

Abstract
Synapsins regulate synaptic transmission by controlling the reserve pool of synaptic vesicles. Each of the three mammalian synapsin genes is subject to alternative splicing, yielding several isoforms whose roles are unknown. To investigate the function of these isoforms, we examined the synaptic effects of introducing each isoform into glutamatergic cultured hippocampal neurons from synapsin triple knock-out mice. Remarkably, we found that synapsin IIa was the only isoform that could rescue the synaptic depression phenotype of the triple knock-out mice; other isoforms examined, including the well-studied synapsin Ia isoform, had no significant effect on the kinetics of synaptic depression. The slowing of synaptic depression by synapsin IIa was quantitatively paralleled by an increase in the density of reserve pool synaptic vesicles, as measured either by fluorescent tagging of the vesicle protein synaptobrevin-2 or by staining with the styryl dye FM4–64 [N-(3-triethylammoniumpropyl)-4-(6-(4-diethylamino)phenyl)-hexatrienyl)pyridinium dibromide]. Our results provide further support for the hypothesis that synapsins define the kinetics of synaptic depression at glutamatergic synapses by controlling the size of the vesicular reserve pool and identify synapsin IIa as the isoform primarily responsible for this task.
Keywords: neurotransmitter release, synaptic vesicle trafficking, synaptic plasticity, alternative splice variants, synaptic physiology, glutamatergic neurons
Introduction
Synapsins serve as central regulators of synaptic vesicle (SV) dynamics in presynaptic terminals (Hilfiker et al., 1999). These proteins are thought to regulate synaptic transmission by controlling the storage and mobilization of SVs in the reserve pool (RP) (Hilfiker et al., 1999), but also may participate at other stages of the SV trafficking cycle, such as regulating the kinetics of SV fusion with the plasma membrane (Hilfiker et al., 1998, 2005; Humeau et al., 2001; Samigullin et al., 2004) and interacting with proteins involved in endocytosis (Evergren et al., 2007). Additionally, synapsins have been reported to play a role in various aspects of neuronal development, including synaptogenesis, synapse maturation and synapse stabilization (Ferreira and Rapoport, 2002; Kao et al., 2002).
Three synapsin genes have been identified in mammals (Südhof et al., 1989; Kao et al., 1999). Deleting each of the three synapsin genes produces different phenotypes, indicating that the various gene products must differ in their function (Li et al., 1995; Rosahl et al., 1995; Feng et al., 2002; Gitler et al., 2004b). In addition, each gene is alternatively spliced to produce related isoforms. The various isoforms possess different combinations of the phosphorylation sites that regulate their binding to SVs and cytoskeletal elements (Greengard et al., 1993; Hilfiker et al., 1999) and also differ in their ability to bind Ca2+ and ATP (Hosaka and Südhof, 1998). However, the physiological function of each synapsin isoform is not clear.
Here, we have defined the function of individual synapsin isoforms by examining their ability to rescue the defects in glutamatergic transmission of hippocampal neurons taken from synapsin triple-knock-out (TKO) mice. We found that none of the five synapsin isoforms considered affected basal synaptic transmission. Remarkably, only synapsin IIa could rescue the defects in synaptic depression kinetics and in SV density within the presynaptic terminal; others, including the most thoroughly studied synapsin isoform, synapsin Ia, had no ability to rescue the synaptic phenotype of TKO neurons. Fluorescence imaging of synaptic vesicle pools revealed that expression of synapsin IIa increased the number of SVs in TKO neurons, specifically increasing the number of vesicles in the RP. Thus, we conclude that synapsin IIa serves the canonical role of maintaining the RP and regulating SV mobilization during synaptic activity at glutamatergic synapses.
Materials and Methods
Cell culture and expression of synapsin isoforms.
Neurons from synapsin TKO mice and their matching “triple”-wild-type (TWT) controls (Gitler et al., 2004b) were cultured as described previously (Gitler et al., 2004a; Nishiki and Augustine, 2004). Green fluorescent protein (GFP)-tagged synapsin isoforms were expressed in cultured neurons via either lipofection or viral infection. In the first approach, neurons were transfected after 2 d in culture, using lipofectamine 2000 (Invitrogen) to introduce mammalian expression plasmids. The plasmids were based on pEGFPC1 (Invitrogen); they encoded EGFP or ECFP fused to the N termini of synapsins Ia, Ib, IIa, IIb, and IIIa, or to the N terminus of synaptobrevin-2 (Gitler et al., 2004a). For viral expression, synapsins Ia, Ib, IIa, IIb, and IIIa fused to enhanced GFP (EGFP) were subcloned into a pFUGW shuttle vector, where the inserted synapsin genes were driven by the human polyubiquitinC promoter. Lentivirus was then prepared as described by Lois et al. (2002). Neurons were infected after 3–4 d in culture, with a 1:3 multiplicity of infection, and studied 7–10 d after infection. Control experiments where EGFP alone was expressed in the neurons indicated that neither the transfection nor viral infection procedures had any effects on synaptic properties (data not shown).
Electrophysiology.
Whole-cell patch-clamp recordings were obtained from single autaptic neurons as described in detail by Gitler et al. (2004b). Neurons were held at a potential of −70 mV and experiments were performed at room temperature (∼25°C). To determine the effect of the expressed synapsin isoforms on synaptic depression, we compared transfected/infected neurons to untransfected/uninfected neurons in the same cultures. Because the introduction of the various isoforms was performed using two different methods (infection and transfection), we were concerned about possible effects of the different expression methods on the results. A two-way ANOVA test followed by Tukey's post hoc analysis indicated that there was no significant effect of the expression method on the time constant of synaptic depression (p = 0.22), allowing us to pool data obtained by the two methods (see Fig. 2). This ANOVA also allowed us to determine the effect of expressing each synapsin isoform on depression kinetics, as described in Results section. The effect of the synapsins on EPSC amplitude, miniature EPSC (mEPSC) amplitude, and mEPSC frequency was assayed in virally infected neurons, because the higher efficiency of viral infection facilitated the generation of larger data sets. These data were analyzed by one-way ANOVAs at a confidence level of p = 0.05, followed by Tukey's post hoc analysis. In all graphs, error bars represent ± 1 SEM, and asterisks represent statistical significance (p < 0.05).
Figure 2.
