

Abstract
The production of ROS (reactive oxygen species) by mammalian mitochondria is important because it underlies oxidative damage in many pathologies and contributes to retrograde redox signalling from the organelle to the cytosol and nucleus. Superoxide (O2•−) is the proximal mitochondrial ROS, and in the present review I outline the principles that govern O2•− production within the matrix of mammalian mitochondria. The flux of O2•− is related to the concentration of potential electron donors, the local concentration of O2 and the second-order rate constants for the reactions between them. Two modes of operation by isolated mitochondria result in significant O2•− production, predominantly from complex I: (i) when the mitochondria are not making ATP and consequently have a high Δp (protonmotive force) and a reduced CoQ (coenzyme Q) pool; and (ii) when there is a high NADH/NAD+ ratio in the mitochondrial matrix. For mitochondria that are actively making ATP, and consequently have a lower Δp and NADH/NAD+ ratio, the extent of O2•− production is far lower. The generation of O2•− within the mitochondrial matrix depends critically on Δp, the NADH/NAD+ and CoQH2/CoQ ratios and the local O2 concentration, which are all highly variable and difficult to measure in vivo. Consequently, it is not possible to estimate O2•− generation by mitochondria in vivo from O2•−-production rates by isolated mitochondria, and such extrapolations in the literature are misleading. Even so, the description outlined here facilitates the understanding of factors that favour mitochondrial ROS production. There is a clear need to develop better methods to measure mitochondrial O2•− and H2O2 formation in vivo, as uncertainty about these values hampers studies on the role of mitochondrial ROS in pathological oxidative damage and redox signalling.
Keywords: complex I, hydrogen peroxide, mitochondrion, reactive oxygen species (ROS), respiratory chain, superoxide
Abbreviations: CoQ, coenzyme Q; CoQH2, reduced CoQ; DPI, diphenyleneiodonium; ETF, electron transfer flavoprotein; α-GPDH, α-glycerophosphate dehydrogenase; HIF-1, hypoxia-inducible factor-1; αKGDH, 2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase); PHD, prolyl hydroxylase; RET, reverse electron transport; ROS, reactive oxygen species; SMP, submitochondrial particle; SOD, superoxide dismutase
INTRODUCTION
Mitochondria are an important source of ROS (reactive oxygen species) within most mammalian cells [1–8]. This ROS production contributes to mitochondrial damage in a range of pathologies and is also important in redox signalling from the organelle to the rest of the cell [3,9]. Consequently, knowledge of how mitochondria produce ROS is vital to understand a range of currently important biomedical topics (Figure 1). The first report that the respiratory chain produced ROS came in 1966 [10], followed by the pioneering work of Chance and colleagues who showed that isolated mitochondria produce H2O2 [4,11,12]. Later, it was confirmed that this H2O2 arose from the dismutation of superoxide (O2•−) generated within mitochondria [13,14]. The parallel discovery that mitochondria contain their own SOD (superoxide dismutase), MnSOD, confirmed the biological significance of mitochondrial O2•− production [15]. Since then, a huge literature has developed on the sources and consequences of mitochondrial ROS production, which I will not attempt to cover systematically. Instead, I shall develop a consensus about how mitochondria produce ROS and indicate current uncertainties and future issues to be addressed. The focus of the present review is the production of the proximal mitochondrial ROS, O2•−, in the mitochondrial matrix by the core metabolic machinery present in the mitochondrial inner membrane and matrix. Other potentially important ROS sources associated with the mitochondrial outer membrane or intermembrane space will not be considered [1,5]. The aim is to provide a useful resource for those working on mitochondrial ROS production, to facilitate the design and interpretation of experiments, and to stimulate new approaches.
Figure 1. Overview of mitochondrial ROS production.

ROS production by mitochondria can lead to oxidative damage to mitochondrial proteins, membranes and DNA, impairing the ability of mitochondria to synthesize ATP and to carry out their wide range of metabolic functions, including the tricarboxylic acid cycle, fatty acid oxidation, the urea cycle, amino acid metabolism, haem synthesis and FeS centre assembly that are central to the normal operation of most cells. Mitochondrial oxidative damage can also increase the tendency of mitochondria to release intermembrane space proteins such as cytochrome c (cyt c) to the cytosol by mitochondrial outer membrane permeabilization (MOMP) and thereby activate the cell's apoptotic machinery. In addition, mitochondrial ROS production leads to induction of the mitochondrial permeability transition pore (PTP), which renders the inner membrane permeable to small molecules in situations such as ischaemia/reperfusion injury. Consequently, it is unsurprising that mitochondrial oxidative damage contributes to a wide range of pathologies. In addition, mitochondrial ROS may act as a modulatable redox signal, reversibly affecting the activity of a range of functions in the mitochondria, cytosol and nucleus.
O2•−, THE PROXIMAL MITOCHONDRIAL ROS
Within mitochondria, O2•− is produced by the one-electron reduction of O2. Therefore it is the kinetic and thermodynamic factors underlying the interaction of potential one-electron donors with O2 that control mitochondrial ROS production. Having two unpaired electrons in antibonding orbitals with parallel spins makes ground-state O2 accept one electron at a time [16]. The standard reduction potential for the transfer of an electron to O2 to form O2•− is −160 mV at pH 7 [16], for a standard state of 1 M O2. As the pKa of O2•− is 4.7 [16], this standard reduction potential is invariant across most biological pH values. The actual reduction potential that determines the thermodynamic tendency of O2 to form O2•−, Eh, will vary with the relative concentrations of O2 and O2•−:
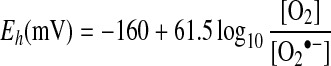 |
(1) |
The [O2] in air-saturated aqueous buffer at 37 °C is approx. 200 μM [17]; however, mitochondria in vivo are exposed to a considerably lower [O2] that varies with tissue and physiological state [18]. This occurs because the mitochondrial [O2] in vivo is largely set by the extracellular [O2] in the tissue, which is itself determined by local O2 delivery and consumption [18]. In addition, [O2] decreases on going from the extracellular environment to the mitochondrion, owing to O2 consumption by cytochrome oxidase [18,19]. Plausible estimates for the mitochondrial [O2] in vivo are in the range 3–30 μM [2]. The steady-state intramitochondrial [O2•−] is difficult to measure accurately, but it is likely to be very low because of micromolar concentrations of MnSOD in the mitochondrial matrix (e.g. 11 μM MnSOD in rat liver mitochondria [20]). This MnSOD will very rapidly (k≈2.3×109 M−1·s−1) catalyse dismutation of O2•− to H2O2 by a process that is first-order with respect to [O2•−] [2,4,5]. Furthermore, by acting as a very effective O2•− sink, MnSOD will favour the reaction of O2 with electron donors to form O2•−, in effect increasing the flux of electrons from electron donors to H2O2 [21,22]. Tentative estimates for [O2•−] within the mitochondrial matrix are in the range 10–200 pM [4,5]. By combining these with a plausible intramitochondrial [O2] of 25 μM [2], eqn (1) gives Eh in the range 150–230 mV for the reduction of O2 to O2•−. Even for a low [O2] of 1 μM and a high [O2•−] of 200 pM, the Eh value obtained is 68 mV.
Therefore, in vivo, the one-electron reduction of O2 to O2•− is thermodynamically favoured, even by relatively oxidizing redox couples, and a wide range of electron donors within mitochondria could potentially carry out this reaction [1]. However, only a small proportion of mitochondrial electron carriers with the thermodynamic potential to reduce O2 to O2•− do so. Under most circumstances, small-molecule electron carriers such as NADH, NADPH, CoQH2 (reduced coenzyme Q) and glutathione (GSH) do not react with O2 to generate O2•−. Instead, mitochondrial O2•− production takes place at redox-active prosthetic groups within proteins, or when electron carriers such as CoQH2 are bound to proteins, and it is the kinetic factors that favour or prevent the one-electron reduction of O2 to O2•− by these that determine mitochondrial O2•− production.
THE CONTROL OF O2•− PRODUCTION BY PROTEIN-BOUND REDOX GROUPS
The factors determining the rate of O2•− production by mitochondria are relatively straightforward. The first is the concentration of the enzyme or protein [E] containing electron carriers that can exist in a redox form able to react with O2 to form O2•−. The second is the proportion (PR) of this enzyme's electron carrier present in a redox form that can react with O2. As many redox-active groups exist only transiently in a state that can react with O2, PR is a time-average. The remaining factors are the local [O2] and the second-order rate constant (kE) for the reaction of that electron carrier with O2 to form O2•−. For a given enzyme or protein (E), the rate of O2•− production is:
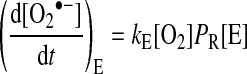 |
(2) |
This can be extended to consider several potential electron-donor sites within mitochondria, and also to take into account multiple electron donor sites within a single protein:
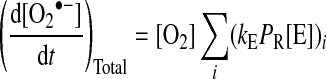 |
(3) |
The concentration of the enzyme responsible for O2•− production, [E], will vary with organism, tissue, state, age or hormonal status, and may underlie many of the changes in maximum ROS production capacity between tissues [23]; for example, complex I content may explain the different maximum capacities of pigeon and rat heart mitochondria [24].
