in UM-SCV-1a, UM-SCV-6 and UT-SCV-2, not detected by MS-MLPA. MSP did not show methylation of TP73 in UM-SCV-4, indicated by MS-MLPA. MSP confirmed methylation of FHIT detected by MS-MLPA for UT-SCV-2 and UT-SCV-4.
DISCUSSION
Epigenetic alterations produce heritable changes in gene expression without a change in the DNA coding sequence itself. Promoter region hypermethylation is known to be an early event in carcinogenesis(7, 8, 24, 25). The consequence of CpG island hypermethylation, especially for those islands associated with tumor suppressor gene (TSG) promoters is the loss of TSG function, which contributes to tumorigenesis (17).
The Methylation-Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) assay is a high throughput quantitative assay permitting the dual detection of genomic alterations of losses and gains and epigenetic events of promoter hypermethylation (7-9, 17, 21). In our study using MS-MLPA, a candidate multi-gene approach, we identified, concomitantly, genetic and epigenetic alterations in SCV.
Aberrant methylation was observed for 9 genes in 11 of 13 SCV cell lines. The most frequently methylated gene was TP73 (9 of 13 cell lines). Decreased TP73 expression as a consequence of promoter hypermethylation was confirmed by RT-PCR in UT-SCV-3, -4 and -6, supporting aberrant methylation of both copies of TP73 by MS-MLPA. TP73, located at 1p36.3 is involved in cell cycle regulation, and is frequently deleted in many types of human tumors(26–28). TP73 codes a product which has significant structural homology to the TP53 gene product in the domains involving transactivation, DNA-binding and oligomerization(29). Functionally, the TP73 gene product is able to activate the TP53-responsive proteins, inhibit cell growth and induce apoptosis(30).
Evaluation of TP73 for epigenetic changes has shown aberrant promoter hypermethylation for oligodendroglial tumors, non-hodgkin’s lymphomas and nasopharyngeal carcinomas. Hypermethylation of TP73 in nasopharyngeal carcinomas has been reported with a frequency of 20%(31). In head and neck squamous cell carcinoma (HNSCC) cell lines, hypermethylation of TP73 occurred as a primary as well as a disease progression event (17).
Expression analysis by RT-PCR of TP73 corroborated aberrant methylation in the SCV-UT cell lines in this study. Reduced mRNA expression has been reported in methylated lymphomas where TP73 abnormalities were mainly found in aggressive tumors with bad response to conventional polychemotherapy suggesting a relation between TP73 inactivation and the aggressiveness of these tumors(32).
IGSF4, DAPK1 and FHIT were aberrantly methylated in 3 of 13 cell lines with concordant loss of expression for IGSF4 in UT-SCV-4, which supported promoter hypermethylation of both copies. IGSF4 is a novel immunoglobulin-like intercellular adhesion molecule first characterized as a tumor suppressor of non-small cell lung cancer and termed TSLC1 (33, 34), where silencing was primarily achieved by allelic loss and promoter methylation. The gene is located at 11q23.2 and encodes a transmembrane glycoprotein of 442 amino acids. TSLC1 (IGSF4) silencing through promoter hypermethylation has been suggested as the major mechanism of epigenetic control in several cancers including non-small cell lung cancer, pancreatic cancer and hepatocellular carcinoma(35).
In esophageal squamous cell carcinoma (ESCC), loss of TSLC1 protein expression as a consequence of promoter hypermethylation, a late stage event in ESCC carcinogenesis, has been implicated in invasion and metastasis and aggressive tumor behavior through the disruption of cell-cell interactions(35). Additionally because expression can be restored by a demethylating agent(35), TSLC1 may offer a promising new therapeutic target in ESCC.
Promoter hypermethylation of the tumor suppressor gene, TSLC1, is also a highly frequent event in cervical cancers where epigenetic silencing of TSLC1 has been implicated in the progression from high-risk HPV-containing, high-grade CIN lesions to invasive cervical cancer(36). Furthermore, TSLC1 silencing was accompanied by complete loss or significant decrease of TSLC1 mRNA expression in these cell lines. In HNSCC, promoter hypermethylation of IGSF4 was a primary as well as a disease progression event, indicating complete abrogation of tumor suppressor function(17).