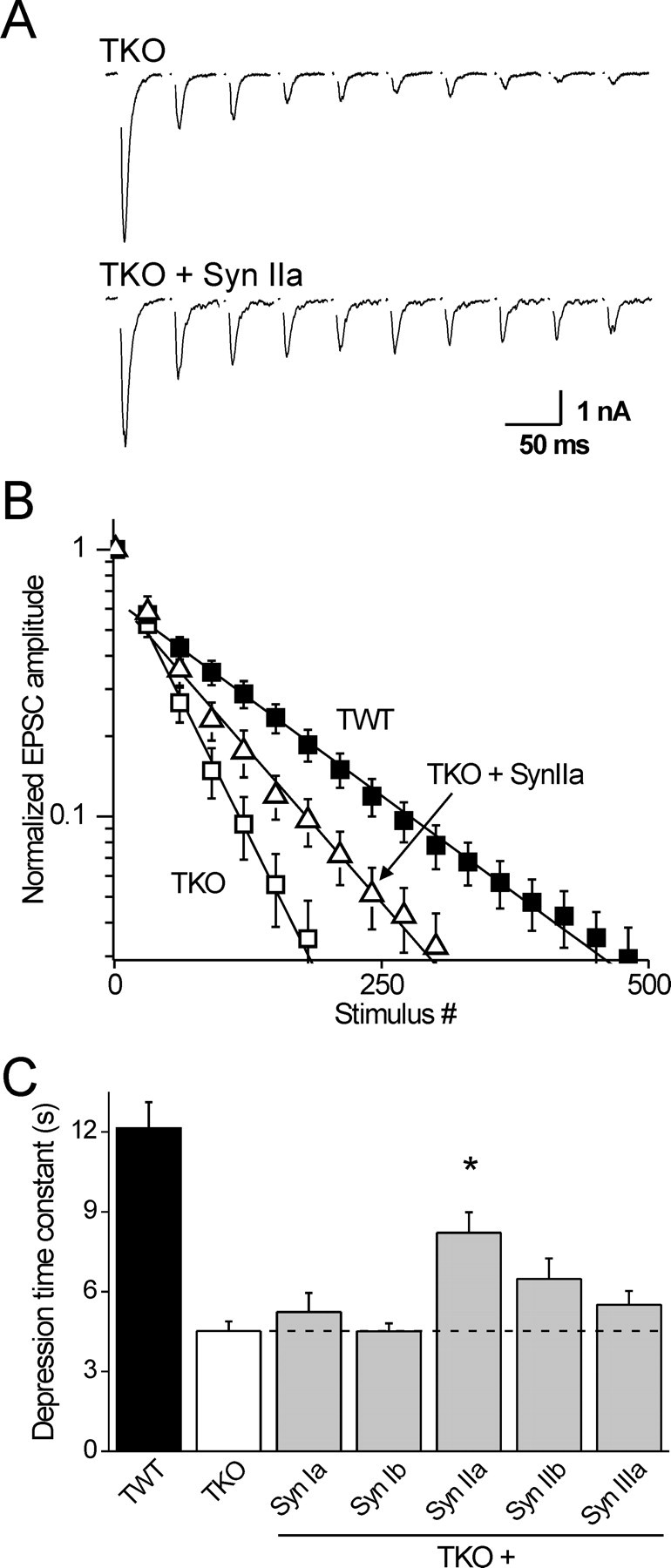
Only synapsin IIa rescues synaptic depression in TKO neurons. A, EPSCs recorded in an untransfected TKO neuron and in a TKO neuron transfected with synapsin IIa. Depression was elicited by 10 Hz trains of stimuli and the traces show every 15th EPSC evoked by the train. B, Mean EPSC amplitudes, normalized to the first response, with every 30th response shown. Lines represent single-exponential functions fitted beginning at the 30th response; note the semilogarithmic scale. C, Mean time constants of synaptic depression of TKO (n = 28) and TWT (n = 28) neurons, and of TKO neurons expressing synapsins Ia (n = 11), Ib (n = 12), IIa (n = 16), IIb (n = 17), and IIIa (n = 18). Only synapsin IIa significantly (*p < 0.05) slows synaptic depression compared with the TKO background.
Microscopy and imaging.
Fluorescence imaging was performed as described previously (Gitler et al., 2004a). To quantify the total SV population, we measured targeting of GFP-synaptobrevin-2 (Gitler et al., 2004b). In brief, neurons were triple transfected with GFP-synaptobrevin-2, Discosoma red 2 (DsRed2) and an ECFP-tagged synapsin. GFP-synaptobrevin clearly targets to presynaptic terminals in both wild-type neurons (Li and Murthy, 2001; Fernández-Alfonso et al., 2006) and in synapsin TKO neurons (Gitler et al., 2004b), permitting reliable identification of synaptic varicosities. Fluorescence intensity in the green (GFP-synaptobrevin-2) and red (DsRed2) channels was measured in identified varicosities and in adjacent axonal segments, and was corrected for background fluorescence outside the transfected axon. The synaptobrevin “targeting factor” was then calculated as follows:
 |
where Synapse and Axon indicate the intensity of GFP or DsRed2 fluorescence in the synaptic varicosities and adjacent axonal segments, respectively. In these experiments, synapsins were labeled with ECFP to identify synapsin-expressing cells. The fluorescence of GFP-synaptobrevin-2 was separated from that of ECFP-synapsin by using a filter cube intended for imaging yellow fluorescent protein (Chroma Technology), in which bleed-through of ECFP fluorescence was negligible (Gitler et al., 2004a).
