

Abstract
Semenogelin (SEMG) I is a Cancer-Testis antigen expressed in myeloma cells. SEMG I expression is upregulated by IL-4, IL-6 and 5-azacytidine. In this study, we set out to define the core promoter sequence needed for the expression of SEMG I in myeloma cells. We found that nucleotide sequences spanning the two putative GATA-1 binding domains are vital for the primary regulation of SEMG I promoter function while the other parts of the promoter sequence are responsible for the fine adjustment of the core promoter function. The core promoter sequence is responsive to the enhancing effect of IL-4 and IL-6.
Keywords: Semenogelin I, Myeloma, GATA-1 binding domains, IL-4, IL-6
INTRODUCTION
SEMG I is a non-glycosylated protein of 50 kD with 439 amino acid residues (1). It is a major protein of semen coagulum and is responsible for inhibiting human sperm capacitation (2). The gene encoding SEMG I is localized to the long arm of chromosome 20 (3), a region of chromosome 20 frequently deleted in myeloproliferative diseases and myelodysplastic syndrome (4). SEMG I shows limited expression in normal tissues except prostate (5) and retinal pigment epithelial cells (6). We previously found that SEMG I was aberrantly expressed in tumor cells from hematologic malignancies, including multiple myeloma (MM) (7). SEMG I expression in tumor cells in hematologic malignancies elicits IgG responses against the SEMG I protein, suggesting the immunogenicity of the protein in the cancer-bearing patients. The combination of being immunogenic in cancer patients and showing limited expression in normal tissues makes SEMG I a potential candidate protein for tumor vaccines.
We recently found that SEMG I expression in MM cells could be upregulated by 5-azacytidine, IL-4 and IL-6 (8). 5-azacytidine induced the expression of SEMG I in MM cells. However, not surprisingly, we were unable to demonstrate any correlation between the single CpG dinucleotide at position −11 with the SEMG I promoter sequence and SEMG I expression since it would be highly unusual for one single CpG dinucleotide within the promoter sequence to be responsible for the regulation of the whole gene. This has led us to hypothesize that the SEMG I gene is probably constitutively active and its regulation and limited expression within normal tissues is controlled via a repressor gene that is regulated by DNA methylation. Therefore, when cells not expressing SEMG I were treated with a DNA hypomethylating agent, the repressor gene is hypomethylated with the consequence of de-repressing the SEMG I gene. IL-4 and IL-6, on the other hand, upregulated SEMG I expression through augmenting SEMG I promoter function. In the present study, we set out to identify the minimal core sequence of SEMG I promoter and determine whether or not IL-4 and IL-6 affect the function of this core promoter sequence. Elucidation of these interactions may shed light on the molecular mechanisms affecting SEMG I expression and offer therapeutic strategies for upregulating the antigen levels for improving the efficacy of SEMG I-based tumor vaccines.
MATERIALS AND METHODS
Generation of truncated and mutant SEMG I promoter sequences
Full length SEMG I promoter sequence was amplified from genomic DNA derived from normal testis and cloned into the TA cloning system for sequence analysis. PCR primers for the amplification of SEMG I promoter were: 5′-GCG GTA CCC ATG AAG GGA AAC TCA CAT TT-3′ and 5′-GCA GAT CTT GAG AGC TGA GCC GAC CTT GT-3′ and amplified a DNA segment of 348 bp spanning the nucleotides from position −1 to −348 from the transcription start site.
A panel of truncated SEMG I promoters was generated by polymerase chain reactions from the 5′ end and then from the 3′ end of the full length genes. These truncated promoters were generated by using a panel of oligonucleotide primers spanning various regions along the full length SEMG I promoter. Successfully amplified DNA fragments were isolated and cloned into the TA cloning system for sequence confirmation prior to being used for subcloning into the pCAT*3-Enhancer vector (Molecular Probes Company, Eugene, OR) between Kpn I and Bgl II.
A pair of mutant SEMG I promoters was also generated from the full length wild-type SEMG I promoter sequence by oligonucleotide-directed mutagenesis, mutating the cytosine or guanine of the CpG dinucleotides at position −11 using GeneEditor (Promega), according to the manufacturer’s instruction. Briefly, the full length wide-type SEMG I promoter template was denatured by 0.2N NaOH solution for 2 minutes and then neutralization by 0.2N NH4Ac (pH 4.5). The denatured DNA template was then precipitated and recovery by ethanol precipitation followed by resuspension in water. The single stranded DNA was annealed to two oligonucleotides, one containing the mutated CpG nucleotides and another to the ampicillin resistant gene. Following polymerization, the plasmid was transformed into BMH 71-18 mutS competent cells and grown on Ampicillin+Selection Mix LB plate. Recombinant colonies were picked and grown in LB. The plasmid was extracted and then transformed into JM109 for selection of mutants. The mutants were confirmed by DNA sequencing.