Death-associated protein kinase 1, DAPK1, located at 9q34.1 encodes a 160-kDa cytoskeletal-associated calcium/calmodulin-dependent serine/threonine kinase which was initially identified as a positive mediator of interferon γ-induced programmed cell death in HeLa cells(37). DAPK1 expression is commonly lost in urinary bladder, breast, B-cell neoplasms and renal cell carcinoma cell lines due to promoter hypermethylation.
Aberrant promoter methylation of DAPK1 has been shown to frequently occur in human head and neck cancers(17, 38), non-small cell lung carcinomas(39), gastric and colorectal carcinomas(40, 41), and uterine cervical carcinomas(42). In HNSCC, DAPK1 promoter hypermethylation has been associated with metastasis to lymph nodes as well as advanced disease stage(38). A study of DAPK1 expression in uterine and ovarian carcinomas showed that aberrant promoter methylation of DAPK1, which occurred frequently in gynecological carcinomas, led to decreased DAPK protein expression suggesting that DAPK1 gene silencing is involved in carcinogenesis of female reproductive organs(43). In bladder cancer, the methylation status of DAPK1 is an important prognostic factor for recurrence(44).
The histidine triad gene, FHIT, is a tumor suppressor gene located at 3p14.2 fragile site and is involved in purine metabolism. Loss of heterozygosity of FHIT has been associated with esophageal, stomach, colon carcinomas, lung cancers(45) as well as HNSCC(46). Promoter hypermethylation of FHIT in squamous cell carcinomas of the esophagus has been reported to be associated with transcriptional inactivation(47). One study of esophageal squamous cell carcinomas detected hypermethylation of FHIT in 50% of tumor cell lines and 45% of primary tumors(48). This same study found that hypermethylation of FHIT occurred frequently in clinical stages I and II of esophageal squamous cell carcinomas suggesting that FHIT hypermethylation may play a role in early carcinogenesis.
The remaining five genes, APC, CDKN2B, VHL, ESR1 and GSTP1, were less frequently methylated, occurring in 2/13, 1/13, 1/13, 2/13 and 1/13 cell lines respectively. Genetic and epigenetic alterations in APC (adenomatosis polyposis coli), a tumor suppressor gene originally implicated in colon cancer have been reported in other malignancies including oral squamous cell carcinomas, gastric cancers and esophageal adenocarcinomas. Uesugi et al(49) previously reported APC as being mutated and/or deleted in primary oral squamous cell carcinoma (OSCC) tissues and suggested that loss of APC function contributes to carcinogenesis in the oral region. Promoter hypermethylation is also an important mechanism of APC inactivation in oral cancers occurring in 25% of OSCC cell(49).
Cyclin-dependent kinase inhibitor 2B (CDKN2B), which is also known as p15, inhibits CDK4 and regulates cell growth by controlling cell cycle G1 progression. The presence of aberrant methylation of p15 and p16 in precancerous oral tissues and lesions of the head and neck(7–9, 17, 50) implicates methylation of p15 and p16 as early events in the pathogenesis of these lesions. In undifferentiated nasopharyngeal carcinoma (NPC), preferential methylation of CDKN2B has been shown to be a useful tumor marker for NPC(31).
The von Hippel –Lindau (VHL) gene is a tumor suppressor gene located at 3p26-p25 and it is responsible for the Von Hippel-Lindau syndrome which is an inherited familial cancer syndrome that makes patients susceptible to a variety of cancers, malignant and benign. Hypermethylation of VHL promoter region in clear-cell renal carcinomas is associated with absence of transcript expression (51). It was also found that treatment of these methylated VHL tumors with a demethylating agent resulted in re-expression of the VHL transcripts(51).
Estrogen receptor 1 (ESR1), mapping to 6q25.1, is important for hormone binding, DNA binding, and activation of transcription(52). ESR1 has metastasis-suppressor properties in breast cancer cells(53), suggesting a tumor-suppressor role for ESR1 (54). In esophageal adenocarcinomas (EAC) abnormal methylation patterns were found in premalignant Barrett’s tissue in addition to adenocarcinoma tissue suggesting that DNA hypermethylation is an early epigenetic event in the progression of EAC(55).