FM dye staining
To visualize recycling synaptic vesicles, cultured neurons were treated for 2 min with a saline containing the fluorescent dye N-(3-triethylammoniumpropyl)-4-(6-(4-diethylamino)phenyl)-hexatrienyl)pyridinium dibromide (FM4-64; 4 μm; Invitrogen) and loaded by a train of 1200 stimuli delivered via field electrodes (1 ms duration, 40 V/cm, 10 Hz). CNQX (10 μm; Tocris Bioscience) was added to this saline to prevent activation of recurrent excitatory synaptic networks. After an additional 2 min in staining solution, neurons were washed with dye-free saline (containing CNQX) for 20 min and then imaged and stimulated as described in Results. Boutons were automatically detected and their fluorescence signals analyzed using algorithms written in IgorPro (WaveMetrics); the number of synapses measured ranged from 350 to 1200 per image. Because the intensity of FM4-64 fluorescence intensity across the population of boutons was not distributed normally (Shapiro–Wilk normality test), the median value of dye staining of boutons was calculated for each image. Medians measured under different conditions were then compared using the Mann–Whitney U test at a confidence level of p = 0.05. To assure an equal contribution of each image to the analysis of the fluorescence intensity distribution, 350 synapses were chosen at random from each image; the distribution histograms and the cumulative probability plots shown in Figure 6 were calculated from this representative subset. FM4-64 and GFP fluorescence were visualized separately by TRITC and GFP filter sets (Chroma Technology).
Figure 6.
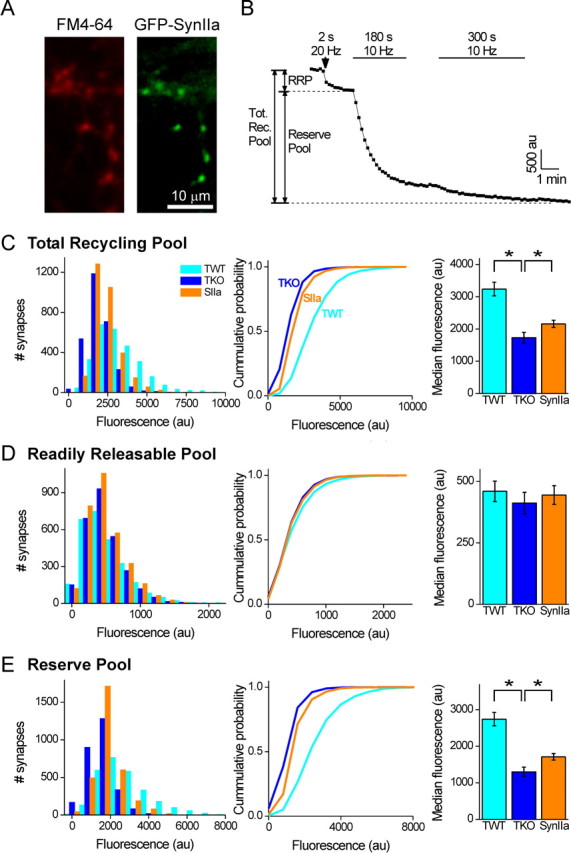
Synapsin IIa affects the size of the RP but not the RRP. FM4-64 unloading was used to assess the effect of reintroduction of synapsin IIa on the size of the RRP and the RP of TKO neurons. A, Representative image of presynaptic terminals of a TKO neuron loaded with FM4-64 by 1200 stimuli delivered at 10 Hz (red, left). These terminals belong to a neuron expressing GFP-synapsin IIa (green, right). B, Example of measurement of FM4-64 unloading. Unloading of FM4-64 by 40 stimuli delivered at 20 Hz served to unload FM4-64 from the RRP, whereas two successive trains of 1800 and 3000 stimuli delivered at 10 Hz subsequently unloaded dye from SVs remaining in the RP. The sum of both signals served as our measure of the total recycling pool. C, Determination of the size of the total recycling pool. Distribution of the magnitude of FM4-64 fluorescence loss during complete unloading (left), cumulative probability derived from the same data (center), and average of the median value determined for each image (right). The median size of the total recycling pool was significantly reduced in TKO terminals compared with TWT terminals. Likewise, reintroduction of synapsin IIa into TKO terminals partially rescued its size (*p < 0.05). D, E, Determination of the size of the RRP and the RP. Deletion of all synapsins had no effect on the size of the RRP (D), but decreased the size of the RP (E). Reintroduction of synapsin IIa into TKO neurons did not affect the RRP, but partially rescued the RP (*p < 0.05). Measurements were performed from 350–1200 synapses in each image of TWT (n = 8), TKO (n = 8), or TKO neurons expressing synapsin IIa (n = 9).
Results
Previous genetic studies of the role of synapsins in synaptic transmission involved deletion of one or more synapsin genes (Li et al., 1995; Rosahl et al., 1995; Gitler et al., 2004b; Hvalby et al., 2006). Consequently, these studies could not distinguish between the roles played by alternative splicing variants from single genes. To avoid such complications, we examined synaptic transmission in hippocampal neurons from synapsin TKO mice and reintroduced each synapsin isoform individually.
The goal of our work was to define the ability of each synapsin isoform to rescue the main TKO phenotype at excitatory synapses, which is a reduction in the size of the RP (Gitler et al., 2004b). We used microisland cultures to ensure that synapsins were reintroduced into all presynaptic terminals innervating a given postsynaptic neuron (Betz et al., 2001; Nishiki and Augustine, 2004; Wojcik et al., 2004). To determine the location of expressed synapsins, as well to identify transfected neurons, each synapsin isoform was fused to the GFP (Gitler et al., 2004a). The exogenous synapsins localized well to presynaptic terminals, as indicated by the punctate distribution of GFP (Fig. 1A). The sole exception was synapsin Ib, which distributed throughout axons as previously reported (Gitler et al., 2004a).
Figure 1.

Synapsins do not determine the properties of basal synaptic transmission. A, A neuron expressing GFP-synapsin IIa. The area marked by a white square in the left is magnified in the right to demonstrate the synaptic localization of synapsin IIa. B, Examples of autaptic EPSCs recorded from TWT and TKO neurons as well as from TKO neurons expressing indicated synapsin isoforms. C, Mean amplitudes of evoked EPSCs in TWT (black bar, n = 46) and TKO neurons (white bar, n = 47) as well as in TKO neurons expressing indicated synapsin isoforms (gray bars). Sample sizes for each isoform are synapsin Ia (n = 34), Ib (n = 25), IIa (n = 22), IIb (n = 23), and IIIa (n = 30). D, Amplitudes of spontaneous synaptic events (mEPSCs) recorded in TWT (n = 23) and TKO neurons (n = 13) as well as in TKO neurons expressing individual synapsin isoforms: synapsin Ia (n = 27), Ib (n = 22), IIa (n = 19), IIb (n = 17), and IIIa (n = 30). EPSC and mEPSC amplitudes recorded in infected neurons were not significantly different from those recorded in uninfected TKO neurons. E, Effects of synapsin isoforms on the frequency of spontaneous synaptic events, presented as in C. Spontaneous glutamate release was twofold higher in synapsin IIb infected neurons than in TKO neurons (*p < 0.05; asterisk), whereas other synapsins had no significant effects on mEPSC frequency.