As the apparent Km of cytochrome oxidase for O2 is very low (<1 μM [25]), changes in [O2] should have little direct effect on mitochondrial function and instead are most likely to interact by affecting O2•− production. The generation of O2•− or H2O2 by isolated respiratory complexes, SMPs (submitochondrial particles) or mitochondria increases when [O2] is raised above the normal atmospheric level of 21% O2, and this increase is roughly proportional to [O2], at least over the lower range of supraphysiological [O2] [4,12,26–28]. Fewer studies have looked at the effects of decreasing [O2], but O2•− production by isolated complex I decreases linearly with lowered [O2] [28], and there is a decrease in H2O2 production by isolated mitochondria when [O2] is lowered below that of air-saturated medium [27,29,30]. However, in one detailed study, the rate of H2O2 production did not change as [O2] was lowered from ∼200 μM to ∼5 μM and only decreased when [O2] was lowered below 5 μM [29]; further investigation of this important issue is required to see whether it arises due to the technical difficulties of measuring low levels of H2O2 production or whether it reflects a genuine phenomenon.
The dependence of mitochondrial H2O2 production on [O2] is likely to be an important factor in the variation of ROS production in vivo because extracellular [O2] varies with physiological state, and there are [O2] gradients from the circulation to the mitochondria where O2 consumption by cytochrome oxidase decreases [O2] locally [18,19]. Thus increasing or decreasing the rate of O2 consumption by mitochondria may be an important aspect for modifying O2•− production in vivo by altering the local [O2] [19]. Furthermore, as O2 is approx. 3-fold more soluble within membranes than in water [31], O2 may be concentrated close to electron carriers within the membrane. Physiological levels of nitric oxide (NO•) compete with O2 for cytochrome oxidase when [O2] is low, effectively raising the apparent Km of this enzyme [32–35], and may thus alter the local [O2] around mitochondria, leading to changes in O2•− production [36–38].
Probably the most important factor that determines O2•− production by mitochondria is the proportion, PR, of a given electron carrier that is reactive with O2 to form O2•−, as PR responds rapidly to a range of biological situations. The relationship between PR and the overall reduction state of an electron carrier group in a protein may be complicated, as the redox form that donates an electron to O2 is not necessarily fully reduced. Instead, a partially reduced form, such as a semiquinone, that can respond to the cumulative reduction of an electron carrier system may be the critical electron donor. For any electron carrier, PR will be affected by changes in the carrier's Em and in its rate of electron supply and release, all of which can be altered by inhibition, damage, mutation or post-translational modification to protein complexes distal or proximal to the site, or to the protein itself. Many other factors can have an impact on PR by affecting these parameters, with changes in Δp (protonmotive force) likely to be particularly important as it significantly affects the PR of electron carriers and it varies rapidly in response to changes in mitochondrial ATP synthesis.
The final factor affecting the rate of O2•− production by electron carriers within proteins is the second-order rate constant (kE) of their reaction with O2. The reaction between protein-bound electron carriers and O2 to form O2•− is generally thought to occur through an outer-sphere mechanism described by the Marcus theory [39,40]. In this mechanism, an electron tunnels from the electron donor to O2, and the rate is very dependent on the distance between O2 and the electron donor [39,40]. This is similar to electron movement down the respiratory chain which occurs by electron tunnelling from carrier to carrier, with a maximum distance of approx. 14 Å (1 Å=0.1 nm) between each carrier for effective tunnelling to occur [41]. Similar distance constraints probably apply to the reaction of protein-bound electron carriers in the respiratory chain with O2 to form O2•−, with the bulk of the protein acting as an insulator to keep O2 at a safe distance from the carriers and thereby minimize O2•− production [41]. Consequently, O2•− production will probably occur at sites where O2 can approach closely to electron carriers, such as at active sites exposed to the aqueous phase or to the membrane core.
The rate of electron transfer from protein electron carriers to O2 has been investigated for flavoenzymes that activate O2, where the reduction of O2 to O2•− is often a precursor to further reactions [42]. For the flavoenzyme glucose oxidase, the rate of O2 reduction to H2O2 is ∼106 M−1·s−1 [39], and, as the rate-limiting step is O2 reduction to O2•− [39], this indicates that O2•− production by protein-bound electron carriers can be rapid. Of course, the production of H2O2 is a consequence of the normal physiological function of glucose oxidase, and for other flavoproteins, where electron transfer to O2 to form O2•− may be a side reaction, the rate varies over five orders of magnitude [43]. Even so, the rate of reduction of O2 by the reduced FMN of complex I to form O2•− is ∼40 O2•−·min−1 [28], corresponding to a second-order rate constant of ∼103 M−1·s−1. Thus the second-order rate constants (kE) for O2•− production as a side reaction by protein-bound electron carriers can be rapid, although it is likely to vary markedly with the environment of the electron donor and its accessibility to O2. Therefore alterations to a protein that enabled O2 to approach more closely to the electron carrier, such as by damage, mutation, post-translational modification, conformational change or quaternary interactions, could lead to dramatic changes in the rate of O2•− production, and potentially play a regulatory function. Such factors determining the second-order rate constant for the reaction of O2 with protein electron carriers could determine mitochondrial O2•− production, but little is known about how kE may be varied by different enzymes within mitochondria.
MEASUREMENT OF O2•− PRODUCTION BY ISOLATED MITOCHONDRIA
Although many purified mitochondrial proteins can be manipulated so as to produce O2•−, the physiological relevance of this is limited. Therefore it is important to understand O2•− production within isolated mitochondria under conditions that mimic those that may arise in vivo under physiological or pathological conditions. A constraint of using isolated mitochondria is that the system is complicated and difficult to manipulate. In particular, the direct measurement of O2•− within mitochondria is challenging due to its rapid dismutation in the presence of ∼10 μM MnSOD (k≈2×109 M−1·s−1 [2,5]), which leads to very low steady-state matrix [O2•−] and competes with O2•− detection systems [44]. {The spontaneous dismutation of O2•− (k≈106 [16]) is less important because it is second-order with respect to [O2•−], which is very low in the presence of MnSOD.} Studies have been carried out on O2•− production by mitochondria lacking MnSOD [44], but there is always the concern that the absence of MnSOD may decrease the flux of O2•− by allowing the back reaction between O2•− and the electron donor [21,22], as well as having the potential to damage the system [45,46]. The use of O2•−-sensitive dyes such as hydroethidine and MitoSOX™ [47–50], measurement of the reaction of O2•− with compounds to form chemiluminescent products [44,51], spin trapping [52] or measurement of the inactivation rate of aconitase [53] do provide useful information on mitochondrial O2•− production. Even so, measurement is challenging, and the quantification of O2•− production by isolated mitochondria is not done routinely.
In contrast with the difficulties of assessing O2•− directly, intramitochondrial O2•− flux can be readily measured in isolated mitochondria following its dismutation to H2O2 by MnSOD and subsequent diffusion from the mitochondria [11,54,55]. There are a number of ways of assaying this H2O2, but typically this is now done by measuring the oxidation of a non-fluorescent dye by H2O2 in conjunction with a peroxidase to form a fluorescent product (Figure 2) [11,55–57]. Although such assays are reliable, sensitive and robust, there are a number of issues to be considered in interpreting these data and in extrapolating from isolated mitochondria to the in vivo situation. First, not all O2•− is necessarily converted into H2O2 in vivo, as some O2•− could react with other electron acceptors or with NO• within mitochondria [58,59]. Although the extent of these reactions is not known, situations may arise where they are significant, particularly as physiological levels of NO• can react very rapidly with O2•− (k≈1010 M−1·s−1) to form peroxynitrite (ONOO−) [58,59]. Secondly, measurement of H2O2 efflux from mitochondria will also be affected by H2O2 produced in the intermembrane space or outer membrane, which can be significant under some conditions [1,52,60]. Thirdly, and most importantly, not all H2O2 produced within the mitochondrial matrix will survive to efflux from the mitochondria, owing to matrix peroxidases that consume H2O2 [1,61–63]. These include peroxiredoxins 3 and 5 [64], catalase [65,66] and glutathione peroxidases 1 and 4 [67], with the peroxiredoxins probably of the greatest significance [64]. Thus the rate of matrix O2•− formation is the sum of the H2O2 measured effluxing from the mitochondria, the O2•− sinks, the rate of H2O2 degradation to water, minus H2O2 production from outside mitochondria (eqn 4) (Figure 2):
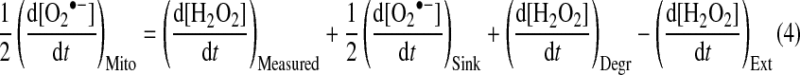 |
(4) |
Figure 2. Measurement of H2O2 production by isolated mitochondria.