Glutathione S-transferase pi (GSTP1), mapping to 11q13 (56), encodes for the glutathione S-transferase pi enzyme which plays an important role in detoxification and has a role in the susceptibility to cancer and other diseases. The pi-class of glutathione S-transferase enzymes has been associated with preneoplastic and neoplastic changes(57). Inactivation of GSTP1 by promoter hypermethylation is characteristic of steroid related neoplasms such as breast, liver, and prostate cancers(57, 58).
Changes in copy number can influence gene expression, resulting in overexpression or underexpression of a gene product, depending on the function of the gene. Employing a genome-wide strategy, we recently reported several losses and gains of individual genes in this same13 SCV cell line cohort (1). In this study to examine simultaneously DNA methylation and gene copy number alterations in tumor suppressor genes, frequent genetic alterations of loss and gain of gene copy number included gain of GSTP1and MEN1, and loss of MFHAS1 and IGSF4 in over 50% of the SCV cell lines. The most frequent alteration was gain in copy of GSTP1 and MEN1in 11 of the 13 cell lines (Table 5). Gain of MEN1, a tumor suppressor (59) associated with the multiple endocrine neoplasia type 1 syndrome located at chromosome 11q13, and loss of the malignant fibrous histiocytoma amplified sequence 1 (MFHAS1) gene, an oncogene located at 8p23.1, most likely reflects chromosomal instability with resultant aneuploid subpopulations. The MEN1 gene encodes the protein menin (60, 61), which interacts with a number of proteins that are involved in transcriptional regulation, genome stability and cell division (62). MFHAS1 expression is enhanced in some malignant fibrous histiocytomas (MFH) (63). Its product is involved in the interaction of proteins related to the cell cycle (64). MFHAS1, also known as MASL1, is a target gene for genomic amplification as well as chromosomal translocation (63).
Epigenetic events of promoter hypermethylation were validated with RT- PCR for TP73 and IGSF4 genes, where reduced mRNA expression corroborated aberrant methylation status. RT-PCR was not in agreement for unmethylated IGSF4 and DAPK1 copy number in UT-SCV-2 and -3 and in UT-SCV-3 and -6, respectively. The latter may be due to heterogeneity reflecting subclonal populations.
MSP of TP73 confirmed aberrant methylation detected by MS-MLPA for UM-SCV-2, UM-SCV-3, UT-SCV-3, -4 and -6. Furthermore, MSP indicated hypermethylation of TP73 in UM-SCV-1A, UM-SCV-6 and UT-SCV-2, not detected by MS-MLPA. Lack of confirmation by MSP of TP73 methylation in UM-SCV-4, detected by MS-MLPA may be primarily due to insufficient amounts of DNA for bisulfite conversion. Repeat bisulfite conversion was not done due to depletion of the DNA sample.
While a distinct advantage of MS-MLPA is the ability to examine aberrant promoter methylation in multiple cancer genes in a single assay run, multiplex PCR of a large number of gene probes (22 unique genes) inherently encounters competitive amplification, in contrast to MSP, which examines only one gene at a time (9), and therefore, is more sensitive than MS-MLPA (9). Additionally, MS-MLPA methylation and quantitation detection algorithms may miss hypermethylation events that do not reach the threshold for detection(17). Regardless, MS-MLPA profiling of multiple genes for aberrantly methylated promoter regions is a valuable screening tool to determine frequency and pattern of gene inactivation in tumorigenesis. These epigenetic signatures, upon subsequent validation as diagnostic or prognostic epigenetic biomarkers, can become reduced to a more definitive candidate gene panel of only a few key genes. The latter would be amenable for increased detection sensitivity by a targeted 3 or 4 MS-MLPA gene probe panel or by MSP.
UMSCV-1A (primary site) and UM-SCV1B (metastasis) cell lines, derived from the same patient had similar if not identical abnormal karyotypes (11) and were concordant for changes in gene copy number as well (1), indicating that genetic events necessary for distant metastasis had evolved at the primary tumor site. There was concordant lack of methylation for 20 of the 22 tumor suppressor genes in the MLPA probe panel in both cell lines, with aberrant methylation of TP73 and DAPK1 in UM-SCV-1A only.