Cell transfection and analysis of Chloramphenicol Acetyl Transferase (CAT) expression
Transfection was carried out using the FuGENE 6 reagent (Roche Molecular Biochemicals). Briefly, the RPMI 8226 cells were seeded into a six-well cluster plate. The cultures were transfected with 2 μg of the recombinant plasmids and assayed for CAT activities after 72 hours. FAST CAT Green (deoxyl) Chloramphenicol Acetyltransferase Assay Kit (Molecular Probes, Eugene, OR) was used to detect CAT activity. The transfectants were first lysed and a cytoplasmic extract prepared. The extract was then incubated with the fluorescent deoxylchloramphenicol substrate and acetyl CoA at 37 °C. The reaction was terminated by the addition of ice-cold ethyl acetate. After drying and dissolution in ethyl acetate, the reaction substrate and product were resolved by thin-layer chromatography (TLC) on silica gel plates and eluted with a chloroform:methanol mixture (85:15 v/v). All experiments were carried out in triplicates. Quantitation of the products was carried out using the Bio-Rad Quantity One software (version 4.5.2) (Bio-Rad Laboratories, Hercules, CA). The amounts of the products from the CAT analysis were calculated; % = (product)/(product + substrate) × 100%.
RESULTS AND DISCUSSIONS
We previously demonstrated that SEMG I expression in myeloma cells can be upregulated by IL-4 and IL-6 (8). Furthermore, we also showed that both of these cytokines stimulated SEMG I expression through their ability to enhance the function of the SEMG I promoter gene in a dose-dependent manner. However, it remained to be determined whether or not the core function of the promoter gene is dependent on the entire 348 bp within the SEMG I promoter or only part of the sequence and whether the flanking sequences exerting a secondary regulatory function. In this study, we, therefore, set out to identify the minimal core promoter sequence of SEMG I promoter gene and determine the functions of the sequences flanking the core promoter gene.
To determine the core promoter sequence, a panel of truncated SEMG I promoters was generated, by PCR (Figure 1). The truncated promoter sequences were subcloned into the pCAT vector and transfected into RPMI 8226 cells. Cell lysates from transfectants were used to measure the CAT activities. We identified that the sequence spanning the two putative GATA-1 binding domains within the SEMG I promoter (from −101 to −214) was crucial for the function of the promoter. Compared to full length promoter sequence (FL), the function of the truncated promoters devoid of these two putative GATA-1 binding domains (T4 and T5) were severely impaired (Figure 2). Truncation of the promoter at the 5′ end (T1) and the 3′ end (T6) resulted in augmentation of the promoter function, suggesting that the sequences within these two regions of the promoter gene provided a suppressor effect to the full length promoter function. However, if truncation of the promoter sequence occurred beyond the putative CAATT binding domain of the promoter (T2), the function of the promoter is greatly reduced, implicating a major stimulatory effect on the function of the SEMG I promoter gene by the putative CAATT binding domain. The results, therefore, suggest that the core promoter sequence is contained within a sequence that included the two putative GATA-1 binding domain of the SEMG I promoter gene with the flanking sequences modulating its function. Based on these results, we propose a functional map for the SEMG I promoter gene, as outlined in Figure 3.
Figure 1.
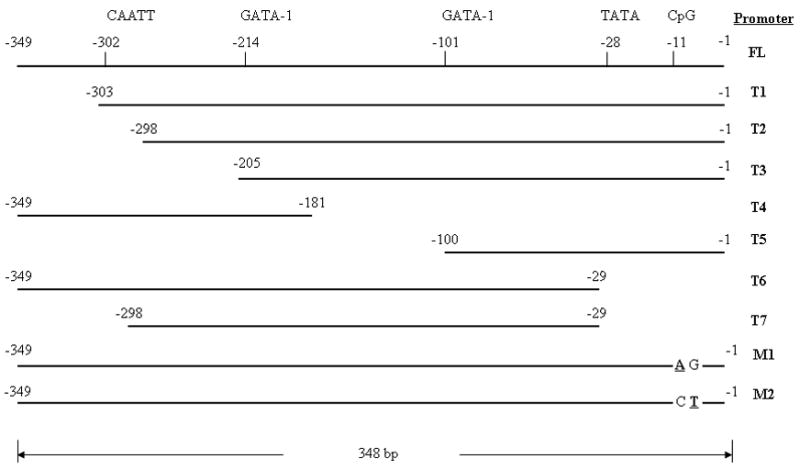
A panel of truncated SEMG I promoters (T1, T2, T3, T4, T5, T6 and T7), including the positions of the putative GATA-1 and CAATT protein binding domains and the CpG dinucleotide, and a pair of mutated (in bold) full length SEMG I promoters (M1 and M2).