The production of O2•− within the mitochondrial matrix, intermembrane space and outer membrane leads to the formation of H2O2 from SOD-catalysed dismutation. Some O2•− can react directly with nitric oxide (NO•) to form peroxynitrite (ONOO−). There are also sources outside mitochondria that produce H2O2 directly. The H2O2 efflux from mitochondria can be measured following reaction with a non-fluorescent substrate such as Amplex Red in conjunction with horseradish peroxidase (HRP) to form a fluorescent product, resorufin. Within mitochondria H2O2 is degraded by glutathione peroxidases (GPx) or peroxiredoxins (Prx) which depend on glutathione (GSH) and thioredoxin-2 (Trx) for their reduction respectively. Glutathione disulfide (GSSG) is reduced back to GSH by glutathione reductase (GR). Trx is reduced by thioredoxin reductase-2 (TrxR). Both enzymes receive reducing equivalents from the NADPH pool, which is kept reduced by the Δp-dependent transhydrogenase (TH), and by isocitrate dehydrogenase (ICDH). Note that many mitochondrial preparations, particularly those from the liver, contain large amounts of catalase contamination. The effects of such extramitochondrial H2O2 sinks are not indicated here as they are usually accounted for by appropriate H2O2 calibration curves in the presence of mitochondria. ox, oxidized; red, reduced. An animated version of this Figure can be seen at http://www.BiochemJ.org/bj/417/0001/bj4170001add.htm.
Consequently, there is a threshold of intramitochondrial O2•− formation before it exceeds scavenging by O2•− sinks and peroxidases, and generates sufficient H2O2 to be measured externally [1,61]. A corollary is that there may be significant O2•− formation within mitochondria that goes undetected, which is supported by the finding that H2O2 efflux from mitochondria increases on inhibition of peroxidases [1,68]. An interesting extension is that the activity of intramitochondrial peroxidases such as peroxiredoxins is decreased on exposure to H2O2 [69–71], and thus the mitochondrial H2O2-degradation rate may vary considerably with condition and history of the organelle. Consistent with this, exposure of mitochondria to H2O2 leads to greater H2O2 efflux [27], and may be one way in which H2O2 efflux from mitochondria is regulated by a feed-forward cycle of ROS-induced ROS efflux. A related factor is that many mitochondrial manipulations affect the activity of H2O2-degradation systems, which are linked to the GSH/GSSG ratio through glutathione peroxidases [67] and to the thioredoxin-2 oxidized/reduced ratio through peroxiredoxins [62,63,68,72,73]. These in turn are affected by the NADPH/NADP+ ratio which is set by the activity of the transhydrogenase and of NADP+-dependent isocitrate dehydrogenase [74,75]. Thus interventions that alter mitochondrial substrate supply or Δp could affect both O2•− production and matrix H2O2 degradation. Finally, it is generally assumed that H2O2 permeates directly through the mitochondrial membrane; however, H2O2 diffusion across the plasma membrane is facilitated by aquaporins [76,77], and the possibility remains that H2O2 from mitochondria may actually be protein-mediated, perhaps leading to another level of control over H2O2 efflux. Therefore, although inferring mitochondrial O2•− production by measuring H2O2 efflux is robust and reliable, a number of points must be borne in mind when interpreting these data.
O2•− PRODUCTION WITHIN ISOLATED MITOCHONDRIA
Sufficient studies have been carried out on the efflux of H2O2 from isolated mitochondria to enable generalizations about the conditions that favour O2•− production and the mitochondrial sources of O2•−. There are two main modes of operation by isolated mitochondria that lead to extensive H2O2 efflux (Figure 3). The first mode occurs when there is a high NADH/NAD+ ratio in the matrix [27,28]. The second mode is when there is a highly reduced CoQ (coenzyme Q) pool, in conjunction with a maximal Δp and no ATP synthesis [57,78,79]. The third mode of mitochondrial operation is when the mitochondria are working normally making ATP (i.e. they are in, or close to, state 3), or using the Δp for other functions such as thermogenesis. In this third mode of operation, H2O2 efflux from mitochondria is negligible compared with modes 1 or 2. Our next focus is to understand in detail the sites and mechanisms of O2•− production under these three modes of mitochondrial operation and thereby infer where and how O2•− is produced in vivo.
Figure 3. Modes of mitochondrial operation that lead to O2•− production.

There are three modes of mitochondrial operation that are associated with O2•− production. In mode 1, the NADH pool is reduced, for example by damage to the respiratory chain, loss of cytochrome c during apoptosis or low ATP demand. This leads to a rate of O2•− formation at the FMN of complex I that is determined by the extent of FMN reduction which is in turn set by the NADH/NAD+ ratio. Other sites such as αKGDH may also contribute. In mode 2, there is no ATP production and there is a high Δp and a reduced CoQ pool which leads to RET through complex I, producing large amounts of O2•−. In mode 3, mitochondria are actively making ATP and consequently have a lower Δp than in mode 2 and a more oxidized NADH pool than in mode 1. Under these conditions, the flux of O2•− within mitochondria is far lower than in modes 1 and 2, and the O2•− sources are unclear.
Sites of O2•− production within isolated mitochondria
Complex I
Mammalian complex I is the entry point for electrons from NADH into the respiratory chain and is a ∼1 MDa complex comprising 45 polypeptides [80,81]. An FMN cofactor accepts electrons from NADH and passes them through a chain of seven FeS (iron–sulfur) centres to the CoQ reduction site, with another FeS centre (N1a) close to the FMN, but not thought to be involved in electron transfer to CoQ [80,81]. The structure of the water-soluble arm of complex I from Thermus thermophilus, which contains the FMN and the FeS centres, is known and is likely to be very similar to that in mammals [80–82]. However, the structure of the CoQ reduction site in the hydrophobic arm is not known and the mechanism of proton pumping by complex I is uncertain [80–82]. The Thermus structure indicates that the seven FeS centres in the hydrophilic arm involved in passing electrons from FMN to CoQ are reasonably well shielded from O2, so that O2 is most likely to access electron carriers at the FMN and CoQ sites, although there are FeS centres at both termini which could also donate electrons to O2 [81].
The first demonstration of ROS production by complex I was in SMPs where reduction of the CoQ pool and generation of a large Δp by succinate led to uncoupler-sensitive H2O2 production [83]. Subsequently, it was shown that isolated complex I in the presence of NADH produces O2•− and that this generation is enhanced by the inhibitor rotenone which binds to the CoQ-binding site [84]. The mechanism of O2•− production by isolated complex I is now reasonably well understood [28,85]. The isolated complex produces O2•− from the reaction of O2 with the fully reduced FMN, and the proportion of the FMN that is fully reduced (PR) is set by the NADH/NAD+ ratio [28,85]. This model explains why inhibition of complex I with rotenone increases O2•− production, as it will lead to a back up of electrons onto FMN which will produce O2•− [86,87]. For complex I within intact mitochondria, the proportion of fully reduced FMN is thought to be set by the NADH/NAD+ ratio, so inhibition of the respiratory chain by damage, mutation, ischaemia, loss of cytochrome c or by the build up of NADH due to low ATP demand and consequent low respiration rate will increase the NADH/NAD+ ratio and lead to O2•− formation [27,28,57,88–90]. In contrast, for most situations where mitochondria are respiring normally on NADH-linked substrates and the NADH/NAD+ ratio is relatively low, only small amounts of O2•− are produced from complex I [87]. An elegant demonstration of the importance of an elevated NADH/NAD+ ratio for O2•− production from complex I is that oxidation of the NADH pool in mammalian mitochondria by expression of a yeast NADH dehydrogenase decreases O2•− production [91].
The other mechanism by which complex I produces large amounts of O2•− is during RET (reverse electron transport) [7,27,87,89]. RET occurs for mitochondria operating in mode 2 when electron supply reduces the CoQ pool, which in the presence of a significant Δp forces electrons back from CoQH2 into complex I, and can reduce NAD+ to NADH at the FMN site [92]. RET was first associated with H2O2 production in SMPs respiring on succinate [83,93]. Later, it was shown that isolated mitochondria respiring on succinate, also generate large amounts of O2•− from complex I by RET [94]. Since then it has become clear that there is extensive O2•− production by RET at complex I in isolated brain, heart, muscle and liver mitochondria under conditions of high Δp with electron supply to the CoQ pool from succinate, α-glycerophosphate or fatty acid oxidation [7,24,87,89]. This O2•− production by RET is abolished by rotenone, confirming that it is due to electrons entering into complex I through the CoQ-binding site(s) [95]. Of particular note, the dependence of RET-associated O2•− production on Δp is very steep and is abolished completely by a small decrease in Δp [79,87], presumably by decreasing the thermodynamic driving force pushing electrons to the O2•−-producing site(s) within complex I. Intriguingly, O2•− production by complex I is more sensitive to changes in the ΔpH (pH gradient) component of the Δp, than to the Δψ (membrane potential) component [79,96].