The HPV status of cell lines UM-SCV 1-7 and UT-SCV 1-3, previously published (65), indicated that only UM-SCV-6 cell line was positive for HPV 16. UM-SCV-6 did not demonstrate any aberrant methylation. HPV status of cell lines UT-SCV 4-6 is not known.
Our novel genome wide strategy identified several genes with aberrant promoter hypermethylation and alterations in copy number. Recurrent genomic aberrations are good indicators of genes that are causally associated with cancer development or progression and either become or reveal gene targets for therapy. Frequently methylated genes in this SCV cohort included TP73, IGSF4, DAPK1 and FHIT. These findings suggest that epigenetic events of DNA hypermethylation may contribute to the underlying pathogenesis of squamous cell carcinoma of the vulva. As a frequent and consistent target of aberrant promoter hypermethylation, TP73 may serve as a therapeutic biomarker for gene reactivation studies in SCV.
Promoter methylation-mediated silencing is a hallmark of many established tumor suppressor genes. An important distinction between genetic and epigenetic changes in cancer is that the latter might be more easily reversed using demethylating therapeutic interventions. Because gene silencing, as a consequence of promoter hypermethylation can be partially relieved by demethylation of the promoter region(66, 67), the molecules that regulate methylation status of DNA are considered promising targets for new cancer therapies. Consistent and SCV-specific epigenetic alterations would signify important biomarkers relevant to the diagnosis, prognosis, and treatment of squamous cell carcinoma of the vulva.
Acknowledgments
Supported by R01 NIH DE 15990 (MJW), DAMD17-02-1-0406 (MJW), DAMD17-00-1-0288 (MJW)
References
- 1.Kunjoonju JP, Raitanen M, Grenman S, et al. Identification of individual genes altered in squamous cell carcinoma of the vulva. Genes Chromosomes Cancer. 2005;44(2):185–93. doi: 10.1002/gcc.20230. [DOI] [PubMed] [Google Scholar]
- 2.Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–63. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 3.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99(6):3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rainier S, Feinberg AP. Genomic imprinting, DNA methylation, and cancer. J Natl Cancer Inst. 1994;86(10):753–9. doi: 10.1093/jnci/86.10.753. [DOI] [PubMed] [Google Scholar]
- 5.Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science. 1981;211(4480):393–6. doi: 10.1126/science.6164095. [DOI] [PubMed] [Google Scholar]
- 6.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–40. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 7.Stephen JK, Vaught LE, Chen KM, et al. An epigenetically derived monoclonal origin for recurrent respiratory papillomatosis. Arch Otolaryngol Head Neck Surg. 2007;133(7):684–92. doi: 10.1001/archotol.133.7.684. [DOI] [PubMed] [Google Scholar]
- 8.Stephen JK, Vaught LE, Chen KM, et al. Epigenetic events underlie the pathogenesis of sinonasal papillomas. Mod Pathol. 2007;20(10):1019–27. doi: 10.1038/modpathol.3800944. [DOI] [PubMed] [Google Scholar]
- 9.Chen K, Sawhney R, Khan M, et al. Methylation of multiple genes as diagnostic and therapeutic markers in primary HNSCC. Arch Otolaryngol Head Neck Surg. doi: 10.1001/archotol.133.11.1131. In Press. [DOI] [PubMed] [Google Scholar]
- 10.Beers M, Berkow R. The Merck Manual of Diagnosis and Therapy. 17. Whitehouse Station: Merck Research Laboratories; 1999. [Google Scholar]
- 11.Grenman SE, Van Dyke DL, Worsham MJ, et al. Phenotypic characterization, karyotype analysis and in vitro tamoxifen sensitivity of new ER-negative vulvar carcinoma cell lines, UM-SCV-1A and UM-SCV-1B. Int J Cancer. 1990;45(5):920–7. doi: 10.1002/ijc.2910450524. [DOI] [PubMed] [Google Scholar]