Synapsins do not affect basal excitatory synaptic transmission
Our primary assay for the status of the RP was the rate of synaptic depression; because the rate of depression depends on the strength of basal synaptic transmission (Betz, 1970; Hilfiker et al., 1998), we first asked whether introduction of synapsins into TKO neurons affected basal transmission. This was examined by measuring the amplitude of EPSCs evoked by low-frequency stimulation (0.066 Hz), conditions that should not elicit depression or other forms of synaptic plasticity (Salin et al., 1996).
Deletion of all three synapsin genes has minimal effects on basal glutamatergic transmission (Gitler et al., 2004b), so we expected that reintroduction of single synapsin isoforms would not affect basal synaptic transmission. Indeed, we found that EPSC amplitude in TKO neurons expressing any of the exogenous synapsin isoforms was comparable to that measured in control TKO neurons from the same cultures (one-way ANOVA at p = 0.05 confidence level followed by Tukey's post hoc analysis) (Fig. 1B,C). Furthermore, the amplitude of spontaneous miniature synaptic events (mEPSCs) remained unchanged compared with TKO neurons (Fig. 1D). The sole difference was a twofold increase in the frequency of spontaneous mEPSCs recorded in neurons transfected with synapsin IIb (p < 0.05) (Fig. 1E). These results indicate that, for the case of glutamatergic synapses, none of the synapsin isoforms examined affect synaptic transmission evoked by single action potentials, permitting interpretation of the effects of these isoforms on synaptic depression.
Synapsin IIa rescues the kinetics of synaptic depression
To probe the role of synapsin isoforms in maintaining the RP of glutamatergic vesicles, we measured the effect of reintroducing each isoform on the rate of synaptic depression. During sustained repetitive activity, postsynaptic responses diminish progressively (Zucker and Regehr, 2002). The initial phase of this progressive depression is attributed to depletion of the readily releasable pool (RRP) of SVs (Dobrunz and Stevens, 1997). Additional activity, beyond the first 25–40 stimuli (Murthy and Stevens, 1998, Stevens and Williams, 2007), mobilizes SVs mainly from the RP. Reducing the size of the RP causes the rate of depression measured during this phase to be accelerated (Gitler et al., 2004b). Therefore, we measured EPSC amplitudes during trains of 500 stimuli applied at 10 Hz (Fig. 2A,B) and measured the depression rate starting at the 30th stimulus. As reported previously (Gitler et al., 2004b), TKO neurons had a time constant of depression that was significantly faster than that of TWT neurons (Fig. 2B). Remarkably, we found that synapsin IIa was the only synapsin isoform that significantly slowed the kinetics of synaptic depression when introduced into TKO neurons (Fig. 2B,C) (Tukey's post hoc analysis of two-way ANOVA at a p = 0.05 confidence level). Synapsin IIb had a similar tendency, which did not reach statistical significance, whereas neither synapsin Ia, synapsin Ib nor synapsin IIIa had any effect on the kinetics of depression (Fig. 2C). The lack of effect of synapsins Ia and Ib is consistent with reports that deletion of the synapsin I gene does not affect the kinetics of synaptic depression in hippocampal neurons (Li et al., 1995; Rosahl et al., 1995), but the synapsin IIIa result contrasts with the effect of deleting the entire synapsin III gene, which slows the initial phase of depression (Feng et al., 2002).
We next determined whether exogenous synapsin IIa could also affect synaptic depression kinetics in TWT neurons, which express endogenous synapsin IIa and have slower depression than observed in TKO neurons (Gitler et al., 2004b). We found (Fig. 3A,B) that overexpression of synapsin IIa in TWT neurons (n = 8) slowed the rate of synaptic depression compared with the rate measured in untransfected TWT neurons (n = 20). This difference was significant, as measured by Tukey's post hoc analysis of one-way ANOVA at a p = 0.05 confidence level. This effect of synapsin IIa was clearly visible as an absence of synaptic depression for the first 100 stimuli or more, in contrast to the case in untransfected TWT neurons where depression was usually evident after 30 stimuli or less (Fig. 3B). Unlike synapsin IIa, overexpression of synapsin Ia had no effect on the kinetics of synaptic depression in TWT neurons (n = 5) (Fig. 3C). We therefore conclude that synapsin IIa serves a unique role in the maintenance of the RP of glutamatergic SVs, as measured by the rate of synaptic depression.
Figure 3.