The complex I site producing O2•− during RET is unclear [85,97]. The simplest possibility is that RET forces electrons right back through complex I to the FMN, and that the site of O2•− production is the same during RET as it is for O2•− production from the reduced FMN in response to an elevated NADH/NAD+ ratio [85,89]. Consistent with this, the flavin inhibitor DPI (diphenyleneiodonium) blocks RET-associated O2•− production by complex I [89]; however, this does not confirm the involvement of the FMN site, as DPI has other interactions with mitochondria [97]. Furthermore, disruption of the CoQ-binding site in complex I under conditions where there was a high ΔpH led to far more extensive O2•− production than reduction of FMN alone, suggesting a role for the CoQ-binding site in RET O2•− production [95]. In addition, there is not a unique relationship between the mitochondrial NADH/NAD+ ratio and O2•− production by complex I under different conditions [98]. This suggests that O2•− production from complex I may occur both by RET and from the reduction of FMN by the NADH pool simultaneously, and that most of the O2•− generated during RET is not produced at the FMN. If O2•− is not produced at FMN during RET, then the focus shifts to the CoQ-binding site, where two electrons are passed from the N2 FeS centre to CoQ, reducing it to CoQH2, while pumping four H+ across the mitochondrial inner membrane by an unknown mechanism. The intriguing pH- and ΔpH-dependence of O2•− production during RET [79] may suggest that critical semiquinones and semiquinolates are formed during proton pumping that react directly with O2 to form O2•− [57,88,95]. However, our lack of knowledge of the full structure of complex I and its exact mechanism of proton pumping hampers progress. More positively, it may be that investigating the mechanism of O2•− production [95], and the unusual interaction of complex I with electron acceptors during RET [44], may shed light on the mechanism of proton pumping by complex I.
To summarize, complex I produces large amounts of O2•− by two mechanisms: when the matrix NADH/NAD+ ratio is high, leading to a reduced FMN site on complex I, and when electron donation to the CoQ pool is coupled with a high Δp leading to RET (Figure 4). Although the site of O2•− production during RET is not known, the rate of O2•− production under RET seems to be the highest that can occur in mitochondria [71,79,95].
Figure 4. Production of O2•− by complex I.

The cartoon of complex I is a chimaera modelled on the hydrophobic arm of Yarrowia lipolytica obtained by electron microscopy [132] and the crystal structure of the hydrophilic arm from Thermus thermophilus [82]. The location of the FMN and the FeS centres in the water-soluble arm are indicated, along with the putative CoQ-binding site. In mode 1, there is extensive O2•− production from the FMN in response to a reduced NADH pool. In mode 2, a high Δp and a reduced CoQ pool lead to RET and a high flux of O2•− from the complex. The site of this O2•− production is uncertain, hence the question mark, but may be associated with the CoQ-binding site(s).
Complex III
Complex III funnels electrons from the CoQ pool to cytochrome c. The monomer is ∼240 kDa and comprises 11 polypeptides, three haems and an FeS centre, and it interacts transiently with CoQ during the Q-cycle at the Qi and Qo sites [99]. Complex III has for a long time been regarded as a source of O2•− within mitochondria [84,100]. When supplied with CoQH2 and when the Qi site is inhibited by antimycin, complex III produces large amounts of O2•− from the reaction of O2 with a ubisemiquinone bound to the Qo site [84,89,100–103]. This O2•− is released from complex III to both sides of the inner membrane [24,27,52,104]. However, in the absence of antimycin, the Qo site ubisemiquinone is not stabilized and O2•− production by complex III is low [105]. Inhibition of respiration at points distal to complex III with cyanide or by loss of cytochrome c does not increase O2•− production by complex III [100], therefore reduction of the CoQ pool is not sufficient to generate O2•− at complex III. It may be that high Δp stabilizes the Qo site ubisemiquinone; however, the very high production of O2•− by complex I during RET in the presence of succinate is abolished completely by rotenone [87,95]. Under these conditions, there is a large Δp and a reduced CoQ pool, suggesting that the maximal O2•− production by uninhibited complex III is negligible compared with that by complex I during RET. However, it is possible that, for mitochondria operating in mode 3, when O2•− production by complex I is low, the contribution of complex III to the overall O2•− flux may then become relatively significant. Under these conditions, factors that affect the stability of the ubisemiquinone radical in the Qo site, such as loss of cytochrome c or changes in Δp or in the redox state of the CoQ and cytochrome c pools, may modulate O2•− production. Therefore, although complex III can be induced to produce O2•− with the inhibitor antimycin, its production in mitochondria under physiological conditions is far lower and is negligible compared with the maximum rates of O2•− production from complex I.
Other sites of O2•− production within mitochondria
Many other sites within mitochondria may also produce O2•− or H2O2. It is convenient to divide most of these into sites that interact with the matrix NADH pool and those that are connected to the CoQ pool within the inner membrane.
When respiratory chain activity is low, or when there is RET, the NADH/NAD+ ratio will increase and this may lead to O2•− production at other sites connected to the NADH pool, in addition to complex I. When complex I is inhibited with rotenone, there is a reduced NADH pool and ROS production from complex I, but addition of 2-oxoglutarate (α-ketoglutarate) as a substrate increases ROS production further, suggesting that the combination of 2-oxoglutarate with a reduced NADH pool may lead to significant ROS production from αKGDH [2-oxoglutarate dehydrogenase (α-ketoglutarate dehydrogenase)] [7,106–108]. One component of αKGDH is dihydrolipoamide dehydrogenase, which contains a flavin that can produce ROS when its electron acceptor NAD+ is limiting [7,106–108]. Decreasing the expression of dihydrolipoamide dehydrogenase in mouse fibroblasts decreased O2•− production in mitochondria isolated from these cells [7,106–108]. A similar situation may exist for other enzymes that contain dihydrolipoamide dehydrogenase, such as pyruvate dehydrogenase, but their capacity for O2•− production seems to be less than that of αKGDH [106]. Therefore, under conditions of high NADH/NAD+ ratio, not only complex I, but also αKGDH, and perhaps other enzymes linked to the NADH pool, may contribute to O2•− production in a manner that depends on the mitochondrial substrates. It may be that when mitochondria are operating in mode 3 and are actively making ATP, O2•− production from complex I is negligible and the contribution from sites such as αKGDH is proportionally more significant [106].
Many potential sites of O2•− production interact with the CoQ pool. Fatty acid oxidation in the matrix reduces ETF (electron transfer flavoprotein) which passes its electrons to CoQ via ETF:CoQ oxidoreductase on the matrix surface of the inner membrane [109]. Oxidation of palmitoyl-CoA by mitochondria leads to H2O2 production [12,90], primarily from RET at complex I; however, ETF:CoQ reductase may itself produce ROS [24]. Dihydro-orotate dehydrogenase on the outer surface of the inner membrane catalyses oxidation of dihydro-orotate to orotate with reduction of CoQ, and dihydro-orotate oxidation is associated with mitochondrial O2•− production [110]; however, it is uncertain whether O2•− is produced by dihydro-orotate dehydrogenase itself. On the outer surface of the inner membrane, αGPDH (α-glycerophosphate dehydrogenase) takes electrons from α-glycerophosphate to CoQ [111–113]. In brown adipose tissue mitochondria [112], and in Drosophila mitochondria [114], oxidation of α-glycerophosphate is associated with ROS production, much of this due to RET from complex I, although some is produced from αGPDH itself on the outer surface of the mitochondrial inner membrane [113]. However, the physiological significance of this is unclear, as αGPDH is expressed at relatively low levels in most mammalian tissues [111,112], although it may be important in the brain [113]. Complex II oxidizes succinate passing electrons to CoQ, but, although the damaged or mutated complex can produce ROS [101,115], it seems that all O2•− production during succinate oxidation arises from complex I by RET. Therefore the activity of a number of CoQ-linked enzymes is associated with increased ROS production, primarily by favouring RET at complex I, but, in some cases, they may produce ROS directly themselves.
In addition to the sites discussed above, there are many other mitochondrial enzymes that can be induced to produce O2•− or H2O2. Some of these are not connected to the NADH or CoQ pools, such as the adrenodoxin reductase/adrenodoxin/cytochrome P450 system in the mitochondrial matrix that receives electrons from the NADPH pool [116,117]. Therefore it is probable that many other sites of mitochondrial O2•− production remain to be discovered, but whether they make a quantitatively significant contribution to mitochondrial ROS production under physiological conditions is unclear [1].
Overview of O2•− production by isolated mitochondria
It is possible to draw some tentative conclusions about O2•− production by isolated mitochondria. Two modes of operation lead mitochondria to produce large amounts of O2•−: mode 1, when there is a build up of NADH, and mode 2, when there is a large Δp and a reduced CoQ pool (Figure 3). In both modes, the predominant site of O2•− production is complex I, in keeping with a growing view that complex I is the major source of O2•− within mitochondria in vivo [1,7,85,118]. Even so, the accumulation of NADH that occurs in modes 1 and 2 may lead to O2•− production from other sites such as αKGDH. The mechanisms of O2•− production from complex I in the two modes are quite distinct, being by reduction of FMN by NADH in mode 1 and by RET in mode 2. Of course, in some situations, both mode 1 and mode 2 may operate simultaneously when a build up of NADH and CoQ coincides with a high Δp.