- 12.Raitanen M, Worsham MJ, Lakkala T, et al. Characterization of 10 vulvar carcinoma cell lines by karyotyping, comparative genomic hybridization and flow cytometry. Gynecol Oncol. 2004;93(1):155–63. doi: 10.1016/j.ygyno.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 13.Worsham MJ, Van Dyke DL, Grenman SE, et al. Consistent chromosome abnormalities in squamous cell carcinoma of the vulva. Genes Chromosomes Cancer. 1991;3(6):420–32. doi: 10.1002/gcc.2870030604. [DOI] [PubMed] [Google Scholar]
- 14.Worsham MJ, Pals G, Schouten JP, et al. Delineating genetic pathways of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2003;129(7):702–8. doi: 10.1001/archotol.129.7.702. [DOI] [PubMed] [Google Scholar]
- 15.Worsham MJ, Pals G, Schouten JP, et al. High-resolution mapping of molecular events associated with immortalization, transformation, and progression to breast cancer in the MCF10 model. Breast Cancer Res Treat. 2006;96(2):177–86. doi: 10.1007/s10549-005-9077-8. [DOI] [PubMed] [Google Scholar]
- 16.Worsham MJ, Chen KM, Tiwari N, et al. Fine-mapping loss of gene architecture at the CDKN2B (p15INK4b), CDKN2A (p14ARF, p16INK4a), and MTAP genes in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2006;132(4):409–15. doi: 10.1001/archotol.132.4.409. [DOI] [PubMed] [Google Scholar]
- 17.Worsham MJ, Chen KM, Meduri V, et al. Epigenetic events of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2006;132(6):668–77. doi: 10.1001/archotol.132.6.668. [DOI] [PubMed] [Google Scholar]
- 18.Saglam O, Shah V, Worsham MJ. Molecular Differentiation of Early and Late Stage Laryngeal Squamous Cell Carcinoma: An Exploratory Analysis. Diagnostic Molecular Pathology. 2007;16(4):218–21. doi: 10.1097/PDM.0b013e3180d0aab5. [DOI] [PubMed] [Google Scholar]
- 19.Carey T. Establishment of epidermoid carcinoma cell lines. In: RW, editor. Head and neck carcinoma. London, UK: John Wiley & Sons, Ltd; 1985. pp. 287–314. [Google Scholar]
- 20.Grenman R, Burk D, Virolainen E, et al. Radiosensitivity of head and neck cancer cells in vitro. A 96-well plate clonogenic cell assay for squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 1988;114(4):427–31. doi: 10.1001/archotol.1988.01860160071024. [DOI] [PubMed] [Google Scholar]
- 21.Nygren AO, Ameziane N, Duarte HM, et al. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005;33(14):e128. doi: 10.1093/nar/gni127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zochbauer-Muller S, Fong KM, Maitra A, et al. 5' CpG Island Methylation of the FHIT Gene Is Correlated with Loss of Gene Expression in Lung and Breast Cancer. Cancer Res. 2001;61:3581–5. [PubMed] [Google Scholar]
- 24.Gasco M, Sullivan A, Repellin C, et al. Coincident inactivation of 14-3-3sigma and p16INK4a is an early event in vulval squamous neoplasia. Oncogene. 2002;21(12):1876–81. doi: 10.1038/sj.onc.1205256. [DOI] [PubMed] [Google Scholar]
- 25.Nuovo GJ, Plaia TW, Belinsky SA, et al. In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci U S A. 1999;96(22):12754–9. doi: 10.1073/pnas.96.22.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagai H, Negrini M, Carter SL, et al. Detection and cloning of a common region of loss of heterozygosity at chromosome 1p in breast cancer. Cancer Res. 1995;55(8):1752–7. [PubMed] [Google Scholar]
- 27.Weith A, Martinsson T, Cziepluch C, et al. Neuroblastoma consensus deletion maps to 1p36.1–2. Genes Chromosomes Cancer. 1989;1(2):159–66. doi: 10.1002/gcc.2870010209. [DOI] [PubMed] [Google Scholar]
- 28.White PS, Maris JM, Beltinger C, et al. A region of consistent deletion in neuroblastoma maps within human chromosome 1p36.2–36.3. Proc Natl Acad Sci U S A. 1995;92(12):5520–4. doi: 10.1073/pnas.92.12.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong S, Pang JC, Hu J, et al. Transcriptional inactivation of TP73 expression in oligodendroglial tumors. Int J Cancer. 2002;98(3):370–5. doi: 10.1002/ijc.10204. [DOI] [PubMed] [Google Scholar]