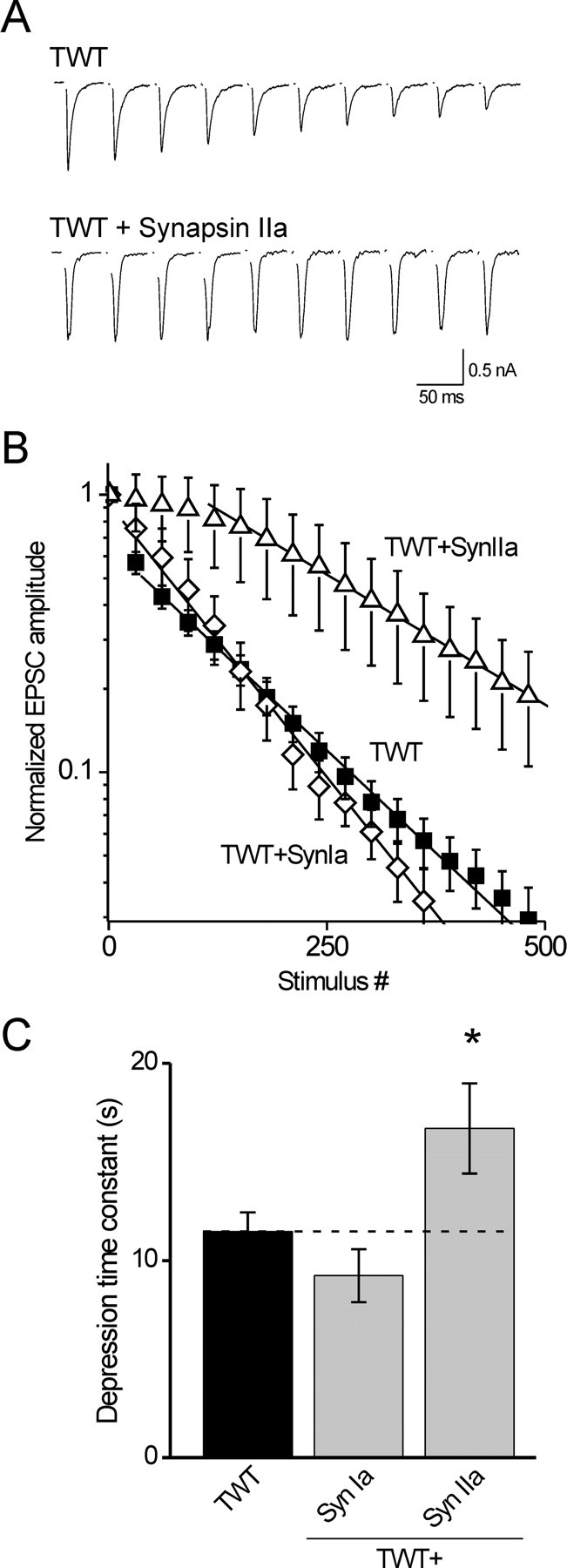
Synapsin IIa slows synaptic depression in TWT neurons. A, EPSCs from an untransfected TWT neuron and a TWT neuron transfected with synapsin IIa. Stimulation and display conditions as in Figure 2. B, Mean EPSC amplitudes, normalized to the first response, with every 30th response shown. Lines represent fits of single-exponential functions beginning at the 30th response, or the 100th response for the case of data from neurons overexpressing synapsin IIa. C, Time constants of synaptic depression in untransfected TWT neurons (n = 20) and in TWT neurons overexpressing synapsin IIa (n = 8) or synapsin Ia (n = 5). Synapsin IIa slowed synaptic depression in TWT neurons, whereas synapsin Ia had no effect. *p < 0.05.
Synapsin IIa rescues synaptic vesicle density
Perturbation of synapsin function is thought to accelerate synaptic depression by reducing the number of SVs within presynaptic terminals, specifically SVs within the RP (Pieribone et al., 1995; Rosahl et al., 1995; Hilfiker et al., 1998; Gitler et al., 2004b). Consistent with this notion, glutamatergic terminals of synapsin TKO neurons have approximately half the number of SVs found in TWT neurons (Gitler et al., 2004b; Siksou et al., 2007). To determine whether synapsin IIa, or any of the other isoforms, could restore SVs to TKO neurons, we examined SV density in TKO neurons expressing each of the synapsin isoforms.
For this purpose, we quantified the enrichment of GFP-tagged synaptobrevin-2, an integral protein of the SV membrane. Previous work established that the relative amount of GFP-synaptobrevin-2 in presynaptic terminals is much lower in synapsin TKO neurons than in TWT neurons and that this decrease is comparable to that observed with other measures of SV number, such as electron microscopic imaging of individual SVs or biochemical measurement of SV protein levels (Gitler et al., 2004b). To correct the measurements of GFP-synaptobrevin-2 fluorescence for variations in cell thickness, the volume marker DsRed2 was coexpressed (Fig. 4A) and GFP fluorescence levels were divided by DsRed2 fluorescence levels. Comparison of such ratios measured in presynaptic terminals and in adjacent axons yields a targeting factor that represents the enrichment of GFP-synaptobrevin in presynaptic terminals relative to axons (see Materials and Methods) (Gitler et al., 2004b).
Figure 4.
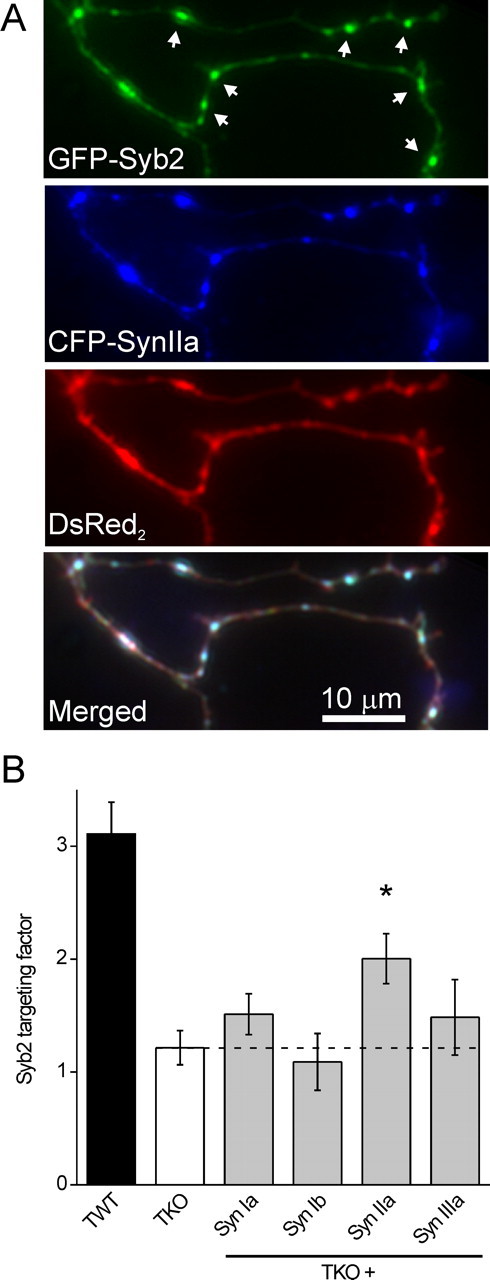
Synapsin IIa increases synaptic vesicle density. A, SV density was assayed in living neurons by measuring GFP synaptobrevin-2 targeting to presynaptic terminals. Shown is a representative image of a TKO neuron cotransfected with GFP-synaptobrevin-2 (green), ECFP-synapsin IIa (blue), and DsRed2 (red). Representative synapses, indicated by accumulation of synaptobrevin-2 and synapsin IIa, are indicated by white arrows in the top panel. B, SV density was assayed in untreated TWT (n = 15) and TKO (n = 8) neurons, as well as TKO neurons cotransfected with synapsin Ia (n = 8), Ib (n = 8), IIa (n = 12), and IIIa (n = 9). Only synapsin IIa significantly (*p < 0.05) increased SV density.