The mode 1 production of O2•− probably occurs in vivo under conditions where damage to the respiratory chain, slow respiration or ischaemia leads to a build up of NADH. This may occur during cytochrome c release in apoptosis and following inhibition of respiration at cytochrome oxidase by NO•. Treatments that oxidize the NADH pool, such as overexpressing NADH oxidases, should decrease the production of O2•− in mode 1, while rotenone and other respiratory inhibitors should increase it (antimycin would not be diagnostic here as it will increase O2•− production at complex III). Whether mode 2 O2•− production occurs in cultured cells or in vivo is not known. If it does, it would be associated with a high Δp and a reduced CoQ pool, and will be very sensitive to mild uncoupling and to inhibition by rotenone. The generation of O2•− by both modes 1 and 2 will be decreased by mild uncoupling, perhaps suggesting that the physiological function of uncoupling proteins-2 and -3 is to modulate mitochondrial O2•− production [118].
Mode 3 occurs when mitochondria are synthesizing ATP or utilizing Δp for other functions. Under these conditions, the lowered Δp and the oxidized NADH pool prevent O2•− production by RET and greatly decrease O2•− production at the FMN of complex I, making H2O2 efflux from mitochondria negligible compared with modes 1 or 2 (e.g. [4,78,89,119]). However, there may still be O2•− production within mitochondria operating in mode 3 that is below the threshold consumed by mitochondrial peroxidases (eqn 4) [68]. Although the sites of this putative O2•− production in mode 3 are unclear, complex I, complex III and matrix enzymes such as αKGDH could all contribute. A further complication is that in vivo substrate supply to the respiratory chain is stringently regulated by factors such as dietary and hormonal status, leading to alterations in the steady-state reduction potential of mitochondrial electron carriers. As mitochondria in vivo probably spend much of their time synthesizing ATP, this mode of O2•− production may account for most of the overall exposure of mitochondria to O2•−. Thus, even though the rate of O2•− production in mode 3 is relatively low and the sources are poorly understood, it may turn out to be the mode of greatest biological importance and be responsible for the long-term accumulation of mitochondrial oxidative damage and for the extensive tissue damage seen in the absence of MnSOD [45,46].
THE PARADOX OF HYPOXIC H2O2 PRODUCTION BY MITOCHONDRIA
Figure 3Figure 3 gives a reasonable description of mitochondrial O2•− production. However, mitochondrial ROS production is also reported to increase under conditions of very low [O2], which is paradoxical and seems to contradict the dependence of mitochondrial O2•− production on [O2] given in eqn (2). These hypoxic effects are seen in cultured cells when ambient O2 is decreased from 21% O2 to 1–3% O2 [120,121]. This corresponds to an equilibrium [O2] of 10–20 μM, although the local [O2] around mitochondria will be lower.
The discovery of increased mitochondrial ROS production during hypoxia arose from investigations of HIF-1 (hypoxia-inducible factor-1), which plays a central role in the response of cells to hypoxia [122,123]. HIF-1 is a heterodimer comprising HIF-1α and HIF-1β that translocates to the nucleus and there, in association with other proteins, initiates transcription of a number of genes in response to hypoxia [122,123]. HIF-1α is constitutively expressed, but, under normoxia, it is rapidly hydroxylated on proline residues by PHD (prolyl hydroxylase), which uses 2-oxoglutarate and O2 as substrates, marking HIF-1α for rapid degradation by the ubiquitin–proteasome system [122,123]. When the [O2] falls, HIF-1α is no longer degraded, allowing the HIF-1 heterodimer to form and induce the transcription of a series of hypoxia-sensitive genes. Further regulation of HIF-1 occurs because PHD activity is sensitive to H2O2, probably through reaction with non-haem iron in its active site [120,121]. Thus the mitochondrial respiratory chain may act as an O2 sensor, releasing H2O2 under hypoxic conditions to decreases the activity of PHD, thereby stabilizing HIF-1α and modulating its response to hypoxia [120,121].
The evidence that hypoxia increases mitochondrial ROS production in cultured cells comes from measurements of cytosolic ROS using several different probes, and from showing that the effects of hypoxia-induced mitochondrial ROS on HIF-1 can be blocked by mitochondria-targeted antioxidants [120,121,124]. Further studies have shown that a functional respiratory chain is required, that loss of cytochrome c, or the Rieske FeS centre of complex III, abolishes this ROS signal [121,125], and that the direct addition of H2O2 overcomes this blockade [126]. These studies have led to the proposal that the source of the ROS is complex III [121], possibly a ubisemiquinone at the Qo site [126]. Although ongoing work may refine details of this model, the question remains of how lowering [O2] can increase H2O2 efflux from mitochondria. When isolated mitochondria were maintained at low [O2], ROS production decreased as the [O2] was lowered from approx. 5 μM O2 to anoxia [29]. This finding makes it unlikely that the decrease in [O2] itself affects O2•− production directly, for example by altering the stability of the ubisemiquinone radical at complex III, the rate of degradation or release of H2O2 from mitochondria or the sidedness of O2•− release from complex III [104,121]. Instead it suggests that the low [O2] environment requires additional factors that occur in the hypoxic cell environment to increase mitochondrial ROS efflux. For example, changes in [NO•] might modulate the response of cytochrome oxidase to low [O2] [127], thereby altering the PR of protein redox groups. Alternatively, hypoxia could act through cell signalling pathways to decrease the activity of mitochondrial matrix or intermembrane space peroxidases, thereby increasing mitochondrial H2O2 efflux. Clearly, more work is required to unravel the mechanism of increased mitochondrial ROS production during hypoxia, but it remains an intriguing puzzle, and our understanding of mitochondrial ROS production will be incomplete until there is a satisfactory explanation for this phenomenon.
HOW MUCH O2•− DO MAMMALIAN MITOCHONDRIA PRODUCE IN VIVO?
To investigate the significance of mitochondrial O2•− production in oxidative damage and redox signalling, it is necessary to know how much O2•− is generated by mitochondria in vivo. The greatest rate of H2O2 production by isolated mitochondria occurs during mode 2 when mitochondria have a high Δp, a reduced CoQ pool and are not making ATP. Under these conditions, ROS production is primarily by RET at complex I, and approx. 1–2% of the O2 consumed by isolated mitochondria under these conditions forms O2•− [1,4,27]. Since it was first published by Chance and colleagues [4,12], this value of 1–2% of respiration going to O2•− has propagated through the literature and has been used erroneously to estimate mitochondrial O2•− production in vivo, even though the original authors made it clear that it only applied to particular experimental conditions [12].
Several factors make extrapolation of H2O2 production by isolated mitochondria to the in vivo situation invalid. First, maximal O2•− production by isolated mitochondria occurs during RET using saturating levels of substrates such as succinate, which are at lower concentrations in vivo. When lower concentrations of succinate are used, approx. 0.4–0.8% of respiration produces H2O2 [57], while use of the physiological substrate palmitoyl-CoA decreases H2O2 production to approx. 0.15% of respiration [24], and, when glutamate/malate are used as substrates, H2O2 production accounts for approx. 0.12% of respiration [27]. Secondly, measurements on isolated mitochondria are generally made using air-saturated medium containing ∼200 μM O2. As mitochondrial O2•− production is probably proportional to [O2] and the physiological [O2] around mitochondria is approx. 10–50 μM, O2•− production may be 5–10-fold lower than for isolated mitochondria in the same state. The third and most important factor limiting extrapolation of in vitro O2•− production to the situation in vivo is that mitochondria in vivo are likely to be making ATP and will thus be operating in mode 3 with a lowered Δp and relatively oxidized NADH and CoQ pools. Consequently, their rates of H2O2 efflux are negligible compared with modes 1 or 2. Therefore, although it is valid to say that 0.12–2% of respiration goes to O2•− in vitro, these values cannot be extrapolated to the in vivo situation where mitochondrial O2•− production will be far, far lower.
Allowing for these caveats, can we estimate roughly how much O2•− is produced by mitochondria in vivo? Unfortunately the answer at the moment is no, because we know little about basic mitochondrial function in vivo. For example, the proportion of time that mitochondria spend actively making ATP (i.e. in state 3) or with a high Δp and a low rate of ATP synthesis (i.e. in state 4) in vivo is not known. The proportion of mitochondria with a reduced NADH pool and whether RET occurs in vivo are also unclear. If, as is probable, mitochondria in vivo spend most of their time close to state 3, then we have very limited knowledge of how much O2•− production occurs in the matrix. Therefore, although there is considerable evidence for the accumulation of oxidative damage within mitochondria in vivo [3,128], from which we can infer that mitochondrial ROS production does occur, it is difficult to estimate the flux of mitochondrial O2•− that leads to this damage.