- 30.Kaghad M, Bonnet H, Yang A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90(4):809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 31.Wong TS, Tang KC, Kwong DL, et al. Differential gene methylation in undifferentiated nasopharyngeal carcinoma. Int J Oncol. 2003;22(4):869–74. [PubMed] [Google Scholar]
- 32.Martinez-Delgado B, Melendez B, Cuadros M, et al. Frequent inactivation of the p73 gene by abnormal methylation or LOH in non-Hodgkin's lymphomas. Int J Cancer. 2002;102(1):15–9. doi: 10.1002/ijc.10618. [DOI] [PubMed] [Google Scholar]
- 33.Kuramochi M, Fukuhara H, Nobukuni T, et al. TSLC1 is a tumor-suppressor gene in human non-small-cell lung cancer. Nat Genet. 2001;27(4):427–30. doi: 10.1038/86934. [DOI] [PubMed] [Google Scholar]
- 34.Murakami Y, Nobukuni T, Tamura K, et al. Localization of tumor suppressor activity important in nonsmall cell lung carcinoma on chromosome 11q. Proc Natl Acad Sci U S A. 1998;95(14):8153–8. doi: 10.1073/pnas.95.14.8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito T, Shimada Y, Hashimoto Y, et al. Involvement of TSLC1 in progression of esophageal squamous cell carcinoma. Cancer Res. 2003;63(19):6320–6. [PubMed] [Google Scholar]
- 36.Steenbergen RD, Kramer D, Braakhuis BJ, et al. TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J Natl Cancer Inst. 2004;96(4):294–305. doi: 10.1093/jnci/djh031. [DOI] [PubMed] [Google Scholar]
- 37.Cohen O, Feinstein E, Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. Embo J. 1997;16(5):998–1008. doi: 10.1093/emboj/16.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez-Cespedes M, Esteller M, Wu L, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60(4):892–5. [PubMed] [Google Scholar]
- 39.Esteller M, Sanchez-Cespedes M, Rosell R, et al. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999;59(1):67–70. [PubMed] [Google Scholar]
- 40.Lee TL, Leung WK, Chan MW, et al. Detection of gene promoter hypermethylation in the tumor and serum of patients with gastric carcinoma. Clin Cancer Res. 2002;8(6):1761–6. [PubMed] [Google Scholar]
- 41.Satoh A, Toyota M, Itoh F, et al. DNA methylation and histone deacetylation associated with silencing DAP kinase gene expression in colorectal and gastric cancers. Br J Cancer. 2002;86(11):1817–23. doi: 10.1038/sj.bjc.6600319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong SM, Kim HS, Rha SH, et al. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001;7(7):1982–6. [PubMed] [Google Scholar]
- 43.Bai T, Tanaka T, Yukawa K, et al. Reduced expression of death-associated protein kinase in human uterine and ovarian carcinoma cells. Oncol Rep. 2004;11(3):661–5. [PubMed] [Google Scholar]
- 44.Tada Y, Wada M, Taguchi K, et al. The association of death-associated protein kinase hypermethylation with early recurrence in superficial bladder cancers. Cancer Res. 2002;62(14):4048–53. [PubMed] [Google Scholar]
- 45.Sozzi G, Veronese ML, Negrini M, et al. The FHIT gene 3p14.2 is abnormal in lung cancer. Cell. 1996;85(1):17–26. doi: 10.1016/s0092-8674(00)81078-8. [DOI] [PubMed] [Google Scholar]
- 46.Virgilio L, Shuster M, Gollin SM, et al. FHIT gene alterations in head and neck squamous cell carcinomas. Proc Natl Acad Sci U S A. 1996;93(18):9770–5. doi: 10.1073/pnas.93.18.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka H, Shimada Y, Harada H, et al. Methylation of the 5' CpG island of the FHIT gene is closely associated with transcriptional inactivation in esophageal squamous cell carcinomas. Cancer Res. 1998;58(15):3429–34. [PubMed] [Google Scholar]
- 48.Kuroki T, Trapasso F, Yendamuri S, et al. Allele loss and promoter hypermethylation of VHL, RAR-beta, RASSF1A, and FHIT tumor suppressor genes on chromosome 3p in esophageal squamous cell carcinoma. Cancer Res. 2003;63(13):3724–8. [PubMed] [Google Scholar]
