

Abstract
Haploinsufficiency of the transcription factor TWIST1 is associated with Saethre-Chotzen Syndrome and is manifested by craniosynostosis, which is the premature closure of the calvaria sutures. Previously, we found that Twist1 forms functional homodimers and heterodimers that have opposing activities. Our data supported a model that within the calvaria sutures Twist1 homodimers (T/T) reside in the osteogenic fronts while Twist/E protein heterodimers (T/E) are in the mid-sutures. Twist1 haploinsufficiency alters the balance between these dimers, favoring an increase in homodimer formation throughout the sutures. The data we present here further supports this model and extends it to integrate the Twist1 dimers with the pathways that are known to regulate cranial suture patency. This data provides the first evidence of a functional link between Twist1 and the FGF pathway, and indicates that differential regulation of FGF signaling by T/T and T/E dimers plays a central role in governing cranial suture patency. Furthermore, we show that inhibition of FGF signaling prevents craniosynostosis in Twist1+/− mice, demonstrating that inhibition of a signaling pathway that is not part of the initiating mutation can prevent suture fusion in a relevant genetic model of craniosynostosis.
Keywords: Twist1, FGFR2, FGF, BMP, bHLH, craniosynostosis, calvaria, sutures, craniofacial, osteoblast differentiation
Introduction
The flat bones of the skull remain separated by openings termed sutures that function as the growth centers of the bones, allowing growth of the skull during fetal and postnatal development. At the edges of these bones are the osteogenic fronts that contain highly proliferative cells expressing osteogenic markers such as Runx2 and alkaline phosphatase (Cohen, 2000; Opperman, 2000). The mid-suture region is composed of mesenchymal precursor cells. In the human, the sutures gradually close from infancy to young adult. The premature closure of sutures, termed craniosynostosis, is a fairly common disorder, occurring in 1 in 2500 births. Non-syndromic craniosynostosis is most common, however approximately 20% of all cases are associated with mutations in the gene TWIST1 or one of the fibroblast growth factor receptor (FGFR) genes (Cunningham et al., 2007; Lenton et al., 2005; Rice, 2005). There is a complex relationship between Twist1 and FGF signaling which is not completely understood. Increases in Twist1 expression and FGF signaling have both been associated with an inhibition of osteoblast differentiation in vitro, yet craniosynostosis occurs due to activating mutations of the FGFR genes and haploinsufficiency of TWIST1 (Cunningham et al., 2007; Lenton et al., 2005; Rice, 2005). Eighty percent of patients with Saethre-Chotzen Syndrome (SCS), which is one of the most common autosomal dominant disorders of craniosynostosis, have mutations in TWIST1, however mutations in FGFR2 and FGFR3 have also been reported in some patients that have phenotypes consistent with SCS, exemplifying this relationship and further indicating that haploinsufficiency of TWIST1 gives a similar phenotype as activation of FGFR signaling (Chun et al., 2002; el Ghouzzi et al., 1997; Howard et al., 1997; Paznekas et al., 1998). Receptor tyrosine kinase signaling by FGF, IGF1, and HGF induces the expression of Twist1 (Dupont et al., 2001; Fang et al., 2001; Isaac et al., 2000; Leshem et al., 2000; Rice et al., 2000), while Twist can affect the expression of FGF receptor genes. In Drosophila and C. elegans, Twist induces the expression of the FGFR homolog DFR1 and egl-15, respectively (Castanon and Baylies, 2002); however, the relationship between Twist1 and FGFR in vertebrates has been less clear. In the cranial sutures, FGFR2 expression is normally only detected in the osteogenic fronts but it extends into the mid-suture of Twist1+/− mice, suggesting that Twist1 may normally inhibit FGFR2 expression (Connerney et al., 2006; Rice et al., 2000). However, calvaria cells isolated from a SCS patient with a mutation in TWIST1 had decreased FGFR2 levels, which were increased when Twist1 was ectopically expressed (Guenou et al., 2005). Therefore, the relationship between Twist1 and FGFR2 may be context dependent. Consistent with this, we recently found that Twist1 forms different dimer complexes that have opposing effects on FGFR2 expression (Connerney et al., 2006).