Although estimates of mitochondrial O2•− production as a proportion of respiration rate are not possible, can we infer from studies of mitochondria ex vivo the extent of O2•− production in vivo? The maximum O2•− production rate in vivo is proportional to the content of respiratory complexes such as complex I, and thus correlates with maximum respiration rate. However, as the actual O2•− production rate depends so closely on factors such as Δp and NADH/NAD+ ratio, which vary markedly in vivo, it is not possible to say that changes in ROS production by mitochondria isolated from animals of different ages or hormonal status bear any relation to differences in mitochondrial ROS production in vivo. Is it at least possible to infer in vivo rates of mitochondrial O2•− production from direct measurements of ROS within living tissue? At the moment, the answer is also no, because quantification is challenging and currently there are no reliable estimates of mitochondrial O2•− production in vivo. A classic estimate of H2O2 production was made in perfused liver where H2O2 production by the whole organ was measured from changes in catalase compound I [129]. Approx. 80 nmol of H2O2/min per g of wet weight was produced in the tissue [127], with about 12 nmol of H2O2/min per g of wet weight estimated to come from mitochondria [90]. However, one of the many assumptions made in this work was that mitochondrial H2O2 production in situ was the same as for isolated mitochondria respiring on succinate, when they would have been producing H2O2 by RET [90]. Therefore we know little about the actual flux of O2•− within mitochondria in vivo, about how it changes under different physiological circumstance, or about its quantititive importance relative to other sources of ROS. More work is required to develop methods to measure mitochondrial ROS production in vivo.
Conclusions
I have outlined the factors that lead to O2•− production within isolated mitochondria and have shown that there are two modes of high O2•− production, predominantly, but not exclusively, from complex I: by RET when the Δp is high and the CoQ pool is reduced, and from the FMN when the NADH/NAD+ ratio is high (Figure 3). When mitochondria are actively making ATP, the rate of O2•− production is far lower and the sites of production are uncertain. This suggests that in vivo conditions leading to RET or an accumulation of NADH will favour O2•− production. However, the extent to which these situations arise in vivo is not known, and, at the moment, it is not possible to estimate the rate of mitochondrial O2•− production in vivo. Even so, the description given here of the factors underlying mitochondrial O2•− production will enable the design and interpretation of experiments to assess the physiological and pathological significance of mitochondrial ROS production in vivo. One area where such knowledge is vital is in the large number of pathologies where mitochondrial disruption leads to oxidative damage [3,5]. This raises the possibility that better understanding of how mitochondria produce ROS will lead to the rational design of therapies to minimize mitochondrial oxidative damage [130]. Also important is the likelihood that ROS production by mitochondria is a redox signal integrating mitochondrial function with that of the rest of the cell [3,9]. Redox signalling can occur by mitochondria releasing H2O2 that modulates the activity of target proteins through the reversible oxidation of critical protein thiols [9,131], thus altering the activity of enzymes, kinases, phosphatases and transcription factors in mitochondria, the cytosol or the nucleus (Figure 5). Although little is known currently about mitochondrial redox signalling, the description of ROS production developed here shows how it can occur and will prove useful in investigating this rapidly developing area. For example, mitochondrial H2O2 efflux could act as a retrograde signal to the cell, reporting on mitochondrial Δp or the redox state of the NADH pool, and thus enable the activity, transcription, translation, import or degradation of mitochondrial components to be adjusted accordingly. It is also likely that external signals can alter O2•− production through the post-translational modification of the respiratory chain, or by altering H2O2 efflux from mitochondria by modulating the activity of matrix peroxidases such as peroxiredoxins. However, it is clear that to address these vital issues, better methods are required to assess mitochondrial Δp and the reduction potential of critical redox couples within mitochondria in vivo, and thus enable the tendency of mitochondria to produce O2•− to be inferred. Furthermore, techniques to measure the production of mitochondrial O2•− and H2O2 directly in vivo also need to be developed. Such innovations are essential to understand the role of mitochondrial O2•− production in redox signalling and in pathological oxidative damage.
Figure 5. Possible mechanisms of mitochondrial redox signalling.

The production of H2O2 from mitochondria is a potential redox signal. H2O2 generated by mitochondria can reversibly alter the activity of proteins with critical protein thiols by modifying them to intra- or inter-protein disulfides, or to mixed disulfides with GSH. These modifications can occur on mitochondrial, cytosolic or nuclear enzymes, carriers or transcription factors, transiently altering their activities. The change in activity can be reversed by reducing the modified protein thiol by endogenous thiol reductants such as GSH or thioredoxin. As the extent of H2O2 production from mitochondria will depend on factors such as Δp or the redox state of the NADH pool, it can act as a retrograde signal to the rest of the cell, reporting on mitochondrial status. This signal can then lead to the short-term modification of, for example, pathways supplying substrates to the mitochondria. Alternatively, longer-term modifications can occur through modifying redox-sensitive transcription factors that adjust the production of mitochondrial components. In addition, external signals may modify O2•− production by the respiratory chain by post-translational modification. Alteration of the activity of mitochondrial peroxidases could also modulate H2O2 efflux from mitochondria to the rest of the cell. It is also possible that secondary redox signals, such as lipid peroxidation products derived from H2O2, could act as secondary redox signals.
Animation
Acknowledgments
I thank Martin Brand, Paul Brookes, Helena Cochemé, Judy Hirst, Thomas Hurd, Andrew James, Adrian Lambert, Paul Schumacker and Jan Trnka for helpful comments and discussions, and Helena Cochemé for producing the figures.
FUNDING
Work is supported by the Medical Research Council (U.K.).
References
- 1.Andreyev A. Y., Kushnareva Y. E., Starkov A. A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 2.Turrens J. F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaban R. S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Chance B., Sies H., Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 5.Cadenas E., Davies K. J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biol. Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 6.Raha S., Robinson B. H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. 2000;25:502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 7.Adam-Vizi V., Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006;27:639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Muller F. The nature and mechanism of superoxide production by the electron transport chain. J. Am. Aging Assoc. 2000;23:227–253. doi: 10.1007/s11357-000-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Droge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 10.Jensen P. K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochim. Biophys. Acta. 1966;122:157–166. doi: 10.1016/0926-6593(66)90057-9. [DOI] [PubMed] [Google Scholar]
- 11.Loschen G., Flohe L., Chance B. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 1971;18:261–264. doi: 10.1016/0014-5793(71)80459-3. [DOI] [PubMed] [Google Scholar]
- 12.Boveris A., Chance B. The mitochondrial generation of hydrogen peroxide: general properties and effect of hyperbaric oxygen. Biochem. J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loschen G., Azzi A., Richter C., Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 14.Forman H. J., Kennedy J. A. Role of superoxide radical in mitochondrial dehydrogenase reactions. Biochem. Biophys. Res. Commun. 1974;60:1044–1050. doi: 10.1016/0006-291x(74)90418-5. [DOI] [PubMed] [Google Scholar]
- 15.Weisiger R. A., Fridovich I. Superoxide dismutase: organelle specificity. J. Biol. Chem. 1973;248:3582–3592. [PubMed] [Google Scholar]
- 16.Sawyer D. T., Valentine J. S. How super is superoxide? Acc. Chem. Res. 1981;14:393–400. [Google Scholar]
- 17.Reynafarje B., Costa L. E., Lehninger A. L. O2 solubility in aqueous media determined by a kinetic method. Anal. Biochem. 1985;145:406–418. doi: 10.1016/0003-2697(85)90381-1. [DOI] [PubMed] [Google Scholar]
- 18.Erecinska M., Silver I. A. Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 2001;128:263–276. doi: 10.1016/s0034-5687(01)00306-1. [DOI] [PubMed] [Google Scholar]
- 19.Skulachev V. P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- 20.Tyler D. D. Polarographic assay and intracellular distribution of superoxide dismutase in rat liver. Biochem. J. 1975;147:493–504. doi: 10.1042/bj1470493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winterbourn C. C., French J. K., Claridge R. F. Superoxide dismutase as an inhibitor of reactions of semiquinone radicals. FEBS Lett. 1978;94:269–272. doi: 10.1016/0014-5793(78)80953-3. [DOI] [PubMed] [Google Scholar]