Such measurements confirmed that SV density is substantially lower in TKO neurons than in TWT neurons (Fig. 4B, left). In addition, we found that synapsin IIa significantly increased SV density in presynaptic terminals of TKO neurons, whereas synapsins Ia, Ib, and IIIa did not (Fig. 4B, right) (Tukey's post hoc analysis of one-way ANOVA at a confidence level of p = 0.05). Thus, synapsin IIa has a unique ability both to rescue synaptic depression and to rescue the number of SVs in presynaptic terminals.
To further define the relationship between these two actions of synapsin IIa, we examined the correlation between them. Considering all of the isoforms, rescue of SV targeting was directly proportional to the actions of these isoforms on the kinetics of synaptic depression (Fig. 5) and the correlation coefficient between the two was very high (r2 = 0.97; p < 0.01). This indicates that the rate of synaptic depression is determined by the density of SVs in the nerve terminal. We further conclude that synapsin IIa is the only isoform capable of rescuing synaptic depression because it is the only isoform that can restore the number of glutamatergic SVs.
Figure 5.

Correlation between synaptic depression kinetics and vesicle population size. Data points are shown for TWT and TKO neurons, as well as TKO neurons transfected with synapsins Ia, Ib, IIa, and IIIa, as indicated. The dashed line represents a linear regression through all points (r2 = 0.97; p < 0.01).
Synapsin IIa increases the size of the reserve pool
The results shown in Figure 4 indicate that the total number of SVs is increased by synapsin IIa and the correlation between the number of SVs and rate of depression shown in Figure 5 suggests that synapsin IIa works by rescuing the RP. To more directly address this possibility, we determined which pools of SVs are affected by the synapsins by using the fluorescent dye FM4-64 (Chi et al., 2001) to selectively label recycling SVs. This approach allowed us to measure the size of the total recycling pool of SVs as well as the size of the RP and RRP components of this pool (as defined by Mozhayeva et al., 2002) (but see Rizzoli and Betz, 2005), while manipulating the synapsin content of the presynaptic terminal.
We stained the total recycling pool of SVs by extensive field stimulation (1200 stimuli, 10 Hz) in the presence of FM4-64. In neurons that were transfected with GFP-tagged synapsin IIa, FM4-64 labeling colocalized with synapsin IIa (Fig. 6A); this confirms that the dye was taken up into presynaptic terminals. We then determined the size of the RRP and the RP by measuring the amount of dye released during stimulation protocols known to differentially trigger exocytosis of vesicles within these two pools. To specifically release the content of SVs in the RRP, neurons were stimulated for 2 s at 20 Hz as in the study by Murthy and Stevens (1998). The dye content of the remaining SVs, consisting of those populating the RP before this stimulus, was then released by exhaustive 10 Hz stimulation (1800 stimuli followed by an additional 3000 stimuli). Quantification of the amount of dye released during these two steps thus provided measurements of the relative size of the RRP and the RP, respectively (Fig. 6B). The sum of these two pools was then used to define the size of the total recycling pool of SVs.
The amount of FM4-64 fluorescence associated with the total recycling pool was smaller in TKO neurons than in TWT neurons (p < 0.05, Mann–Whitney U test) (Fig. 6C), indicating a reduced number of recycling SVs in TKO neurons. This effect of synapsins on the total recycling pool was partially rescued by expression of synapsin IIa (p < 0.05) (Fig. 6C). These effects of synapsins on the size of the total recycling SV pool were comparable to their effects on the total number of SVs, as measured by presynaptic targeting of GFP-synaptobrevin-2 (Fig. 4B).
To determine whether these effects on recycling SVs were caused by changes in the RRP or the RP, we separately considered the contribution of each pool. Brief stimuli used to selectively release FM4-64 from the RRP produced small changes in TKO and TWT neurons, as well as in TKO neurons expressing synapsin IIa (Fig. 6D). The changes were very similar under all three conditions, indicating that the effect of synapsins on the total recycling pool is not attributable to changes in the RRP. Instead, the changes in total recycling pool should be produced by changes in the size of the RP. Our measurements revealed that loss of synapsins indeed affected the RP preferentially, because the amount of FM4-64 fluorescence associated with the RP was smaller for TKO neurons than for TWT neurons (p < 0.05) (Fig. 6E). Likewise, expression of synapsin IIa in TKO neurons increased the size of the RP (p < 0.05) (Fig. 6E), in agreement with the observed effect of synapsin IIa on the kinetics of synaptic depression (Fig. 2). Assuming an RRP size of 10 SVs (Rizzoli and Betz, 2005), our data indicate that on average an additional 60 and 32 SVs cycled during strong stimulation in TWT and TKO neurons, respectively. Similar calculations indicate that expression of synapsin IIa in TKO neurons increased the RP by seven vesicles. We conclude that synapsin IIa affects the kinetics of synaptic depression by increasing the number of SVs, specifically the number of vesicles in the RP.
Discussion
Synapsins have long been implicated in control of SV number in presynaptic terminals and, consequently, in regulation of the kinetics of SV mobilization and exocytosis during synaptic depression. However, until now it was not clear which of the synapsin isoforms are responsible for this function. Our results indicate that synapsin IIa is the main isoform involved at glutamatergic synapses: this isoform was the only one capable of rescuing the defects in either synaptic depression or SV density in cultured hippocampal neurons from synapsin TKO mice. Our data suggest that the related isoform, synapsin IIb, may have a similar but weaker ability to rescue, although this was not corroborated by statistical analysis. Remarkably, synapsins Ia, Ib, and IIIa had no effect.