Twist1 is a basic-Helix-Loop-Helix (bHLH) transcription factor that plays both positive and negative roles in the regulation of early morphogenesis and differentiation of mesenchymal tissues (O'Rourke and Tam, 2002). We found that, unlike most other bHLH proteins, Twist1 can form functional homodimers (T/T) as well as heterodimers with ubiquitously expressed bHLH E proteins (T/E). These dimers have distinct activities and regulate the expression of different sets of genes. The ratio between these dimers is determined by the relative levels of Twist1 and the HLH Id proteins (Connerney et al., 2006). Id proteins preferentially dimerize with E proteins and disrupt functional Class I/II bHLH heterodimers from forming (Massari and Murre, 2000). We found that Twist1 formed T/E dimers in the absence of Id, and formed T/T dimers when Id levels were increased (Connerney et al., 2006). Consistent with this, we found that in the sutures, genes that are regulated by T/T dimers, such as FGFR2 and periostin, are expressed in the osteogenic fronts where Twist1 and Id1 are co-expressed, while T/E regulated genes, such as thrombospondin 1 (TSP-1), are expressed in the mid-sutures where only Twist1 is expressed. In the sutures of Twist1+/− mice, the ratio between these dimers is altered, favoring an increase in homodimers and an expansion into the mid-suture of the expression of T/T-regulated genes, and a complete absence of T/E-regulated genes. Furthermore, we were able to inhibit craniosynostosis in Twist1+/− mice by modulating the balance between these dimers toward T/E formation, by either increasing the expression of E2A E12 or by decreasing Id1 expression (Connerney et al., 2006).
Here we propose a model that integrates Twist1 dimer formation with the signaling pathways known to regulate cranial suture patency (Fig. 1A). Our model predicts that the decrease in Twist1 expression due to Twist1 haploinsufficiency results in a higher T/T to T/E ratio extending beyond the osteogenic fronts due to Id1 levels out-competing Twist1 for dimerization with E proteins. The increase in T/T dimers expands the expression of FGFR2, resulting in increased FGF signaling. T/E dimers have recently been shown to inhibit BMP signaling (Hayashi et al., 2007), which is predominantly active in the osteogenic fronts (Warren et al., 2003), and therefore the decrease in T/E dimers in Twist+/− sutures, would allow for an expansion of BMP signaling as well. BMP signaling induces Id expression (Rice et al., 2000), which would further promote T/T formation. This positive feedback loop then causes the sutures to close prematurely, resulting in craniosynostosis. Our data support this hypothesis and illustrate that the sutures of Twist1+/− mice have increased FGF and BMP signaling, and that inhibition of FGF signaling prevents craniosynostosis in Twist+/− mice. Furthermore, enhanced T/T expression on a wild type background leads to a similar suture closure phenotype as Twist1+/− mice, indicating that T/T dimers play an active role in promoting craniosynostosis, and that the balance between the Twist1 dimers dictates suture patency.
Figure 1.
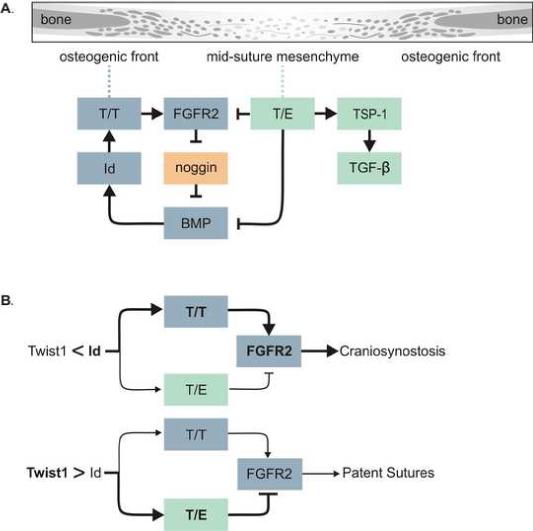
Model. (A) Drawing showing parts of a suture going from a more differentiated bone to a less differentiated mid-suture mesenchyme. The model shows T/T dimers up-regulating FGFR2 expression resulting in increased FGF signaling toward the middle of the suture. FGF signaling inhibits the expression of the BMP antagonist noggin, resulting in increased BMP signaling which up-regulates Id1 expression, further enhancing T/T homodimer formation. On the other hand, T/E dimers inhibit FGFR2 expression, as well as BMP signaling through binding to Smad proteins. T/E dimers also up-regulate TSP1 expression, which activates latent TGFβ. This could have differing outcomes dependent on which TGFβ isoform is present. (B) The ratio of T/T to T/E determines the functional output of Twist1 expression, and this is determined by the relative expression of Twist1 and Id1 proteins. When Id1 levels are greater that Twist1 this balance is shifted toward T/T formation resulting in up-regulation of FGFR2 leading to craniosynostosis. When Twist1 levels are higher than Id1 the balance is toward an increase in T/E formation, inhibiting FGFR2 expression and resulting in patent sutures.