- 22.Schafer F. Q., Buettner G. R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radical Biol. Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 23.Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- 24.St-Pierre J., Buckingham J. A., Roebuck S. J., Brand M. D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 25.Wikstrom M., Krab K., Saraste M. London: Academic Press; 1981. Cytochrome Oxidase: a Synthesis. [Google Scholar]
- 26.Turrens J. F., Freeman B. A., Levitt J. G., Crapo J. D. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch. Biochem. Biophys. 1982;217:401–410. doi: 10.1016/0003-9861(82)90518-5. [DOI] [PubMed] [Google Scholar]
- 27.Kudin A. P., Bimpong-Buta N. Y., Vielhaber S., Elger C. E., Kunz W. S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 28.Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman D. L., Salter J. D., Brookes P. S. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H101–H108. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez S., Valdez L. B., Zaobornyj T., Boveris A. Oxygen dependence of mitochondrial nitric oxide synthase activity. Biochem. Biophys. Res. Commun. 2003;305:771–775. doi: 10.1016/s0006-291x(03)00818-0. [DOI] [PubMed] [Google Scholar]
- 31.Shiva S., Brookes P. S., Patel R. P., Anderson P. G., Darley-Usmar V. M. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown G. C., Cooper C. E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 33.Brown G. C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- 34.Cleeter M. J. W., Cooper J. M., Darley-Usmar V. M., Moncada S., Schapira A. H. V. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of mitochondrial respiratory chain, by nitric oxide. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. [DOI] [PubMed] [Google Scholar]
- 35.Brown G. C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 36.Mateo J., García-Lecea M., Cadenas S., Hernández C., Moncada S. Regulation of hypoxia-inducible factor-1α by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J. 2003;376:537–544. doi: 10.1042/BJ20031155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hagen T., Taylor C. T., Lam F., Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1α. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 38.Moncada S., Erusalimsky J. D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. [DOI] [PubMed] [Google Scholar]
- 39.Klinman J. P. How do enzymes activate oxygen without inactivating themselves? Acc. Chem. Res. 2007;40:325–333. doi: 10.1021/ar6000507. [DOI] [PubMed] [Google Scholar]
- 40.Marcus R. A., Sutin N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 41.Moser C. C., Farid T. A., Chobot S. E., Dutton P. L. Electron tunneling chains of mitochondria. Biochim. Biophys. Acta. 2006;1757:1096–1109. doi: 10.1016/j.bbabio.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 42.Massey V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 43.Imlay J. A. A metabolic enzyme that rapidly produces superoxide, fumarate reductase of Escherichia coli. J. Biol. Chem. 1995;270:19767–19777. [PubMed] [Google Scholar]
- 44.Cochemé H. M., Murphy M. P. Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 2008;283:1786–1798. doi: 10.1074/jbc.M708597200. [DOI] [PubMed] [Google Scholar]
- 45.Li Y., Huang T.-T., Carlson E. J., Melov S., Ursell P. C., Olson J. L., Noble L. J., Yoshimura M. P., Berger C., Chan P. H., et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 46.Lebovitz R. M., Zhang H., Vogel H., Cartwright J., Dionne L., Lu N., Huang S., Matzuk M. M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao H., Joseph J., Fales H. M., Sokoloski E. A., Levine R. L., Vasquez-Vivar J., Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. U.S.A. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson K. M., Janes M. S., Pehar M., Monette J. S., Ross M. F., Hagen T. M., Murphy M. P., Beckman J. S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. U.S.A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zielonka J., Vasquez-Vivar J., Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat. Protoc. 2008;3:8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- 50.Zielonka J., Srinivasan S., Hardy M., Ouari O., Lopez M., Vasquez-Vivar J., Avadhani N. G., Kalyanaraman B. Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: identification of homo- and heterodimers. Free Radical Biol. Med. 2008;44:835–846. doi: 10.1016/j.freeradbiomed.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lucas M., Solano F. Coelenterazine is a superoxide anion-sensitive chemiluminescent probe: its usefulness in the assay of respiratory burst in neutrophils. Anal. Biochem. 1992;206:273–277. doi: 10.1016/0003-2697(92)90366-f. [DOI] [PubMed] [Google Scholar]
- 52.Han D., Antunes F., Canali R., Rettori D., Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 53.Gardner P. R. Aconitase: sensitive target and measure of superoxide. Methods Enzmol. 2002;349:9–23. doi: 10.1016/s0076-6879(02)49317-2. [DOI] [PubMed] [Google Scholar]
- 54.Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984;105:429–435. doi: 10.1016/s0076-6879(84)05060-6. [DOI] [PubMed] [Google Scholar]
- 55.Barja G. Methods in Aging Research. Boca Raton: CRC Press; 1999. Kinetic measurement of mitochondrial oxygen radical production; pp. 533–548. [Google Scholar]
- 56.Sohal R. S. Hydrogen peroxide production by mitochondria may be a biomarker of aging. Mech. Ageing Dev. 1991;60:189–198. doi: 10.1016/0047-6374(91)90130-r. [DOI] [PubMed] [Google Scholar]
- 57.Hansford R. G., Hogue B. A., Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 58.Szabo C., Ischiropoulos H., Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 59.Packer M. A., Porteous C. M., Murphy M. P. Mitochondrial superoxide production in the presence of nitric oxide leads to the formation of peroxynitrite. Biochem. Mol. Biol. Int. 1996;40:527–534. doi: 10.1080/15216549600201103. [DOI] [PubMed] [Google Scholar]
- 60.Giorgio M., Migliaccio E., Orsini F., Paolucci D., Moroni M., Contursi C., Pelliccia G., Luzi L., Minucci S., Marcaccio M., et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 61.Zoccarato F., Cavallini L., Alexandre A. Respiration-dependent removal of exogenous H2O2 in brain mitochondria: inhibition by Ca2+ J. Biol. Chem. 2004;279:4166–4174. doi: 10.1074/jbc.M308143200. [DOI] [PubMed] [Google Scholar]
- 62.Rhee S. G., Yang K. S., Kang S. W., Woo H. A., Chang T. S. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid. Redox Signaling. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 63.Hurd T. R., Costa N. J., Dahm C. C., Beer S. M., Brown S. E., Filipovska A., Murphy M. P. Glutathionylation of mitochondrial proteins. Antioxid. Redox Signaling. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- 64.Rhee S. G., Kang S. W., Chang T. S., Jeong W., Kim K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life. 2001;52:35–41. doi: 10.1080/15216540252774748. [DOI] [PubMed] [Google Scholar]
- 65.Salvi M., Battaglia V., Brunati A. M., La Rocca N., Tibaldi E., Pietrangeli P., Marcocci L., Mondovi B., Rossi C. A., Toninello A. Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem. 2007;282:24407–24415. doi: 10.1074/jbc.M701589200. [DOI] [PubMed] [Google Scholar]
- 66.Radi R., Turrens J. F., Chang L. Y., Bush K. M., Crapo J. D., Freeman B. A. Detection of catalase in rat heart mitochondria. J. Biol. Chem. 1991;266:22028–22034. [PubMed] [Google Scholar]
- 67.Imai H., Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radical Biol. Med. 2003;34:145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 68.Rigobello M. P., Folda A., Scutari G., Bindoli A. The modulation of thiol redox state affects the production and metabolism of hydrogen peroxide by heart mitochondria. Arch. Biochem. Biophys. 2005;441:112–122. doi: 10.1016/j.abb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 69.Rhee S. G., Kang S. W., Jeong W., Chang T. S., Yang K. S., Woo H. A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 70.Cox A. G., Pullar J. M., Hughes G., Ledgerwood E. C., Hampton M. B. Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis. Free Radical Biol. Med. 2008;44:1001–1009. doi: 10.1016/j.freeradbiomed.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 71.Hurd T. R., Prime T. A., Harbour M. E., Lilley K. S., Murphy M. P. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J. Biol. Chem. 2007;282:22040–22051. doi: 10.1074/jbc.M703591200. [DOI] [PubMed] [Google Scholar]
- 72.Holmgren A. Thioredoxin. Annu. Rev. Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 73.Hurd T. R., Filipovska A., Costa N. J., Dahm C. C., Murphy M. P. Disulphide formation on mitochondrial protein thiols. Biochem. Soc. Trans. 2005;33:1390–1393. doi: 10.1042/BST0331390. [DOI] [PubMed] [Google Scholar]
- 74.Rydström J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta. 2006;1757:721–726. doi: 10.1016/j.bbabio.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 75.Sazanov L. A., Jackson J. B. Proton-translocating transhydrogenase and NAD- and NADP-linked isocitrate dehydrogenases operate in a substrate cycle which contributes to fine regulation of the tricarboxylic acid cycle activity in mitochondria. FEBS Lett. 1994;344:109–116. doi: 10.1016/0014-5793(94)00370-x. [DOI] [PubMed] [Google Scholar]
- 76.Bienert G. P., Moller A. L., Kristiansen K. A., Schulz A., Moller I. M., Schjoerring J. K., Jahn T. P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 77.Bienert G. P., Schjoerring J. K., Jahn T. P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 78.Korshunov S. S., Skulachev V. P., Starkov A. A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 79.Lambert A. J., Brand M. D. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hirst J., Carroll J., Fearnley I. M., Shannon R. J., Walker J. E. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim. Biophys. Acta. 2003;1604:135–150. doi: 10.1016/s0005-2728(03)00059-8. [DOI] [PubMed] [Google Scholar]