Figure 2.
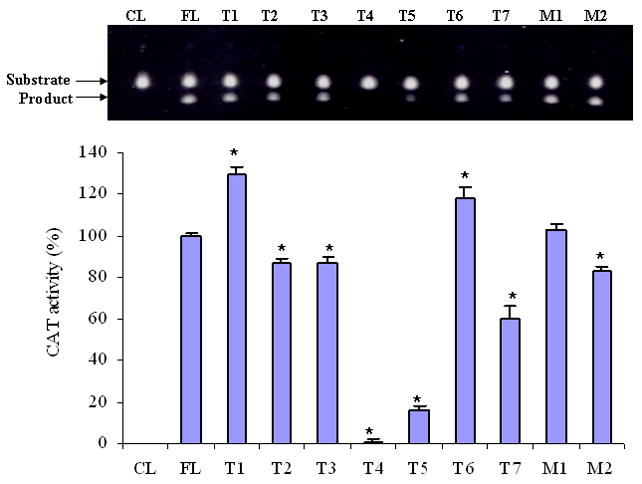
Functions of the truncated SEMG I and mutated full length SEMG I promoters, as measured by CAT activities. The promoter function was essentially abolished if the both of the putative GATA-1 binding domains were not included. Mutation of the guanine at the CpG nucleotide also resulted in a change in the promoter function (All results were expressed as mean ± SD of three readings) (Results are from one of two similar experiments) (* = p < 0.001).
Figure 3.
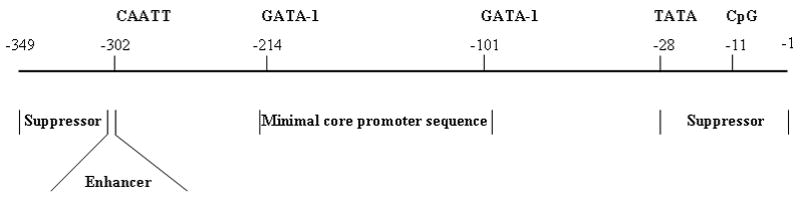
Proposed functional map of SEMG I promoter gene.
There is a CpG dinucleotide within the putative SEMG I promoter at position −11. We previously found that there was not a correlation between the methylation status of the SEMG I CpG dinucleotide and SEMG I gene expression. In the present study, we determined whether or not the CpG dinucleotide plays any regulatory function in the SEMG I promoter independent of DNA methylation. To do so, a pair of mutant SEMG I promoters was generated by site-directed mutagenesis (Figure 1), sequentially mutating the cytosine (M1) or guanine (M2) of the CpG dinucleotides and determined for any changes in the function of the mutant promoters by CAT analysis. Mutation of the cytosine of the CpG dinucleotide at position −11 (M1) did not significant alter the function of the full length SEMG I promoter sequence but mutation of the guanine resulted in a much reduced function of the SEMG I promoter (Figure 2). These results, therefore, suggest that, although the methylation status of the single CpG dinucleotide of the full length SEMG I promoter at position −11 is not involved in the regulation of SEMG I gene expression (8), the guanine within the CpG modulate the function of the promoter gene.
Having previously shown that IL-4 and IL-6 were able to upregulate SEMG I expression through their effect on the SEMG I full length promoter gene (8), we then determined in this study whether or not these cytokines enhance SEMG I expression, at least in part, through their effect on the core SEMG I promoter sequence. RPMI 8226 cells transfected with the pCAT*3 Enhancer-SEMG I-core promoter vectors were pre-incubated with these cytokines for 96 hours and then tested for the function of the promoter in inducing chloramphenicol acetyltransferase activities in the cell lysates of the transfectants. We first demonstrated that the function of the core SEMG I promoter was comparable to that of full length SEMG I promoter gene (Figure 4). In vitro methylation of the promoter gene with Sss I enzyme resulted in the abrogation of the SEMG I promoter function. Furthermore, both IL-4 and IL-6 were able to upregulate the function of the core SEMG I promoter sequence in these transfectants (Figure 4). The effects of IL-4 and IL-6 on the function of the SEMG I core promoter were dose dependent (Figures 4). These results, therefore, support the notion that IL-4 and IL-6 enhance SEMG 1 expression, at least in part, by altering the function of the core SEMG I promoter gene. These results, however, do not exclude any additional effect of these cytokines on the suppressor/stimulatory effects of the sequences flanking the core promoter sequences.