Materials and Methods
Transgenic mice
Twist1+/− mice were obtained from the Jackson Laboratory, Bar Harbor, ME. Cre-responsive transgenes CC-TT, CC-TE, and CG-Spry1 were constructed by modification of the CAG-CAT-Z construct, which contains a CMV enhancer-chicken β-actin gene (CAG) promoter-loxP1-chloramphenicol acetyltransferase (CAT) gene-loxP1-LacZ gene (Araki et al., 1995). The CC-TT and CC-TE constructs replaced the LacZ gene with a cassette from pIREShrGFP (Stratagene, La Jolla, CA) constructs containing the myc-tagged TT and TE tethered dimers (Connerney et al., 2006) upstream of an IRES-hrGFP. The CC-TT and CC-TE constructs constitutively express CAT, and Cre recombinase-induced expression of either TT or TE and hrGFP, although the hrGFP expression is weak. The CG-Spry1 construct was generated by replacing the CAT gene with the GFP gene, and replacing LacZ with a myc/his-tagged mouse Spry1 open reading frame, generated by PCR using gene specific primers. The CG-Spry1 construct has constitutive GFP expression, and Cre recombinase-induced activation of Spry1 expression with concomitant loss of the GFP marker (Yang, et al, in preparation). Transgene activation by Cre recombinase was validated in Cre8 cells in vitro by immunoblotting using anti-myc antibodies (Upstate Biotech, Temecula, CA). The purified linearized construct was introduced by pronuclear injection on the FVB background. Resultant mice were screened for CAT or GFP expression, respectively, and founder lines validated by genotyping using CAT or GFP-specific primers. CAT primers: 5’-TGTGGTATGGCTGATTATGATCTC (Forward) and 3’-GCAGTGGTGGAATGCCTTTA– 3’ (Reverse), which amplify a 460 bp fragment; GFP primers: 5’-CTCGAGCCACCATGAGTAAAGGAGAAGAAC-3’ (Forward) and 5’-CTTCACTATTTGTAGAGTTCATCCATGC-3’ (Reverse), which amplify a 733 bp fragment.
We crossed these transgenic mice with CAG-estrogen receptor/Cre recombinase (CreERT2) mice (Hayashi and McMahon, 2002). Activation of CreERT2 was achieved by injection of tamoxifen (10mg/ml corn oil). To harvest embryos for calvarial explants, CC-TT and CC-TE females were crossed with CAG-CreERT2 males. Females were injected with 8mg/40g body weight tamoxifen at embryonic day 15.5 (E15.5) and embryos were harvested at E16.5. To activate transgenes after birth, 3-day-old pups were injected subcutaneously above the cranial sutures with 8μg/g weight and the mice were sacrificed at 5 weeks.
Immunohistochemistry
Skulls from newborn mice (P1) were dissected in ice-cold PBS and transferred immediately to cold 8% paraformaldehyde as in (Corson et al., 2003). Heads were de-skinned and dissected free from the underlying brain. Samples were embedded in paraffin and serial 5μm sections were processed for immunohistochemistry using the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA). The antibodies used in this process were anti-phospho-p44 (Cell Signaling Technology, Danvers, MA), anti-Bek C-17 and anti-Id1 sc-734 (Santa Cruz Biotechnology, Santa Cruz, CA), and anti-phospho-Smad1/5/8 (LeClair et al., 2007). Unbaked sections were stained for alkaline phosphatase with BM Purple.
In situ hybridization analysis
Skulls from newborn mice (P1) were fixed in 4% paraformaldehyde, cryosectioned and analyzed by in situ hybridization as in (Yoshida et al., 2005). In situ hybridization probes were against erm (gift from B. Hogan, Duke University), and noggin. The noggin probe was generated by PCR using the following primers: CGAGGATCCATGGAGCGCTGCCCCAGC, and CCGCTCGAGCTAGCAGGAACACTTACAC.
Western blot analysis
Individual frontal, coronal, and sagittal sutures were dissected from the skulls of two P1 pups and were pooled and homogenized in passive lysis buffer (Promega) containing Complete Mini protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN) using an Autodisruptor 24 (Autogen, Holliston, MA) following the recommended protocol from the manufacturer. Western blot analysis was performed as previously described (Leshem et al., 2000), using anti-phospho-Erk1/2 (Cell Signaling Technology, Danvers, MA) and anti-β-actin (Sigma-Aldrich, St. Louis, MO). The gels were scanned and quantified using ImageQuant 5.2 program (GE Healthcare, Piscataway, NJ), and fold induction between wild type and Twist1+/− sutures was determined after normalizing each band to the level of β-actin for that sample.
Preparation of calvaria explants
Female pregnant mice were given intra-peritoneal injections of tamoxifen at E15.5 and embryos were harvested at E16.5 for explant culture. Calvarias were prepared as previously described (Connerney et al., 2006). Briefly, whole calvaria were de-skinned and sectioned horizontally below the nose. The calvaria explants, containing the brain and dura mater, were then placed on Falcon cell culture inserts (PET pore size 0.4μm) in 12-well dishes containing DMEM, 10mg/ml L-ascorbic acid, 500mM β-glycerolphosphate, 10% FBS, 1% penicillin-streptomycin. Explants were incubated in a CO2 incubator at 37°C with media changes daily.