- 49.Uesugi H, Uzawa K, Kawasaki K, et al. Status of reduced expression and hypermethylation of the APC tumor suppressor gene in human oral squamous cell carcinoma. Int J Mol Med. 2005;15(4):597–602. [PubMed] [Google Scholar]
- 50.Shintani S, Nakahara Y, Mihara M, et al. Inactivation of the p14(ARF), p15(INK4B) and p16(INK4A) genes is a frequent event in human oral squamous cell carcinomas. Oral Oncol. 2001;37(6):498–504. doi: 10.1016/s1368-8375(00)00142-1. [DOI] [PubMed] [Google Scholar]
- 51.Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A. 1994;91(21):9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ponglikitmongkol M, Green S, Chambon P. Genomic organization of the human oestrogen receptor gene. Embo J. 1988;7(11):3385–8. doi: 10.1002/j.1460-2075.1988.tb03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia M, Derocq D, Freiss G, et al. Activation of estrogen receptor transfected into a receptor-negative breast cancer cell line decreases the metastatic and invasive potential of the cells. Proc Natl Acad Sci U S A. 1992;89(23):11538–42. doi: 10.1073/pnas.89.23.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Issa JP, Ottaviano YL, Celano P, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7(4):536–40. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 55.Eads CA, Lord RV, Kurumboor SK, et al. Fields of aberrant CpG island hypermethylation in Barrett's esophagus and associated adenocarcinoma. Cancer Res. 2000;60(18):5021–6. [PubMed] [Google Scholar]
- 56.Moscow JA, Townsend AJ, Goldsmith ME, et al. Isolation of the human anionic glutathione S-transferase cDNA and the relation of its gene expression to estrogen-receptor content in primary breast cancer. Proc Natl Acad Sci U S A. 1988;85(17):6518–22. doi: 10.1073/pnas.85.17.6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Esteller M, Corn PG, Urena JM, et al. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 1998;58(20):4515–8. [PubMed] [Google Scholar]
- 58.Lee WH, Morton RA, Epstein JI, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A. 1994;91(24):11733–7. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chandrasekharappa SC, Teh BT. Functional studies of the MEN1 gene. J Intern Med. 2003;253(6):606–15. doi: 10.1046/j.1365-2796.2003.01165.x. [DOI] [PubMed] [Google Scholar]
- 60.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276(5311):404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 61.Lemmens I, Van de Ven WJ, Kas K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6(7):1177–83. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- 62.Agarwal SK, Kennedy PA, Scacheri PC, et al. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res. 2005;37(6):369–74. doi: 10.1055/s-2005-870139. [DOI] [PubMed] [Google Scholar]
- 63.Tagawa H, Karnan S, Kasugai Y, et al. MASL1, a candidate oncogene found in amplification at 8p23.1, is translocated in immunoblastic B-cell lymphoma cell line OCI-LY8. Oncogene. 2004;23(14):2576–81. doi: 10.1038/sj.onc.1207352. [DOI] [PubMed] [Google Scholar]
- 64.Sakabe T, Shinomiya T, Mori T, et al. Identification of a novel gene, MASL1, within an amplicon at 8p23.1 detected in malignant fibrous histiocytomas by comparative genomic hybridization. Cancer Res. 1999;59(3):511–5. [PubMed] [Google Scholar]
- 65.Rantanen V, Engblom P, Raitanen M, et al. Mutations of TP53 do not correlate with the sensitivity to paclitaxel--a study using 27 gynaecological cancer cell lines. Eur J Cancer. 2002;38(13):1783–91. doi: 10.1016/s0959-8049(02)00119-3. [DOI] [PubMed] [Google Scholar]
- 66.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16(4):168–74. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 67.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21(2):163–7. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]