- 81.Sazanov L. A. Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry. 2007;46:2275–2288. doi: 10.1021/bi602508x. [DOI] [PubMed] [Google Scholar]
- 82.Sazanov L. A., Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 83.Hinkle P. C., Butow R. A., Racker E., Chance B. Partial resolution of the enzymes catalyzing oxidative phosphorylation. XV. Reverse electron transfer in the flavin-cytochrome β region of the respiratory chain of beef heart submitochondrial particles. J. Biol. Chem. 1967;242:5169–5173. [PubMed] [Google Scholar]
- 84.Cadenas E., Boveris A., Ragan C. I., Stoppani A. O. Production of superoxide radicals and hydrogen peroxide by NADH–ubiquinone reductase and ubiquinol–cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 1977;180:248–257. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- 85.Hirst J., King M. S., Pryde K. R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008;36:976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 86.Takeshige K., Minakami S. NADH- and NADPH-dependent formation of superoxide anions by bovine heart submitochondrial particles and NADH–ubiquinone reductase preparation. Biochem. J. 1979;180:129–135. doi: 10.1042/bj1800129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Votyakova T. V., Reynolds I. J. Δψm-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 88.Kushnareva Y., Murphy A. N., Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation–reduction state. Biochem. J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y., Fiskum G., Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 90.Boveris A., Oshino N., Chance B. The cellular production of hydrogen peroxide. Biochem. J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seo B. B., Marella M., Yagi T., Matsuno-Yagi A. The single subunit NADH dehydrogenase reduces generation of reactive oxygen species from complex I. FEBS Lett. 2006;580:6105–6108. doi: 10.1016/j.febslet.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 92.Chance B., Hollunger G. The interaction of energy and electron transfer reactions in mitochondria. I. General properties and nature of the products of succinate-linked reduction of pyridine nucleotide. J Biol. Chem. 1961;236:1534–1543. [PubMed] [Google Scholar]
- 93.Krishnamoorthy G., Hinkle P. C. Studies on the electron transfer pathway, topography of iron–sulfur centers, and site of coupling in NADH-Q oxidoreductase. J. Biol. Chem. 1988;263:17566–17575. [PubMed] [Google Scholar]
- 94.Cino M., Del Maestro R. F. Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch. Biochem. Biophys. 1989;269:623–638. doi: 10.1016/0003-9861(89)90148-3. [DOI] [PubMed] [Google Scholar]
- 95.Lambert A. J., Brand M. D. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 96.Liu S. S. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci. Rep. 1997;17:259–272. doi: 10.1023/a:1027328510931. [DOI] [PubMed] [Google Scholar]
- 97.Lambert A. J., Buckingham J. A., Boysen H. M., Brand M. D. Diphenyleneiodonium acutely inhibits reactive oxygen species production by mitochondrial complex I during reverse, but not forward electron transport. Biochim. Biophys. Acta. 2008;1777:397–403. doi: 10.1016/j.bbabio.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 98.Lambert A. J., Buckingham J. A., Brand M. D. Dissociation of superoxide production by mitochondrial complex I from NAD(P)H redox state. FEBS Lett. 2008;582:1711–1714. doi: 10.1016/j.febslet.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 99.Iwata S., Lee J. W., Okada K., Lee J. K., Iwata M., Rasmussen B., Link T. A., Ramaswamy S., Jap B. K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 100.Turrens J. F., Alexandre A., Lehninger A. L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 101.Zhang L., Yu L., Yu C. A. Generation of superoxide anion by succinate–cytochrome c reductase from bovine heart mitochondria. J. Biol. Chem. 1998;273:33972–33976. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]
- 102.Rich P. R., Bonner W. D. The sites of superoxide anion generation in higher plant mitochondria. Arch. Biochem. Biophys. 1978;188:206–213. doi: 10.1016/0003-9861(78)90373-9. [DOI] [PubMed] [Google Scholar]
- 103.Grigolava I. V., Ksenzenko M., Konstantinob A. A., Tikhonov A. N., Kerimov T. M. Tiron as a spin-trap for superoxide radicals produced by the respiratory chain of submitochondrial particles. Biochemisty (Moscow) 1980;45:75–82. [PubMed] [Google Scholar]
- 104.Muller F. L., Liu Y., Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 105.Forman H. J., Azzi A. On the virtual existence of superoxide anions in mitochondria: thoughts regarding its role in pathophysiology. FASEB J. 1997;11:374–375. doi: 10.1096/fasebj.11.5.9141504. [DOI] [PubMed] [Google Scholar]
- 106.Starkov A. A., Fiskum G., Chinopoulos C., Lorenzo B. J., Browne S. E., Patel M. S., Beal M. F. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tretter L., Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J. Neurosci. 2004;24:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bunik V. I., Sievers C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 2002;269:5004–5015. doi: 10.1046/j.1432-1033.2002.03204.x. [DOI] [PubMed] [Google Scholar]
- 109.Eaton S. Control of mitochondrial β-oxidation flux. Prog. Lipid Res. 2002;41:197–239. doi: 10.1016/s0163-7827(01)00024-8. [DOI] [PubMed] [Google Scholar]
- 110.Forman H. J., Kennedy J. Dihydroorotate-dependent superoxide production in rat brain and liver: a function of the primary dehydrogenase. Arch. Biochem. Biophys. 1976;173:219–224. doi: 10.1016/0003-9861(76)90252-6. [DOI] [PubMed] [Google Scholar]
- 111.Koza R. A., Kozak U. C., Brown L. J., Leiter E. H., MacDonald M. J., Kozak L. P. Sequence and tissue-dependent RNA expression of mouse FAD-linked glycerol-3-phosphate dehydrogenase. Arch. Biochem. Biophys. 1996;336:97–104. doi: 10.1006/abbi.1996.0536. [DOI] [PubMed] [Google Scholar]
- 112.Drahota Z., Chowdhury S. K., Floryk D., Mracek T., Wilhelm J., Rauchova H., Lenaz G., Houstek J. Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J. Bioenerg. Biomembr. 2002;34:105–113. doi: 10.1023/a:1015123908918. [DOI] [PubMed] [Google Scholar]
- 113.Tretter L., Takacs K., Hegedus V., Adam-Vizi V. Characteristics of α-glycerophosphate-evoked H2O2 generation in brain mitochondria. J. Neurochem. 2007;100:650–663. doi: 10.1111/j.1471-4159.2006.04223.x. [DOI] [PubMed] [Google Scholar]
- 114.Miwa S., St-Pierre J., Partridge L., Brand M. D. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radical Biol. Med. 2003;35:938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 115.Guzy R. D., Sharma B., Bell E., Chandel N. S., Schumacker P. T. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hanukoglu I. Antioxidant protective mechanisms against reactive oxygen species (ROS) generated by mitochondrial P450 systems in steroidogenic cells. Drug Metab. Rev. 2006;38:171–196. doi: 10.1080/03602530600570040. [DOI] [PubMed] [Google Scholar]
- 117.Hanukoglu I., Rapoport R., Weiner L., Sklan D. Electron leakage from the mitochondrial NADPH–adrenodoxin reductase–adrenodoxin–P450scc (cholesterol side chain cleavage) system. Arch. Biochem. Biophys. 1993;305:489–498. doi: 10.1006/abbi.1993.1452. [DOI] [PubMed] [Google Scholar]
- 118.Brand M. D., Affourtit C., Esteves T. C., Green K., Lambert A. J., Miwa S., Pakay J. L., Parker N. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radical Biol. Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 119.Starkov A. A., Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 120.Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., Schumacker P. T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guzy R. D., Schumacker P. T. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006;91:807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 122.Schofield C. J., Ratcliffe P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 123.Semenza G. L. O2-regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF-1. J. Appl. Physiol. 2004;96:1173–1177. doi: 10.1152/japplphysiol.00770.2003. [DOI] [PubMed] [Google Scholar]
- 124.Sanjuan-Pla A., Cervera A. M., Apostolova N., Garcia-Bou R., Victor V. M., Murphy M. P., McCreath K. J. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1α. FEBS Lett. 2005;579:2669–2674. doi: 10.1016/j.febslet.2005.03.088. [DOI] [PubMed] [Google Scholar]
- 125.Guzy R. D., Hoyos B., Robin E., Chen H., Liu L., Mansfield K. D., Simon M. C., Hammerling U., Schumacker P. T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 126.Bell E. L., Klimova T. A., Eisenbart J., Moraes C. T., Murphy M. P., Budinger G. R., Chandel N. S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Palacios-Callender M., Quintero M., Hollis V. S., Springett R. J., Moncada S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7630–7635. doi: 10.1073/pnas.0401723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Beckman K. B., Ames B. N. The free radical theory of aging matures. Physiol. Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 129.Oshino N., Jamieson D., Chance B. The properties of hydrogen peroxide production under hyperoxic and hypoxic conditions of perfused rat liver. Biochem. J. 1975;146:53–65. doi: 10.1042/bj1460053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Murphy M. P., Smith R. A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 131.Hurd T. R., Prime T. A., Harbour M. E., Lilley K. S., Murphy M. P. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J. Biol. Chem. 2007;282:22040–22051. doi: 10.1074/jbc.M703591200. [DOI] [PubMed] [Google Scholar]
- 132.Radermacher M., Ruiz T., Clason T., Benjamin S., Brandt U., Zickermann V. The three-dimensional structure of complex I from Yarrowia lipolytica: a highly dynamic enzyme. J. Struct. Biol. 2006;154:269–279. doi: 10.1016/j.jsb.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.