Our experiments provide new results consistent with the hypothesis that synapsins participate in neurotransmitter release by maintaining SVs in a RP. First, we found that the kinetics of a late phase of synaptic depression usually attributed to mobilization of SVs from the RP depends on the synapsin isoform present in the presynaptic terminal. Second, we found that the number of SVs, as measured by GFP-synaptobrevin imaging, also depends on the synapsin isoform present. Third, with FM4-64 labeling we observed that synapsin IIa augmented the RP in TKO neurons, without affecting the size of the RRP. Finally, we found that the time constant of synaptic depression was well correlated with the SV population size, independent of the synapsin isoform present. The close correspondence of these measures strongly suggests that GFP-synaptobrevin provides a reliable estimate of the size of the RP, consistent with previous work (Gitler et al., 2004b) (but see Fernández-Alfonso et al., 2006). This correspondence argues against the alternative explanation that synapsins slow synaptic depression by altering the release probability of vesicles in the RRP. Another argument against this possibility is that synapsin IIa had no effect on the size of EPSCs evoked by single action potentials: EPSC size would have to change if release probability was affected, given that the RRP size was unchanged.
The mechanism underlying the rescue of the RP size by synapsin IIa is unclear. A likely explanation is that synapsins maintain SVs in the RP by cross-linking vesicles to the cytoskeleton (Greengard et al., 1993). However, the fact that the total number of vesicles is reduced in synapsin TKO neurons (Fig. 4) (Gitler et al., 2004b; Siksou et al., 2007) suggests that synapsins may also directly or indirectly regulate SV biogenesis and/or degradation.
Our results identify synapsin IIa as the main modulator of glutamatergic SV pools and extend previous evidence that the three synapsin genes are not equivalent in function (Li et al., 1995; Rosahl et al., 1995; Feng et al., 2002). Two previous genetic studies implicated the synapsin II gene (SynII) in regulating SV population sizes. Rosahl et al. (1995) found that synaptic depression at glutamatergic hippocampal Schaeffer collateral synapses was accelerated by deletion of SynII, but not SynI. However, because their study probed neither the absolute amount of glutamate release nor the sizes of vesicle pools, the underlying mechanism remained undetermined. More recently, it was shown that deleting SynII accelerates synaptic depression and reduces the number of RP vesicles at the cholinergic neuromuscular synapse, without affecting basal release at physiological extracellular Ca2+ concentrations (Samigullin et al., 2004) (but see Gaffield and Betz, 2007). Our results reveal that these effects on synaptic depression are caused by loss of synapsin IIa and the resultant reduction in the size of the RP. The identification of synapsin IIa as the main modulator of glutamatergic SV pools is especially intriguing because this isoform lacks two calcium/calmodulin-dependent protein kinase II (CaMKII) phosphorylation sites proposed to control SV mobilization (Llinás et al., 1991; Benfenati et al., 1992; Chi et al., 2003). Thus, regulation of mobilization of the synapsin IIa-dependent RP may occur via other means, such as the PKA/CaMKI phosphorylation site common to all synapsins (Hosaka et al., 1999) or, alternatively, by CaMKII phosphorylating a substrate other than synapsin IIa.
Although synapsin IIa was the only isoform that significantly rescued depression and SV content, this rescue was incomplete. Probably this partial rescue was not caused by expression of insufficient amounts of synapsin IIa, because very similar rescue levels were observed with two different protein expression techniques (viral infection and lipofection), and two promoters (cytomegalovirus and polyubiquitinC) that should yield different amounts of synapsin IIa protein. Even at the levels of synapsin IIa found in TWT neurons, overexpressing synapsin IIa in TWT neurons led to further slowing of synaptic depression (Fig. 3). This indicates that the mechanism responsible for enhancing the size of the RP is not saturated even at physiological levels of synapsin IIa. It is possible that the rescue in TKO neurons was incomplete because of the absence of other synapsin isoforms; synapsin IIa heterodimerizes with other synapsin isoforms (Hosaka and Südhof, 1999; Gitler et al., 2004a), and such interactions could influence the ability of synapsin IIa to maintain the RP. Additional work will be needed to determine whether other synapsin isoforms can enhance the actions of synapsin IIa. In any case, our observations indicate that changing the level of expression of synapsin IIa in presynaptic terminals can serve as a means of regulating RP size.
In this study, we observed that introduction of the synapsins into TKO neurons did not affect basal excitatory transmission, except for an increase in mEPSC frequency by synapsin IIb (Fig. 1E). Because this effect was not accompanied by an increase in EPSC amplitude, it is unlikely that it reflects an increase in the number of synapses, a parameter that is not altered by loss of synapsins (Gitler et al., 2004b) (but see Ferreira and Rapoport, 2002). Although a previous study reported a difference in mEPSC amplitude between TWT and TKO neurons (Gitler et al., 2004b), this difference was not observed in the current measurements (Fig. 1D). We suspect that this may result from small differences in experimental conditions between the two sets of experiments. These effects of synapsins on the frequency and amplitude of spontaneous transmission remain open questions for further investigation.
Previous results indicated that domain E, a domain found only in “a-type” synapsin splice variants (synapsins Ia, IIa, and IIIa), is important for maintaining the RP of glutamatergic SVs (Hilfiker et al., 1998). Thus, we were not surprised to find that an a-type synapsin variant is involved in maintaining the RP. There have been several suggestions that synapsin Ia, the synapsin isoform most often studied biochemically (Hilfiker et al., 1999), may play such a role in SV trafficking in glutamatergic terminals. For example, overexpression of versions of synapsin Ia with mutations in various phosphorylation sites affects SV mobilization in wild-type neurons (Chi et al., 2001, 2003), and deletion of SynI reduces total release of glutamate and slows recovery of synaptic strength after intense activity (Li et al., 1995). In addition, the number of SVs and the quantity of SV proteins is reduced in SynI knock-out mice, although to a lesser degree than in SynII or SynI/SynII knock-out mice (Li et al., 1995; Rosahl et al., 1995; Takei et al., 1995). Thus, we were surprised that synapsin Ia had no ability to rescue the phenotype of glutamatergic TKO synapses and that synapsin IIa is the a-type synapsin isoform responsible for maintaining the RP. Our results do not exclude the possibility that synapsin Ia may secondarily regulate presynaptic function by interacting with synapsin IIa or other synapsin isoforms, as postulated above.