Figure 4.
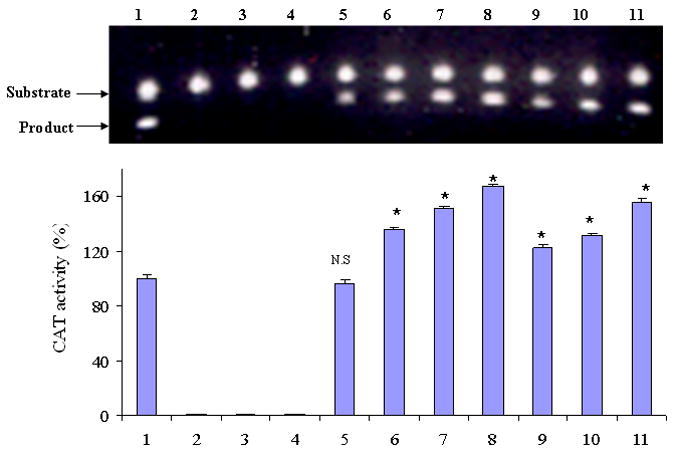
Effects of IL-4 and IL-6 on the function of the core SEMG I promoter sequence, as measured by CAT activities (1 = Full length (FL) SEMG I promoter; 2 = Negative control consisted of methylated vector containing FL; 3 = Negative control consisted of methylated vector containing FL and incubated in IL-4; 4 = Negative control consisted of methylated vector containing FL and incubated in IL-6; 5 = Core promoter sequence (CPS); 6 = CPS with 1 U/ml IL-4; 7 = CPS with 10 U/ml IL-4; 8 = CPS with 100 U/ml IL-4; 9 = CPS with 0.01 ng/ml IL-6; 10 = CPS with 0.1 ng/ml IL-6; 11 = CPS with 1 ng/ml IL-6) (All results were expressed as mean ± SD of three readings) (Results are from one of two similar experiments) (* = p < 0.001; NS = not significant).
In conclusion, we have demonstrated in the present study that SEMG I promoter gene consists of a core sequence and a regulatory element; the core element resides within the sequence spanning the two putative GATA-1 binding sites at positions −101 and −214 and the sequences flanking this length of DNA provide the secondary effects on the function of the SEMG I promoter.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lilja H, Abrahamsson PA, Lundwall A. Semenogelin, the predominant protein in human semen. Primary structure and identification of closely related protein in the male accessory sex glands and on the spermatozoa. J Biol Chem. 1989;264:1894–1900. [PubMed] [Google Scholar]
- 2.De Lamirande E, Yoshida K, Yoshiike TM, Iwamoto T, Gagnon C. Semenogelin, the main protein of semen coagulum, inhibits human sperm capacitation by interfering with the superoxide anion generated during this process. J Androl. 2001;22:672–679. [PubMed] [Google Scholar]
- 3.Ulvsback M, Lazure C, Lilja H, Spurr NK, Rao VV, Loffler C, Hansmann I, Lundwall A. Gene structure of semenogelin I and II. The predominant proteins in human semen are encoded by two homologous genes on chromosome 20. J Biol Chem. 1992;267:18080–18084. [PubMed] [Google Scholar]
- 4.Asimakopoulos FA, White NJ, Nacheva E, Green AR. Molecular analysis of chromosome 20q deletions associated with myeloproliferative disorders and myelodysplastic syndromes. Blood. 1994;84:3086–3094. [PubMed] [Google Scholar]
- 5.Lundwall A, Bjartell A, Olsson AY, Malm J. Semenogelin I and II, the predominant human seminal plasma proteins, are also expressed in non-genital tissues. Mol Human Reprod. 2002;8:805–810. doi: 10.1093/molehr/8.9.805. [DOI] [PubMed] [Google Scholar]
- 6.Boniha VL, Rayborn ME, Shadrach K, Lundwall A, Malm J, Bhattacharya SK, Crabb JW, Hollyfield JG. Characterization of semenogelin proteins in the human retina. Exp Eye Res. 2006;83:120–127. doi: 10.1016/j.exer.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Wang Z, Liu H, Giles FJ, Lim SH. Pattern of gene expression and immune responses to Semenogelin 1 in chronic hematologic malignancies. J Immunother. 2003;26:461–467. doi: 10.1097/00002371-200311000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Wang Z, Zhang J, Farmer B, Lim SH. Semenogelin I expression in myeloma cells can be upregulated pharmacologically. Leuk Res. doi: 10.1016/j.leukres.2008.03.036. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]