Loss of synapsins differentially affects transmission in different types of neurons. In synapsin TKO mice, the kinetics of depression and basal amounts of evoked transmitter release at GABAergic neurons and glutamatergic neurons are differentially affected (Gitler et al., 2004b). Furthermore, transmission at two different glutamatergic synapses is differentially affected by simultaneous deletion of SynI and SynII (Kielland et al., 2006). Given this, as well as the observation that synapses contain different complements of synapsin isoforms (Südhof et al., 1989), the other synapsin isoforms might have important functions at presynaptic terminals other than the glutamatergic hippocampal synapses that we studied. For example, synapsin I isoforms may be more important in inhibitory interneurons: in hippocampal neurons, deletion of SynI has much larger effects on inhibitory transmission than on excitatory transmission (Rosahl et al., 1995; Terada et al., 1999; Baldelli et al., 2007). Cell-specific expression of different isoforms also has been described for the Munc13 family of presynaptic proteins (Varoqueaux et al., 2002), so that isoform heterogeneity may be a common mechanism for achieving diversity in presynaptic properties.
In conclusion, we found that synapsin isoforms differ in their functions and that synapsin IIa serves as the prototypical synapsin, both slowing synaptic depression during high-frequency stimulation and increasing the size of the RP component of the SV population. This is the first synapsin isoform whose physiological function has been unambiguously identified. The roles played by the other synapsin isoforms, as well as the relationship between the biochemical properties of these isoforms and their functions, remain to be determined.
Footnotes
This work was supported by the National Institutes of Health (G.J.A.), Israel Science Foundation Grant 452/06, a grant from the Marc Rich Foundation, Lausanne, Switzerland, a European Molecular Biology Organization long-term postdoctoral fellowship, and the Pfizer postdoctoral fellowship of the Life Sciences Research Foundation (D.G.). We thank Ken Berglund for developing the automated synapse detection procedure.
References
- Baldelli P, Fassio A, Valtorta F, Benfenati F. Lack of synapsin I reduces the readily releasable pool of synaptic vesicles at central inhibitory synapses. J Neurosci. 2007;27:13520–13531. doi: 10.1523/JNEUROSCI.3151-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N. Functional interaction of the active zone proteins Munc13–1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- Betz WJ. Depression of transmitter release at the neuromuscular junction of the frog. J Physiol. 1970;206:629–644. doi: 10.1113/jphysiol.1970.sp009034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Evergren E, Benfenati F, Shupliakov O. The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res. 2007;85:2648–2656. doi: 10.1002/jnr.21176. [DOI] [PubMed] [Google Scholar]
- Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, Takahashi RH, Kao HT, McEwen BS, Ryan TA, Augustine GJ, Greengard P. Regulation of neurotransmitter release by synapsin III. J Neurosci. 2002;22:4372–4380. doi: 10.1523/JNEUROSCI.22-11-04372.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Alfonso T, Kwan R, Ryan TA. Synaptic vesicles interchange their membrane proteins with a large surface reservoir during recycling. Neuron. 2006;51:179–186. doi: 10.1016/j.neuron.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Rapoport M. The synapsins: beyond the regulation of neurotransmitter release. Cell Mol Life Sci. 2002;59:589–595. doi: 10.1007/s00018-002-8451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci. 2007;27:13691–13700. doi: 10.1523/JNEUROSCI.3910-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004a;24:3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004b;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Benfenati F, Doussau F, Nairn AC, Czernik AJ, Augustine GJ, Greengard P. Structural domains involved in the regulation of transmitter release by synapsins. J Neurosci. 2005;25:2658–2669. doi: 10.1523/JNEUROSCI.4278-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC. Synapsins I and II are ATP-binding proteins with differential Ca2+ regulation. J Biol Chem. 1998;273:1425–1429. doi: 10.1074/jbc.273.3.1425. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC. Homo- and heterodimerization of synapsins. J Biol Chem. 1999;274:16747–16753. doi: 10.1074/jbc.274.24.16747. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Hammer RE, Südhof TC. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron. 1999;24:377–387. doi: 10.1016/s0896-6273(00)80851-x. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J Neurosci. 2001;21:4195–4206. doi: 10.1523/JNEUROSCI.21-12-04195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvalby O, Jensen V, Kao HT, Walaas SI. Synapsin-regulated synaptic transmission from readily releasable synaptic vesicles in excitatory hippocampal synapses in mice. J Physiol. 2006;571:75–82. doi: 10.1113/jphysiol.2005.100685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HT, Porton B, Hilfiker S, Stefani G, Pieribone VA, DeSalle R, Greengard P. Molecular evolution of the synapsin gene family. J Exp Zool. 1999;285:360–377. [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P. A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci. 2002;5:431–437. doi: 10.1038/nn840. [DOI] [PubMed] [Google Scholar]
- Kielland A, Erisir A, Walaas SI, Heggelund P. Synapsin utilization differs among functional classes of synapses on thalamocortical cells. J Neurosci. 2006;26:5786–5793. doi: 10.1523/JNEUROSCI.4631-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P, Andersen P. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Murthy VN. Visualizing postendocytic traffic of synaptic vesicles at hippocampal synapses. Neuron. 2001;31:593–605. doi: 10.1016/s0896-6273(01)00398-1. [DOI] [PubMed] [Google Scholar]
- Llinás R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca2+-calmodulin-dependent protein kinase II of transmitter release in squid giant synapse. J Physiol. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci. 2004;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigullin D, Bill CA, Coleman WL, Bykhovskaia M. Regulation of transmitter release by synapsin II in the mouse motor terminal. J Physiol. 2004;23:23. doi: 10.1113/jphysiol.2004.073494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siksou L, Rostaing P, Lechaire JP, Boudier T, Ohtsuka T, Fejtová A, Kao HT, Greengard P, Gundelfinger ED, Triller A, Marty S. Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci. 2007;27:6868–6877. doi: 10.1523/JNEUROSCI.1773-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Williams JH. Discharge of the readily releasable pool with action potentials at hippocampal synapses. J Neurophysiol. 2007;98:3221–3229. doi: 10.1152/jn.00857.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, Greengard P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- Takei Y, Harada A, Takeda S, Kobayashi K, Terada S, Noda T, Takahashi T, Hirokawa N. Synapsin I deficiency results in the structural change in the presynaptic terminals in the murine nervous system. J Cell Biol. 1995;131:1789–1800. doi: 10.1083/jcb.131.6.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada S, Tsujimoto T, Takei Y, Takahashi T, Hirokawa N. Impairment of inhibitory synaptic transmission in mice lacking synapsin I. J Cell Biol. 1999;145:1039–1048. doi: 10.1083/jcb.145.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]