Heparin-acrylic beads (Sigma-Aldrich, St. Louis, MO) were prepared as described (Rice et al., 2000). Briefly, beads were incubated in 25 μg/ml of recombinant FGF2 protein or 25 μg/ml bovine serum albumin (BSA) at 37°C for 1 hour and then washed with growth media. Beads were placed either on the osteogenic fronts or in the mid-suture mesenchyme and the calvarial explants were cultured for 5 or 7 days as indicated, changing media daily. After culture, calvaria were fixed and subjected to alcian blue and alizarin red staining to analyze the extent of suture closure.
Results
Increased FGF signaling in the sutures of Twist1+/− mice
Although FGFR2 expression is expanded in the sutures of Twist1+/− mice (Connerney et al., 2006; Rice et al., 2000), it is unclear if this plays a functional role in promoting craniosynostosis in these mice. Therefore, we investigated if this expanded FGFR2 expression resulted in increased FGF signaling and/or responsiveness within the sutures. Similar to FGFR2 gene expression (Connerney et al., 2006), FGFR2 protein was detected throughout the sagittal sutures of Twist1+/− mice, while its expression was primarily restricted to the osteogenic fronts in wild type mice (Fig. 2a). A similar enhancement of FGFR2 expression was also observed in the coronal sutures. To determine the extent of FGF activation in the sutures, we used an antibody specific for the activated forms of Erk1/2. As can be seen in a serial section, activation of Erk1/2 correlated very closely with FGFR2 expression. Phospho-Erk1/2 was restricted to the osteogenic fronts of wild type mice, and was expanded to the mid-suture of Twist1+/− mice, which was particularly evident in the coronal sutures (Fig. 2A), suggesting that there is enhanced FGF signaling in the sutures of these mice. Western analysis of individual sutures from wild type and Twist1+/− mice also indicated that there was an increase in activated Erk1/2 in the Twist1+/− sutures (Fig. 2B). Consistent with the immunostaining, the most pronounced increase in Erk1/2 activation was seen in the coronal sutures, which are the sutures that normally fuse due to Twist1 haploinsufficiency. Interestingly, there were significant levels of activated Erk1/2 in the frontal sutures of wild type mice, which is the only cranial suture that normally fuses. This is consistent with FGF activity being associated with fusing sutures (Warren et al., 2003). Because Erk1/2 activity is not specific to FGF signaling, we also monitored the expression of erm, whose expression is primarily associated with FGF8 expression, but is also found in some other FGF signaling centers (Brent and Tabin, 2004; Firnberg and Neubuser, 2002; Lunn et al., 2007; Raible and Brand, 2001). Consistent with its regulation by FGF, erm expression was restricted to the osteogenic fronts in wild type sagittal and coronal sutures, and its expression increased throughout the sutures in Twist1+/− mice (Fig. 2B).
Figure 2.
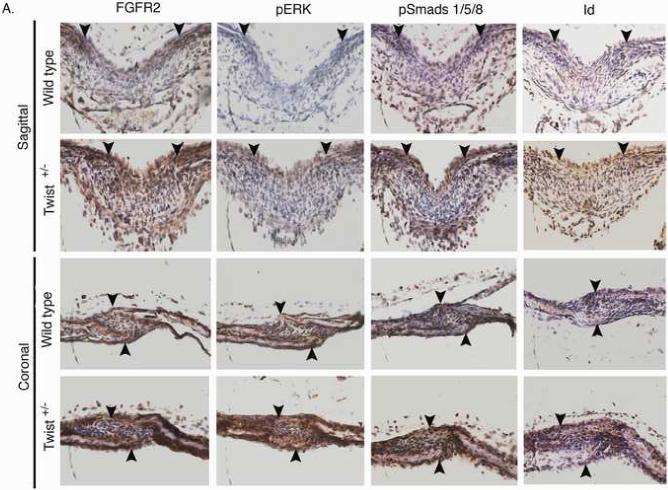
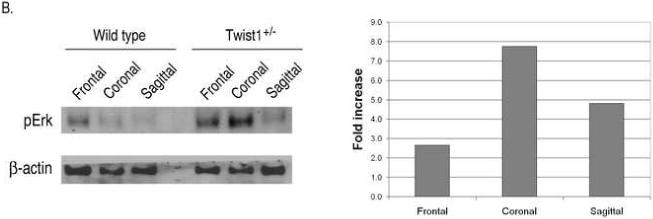
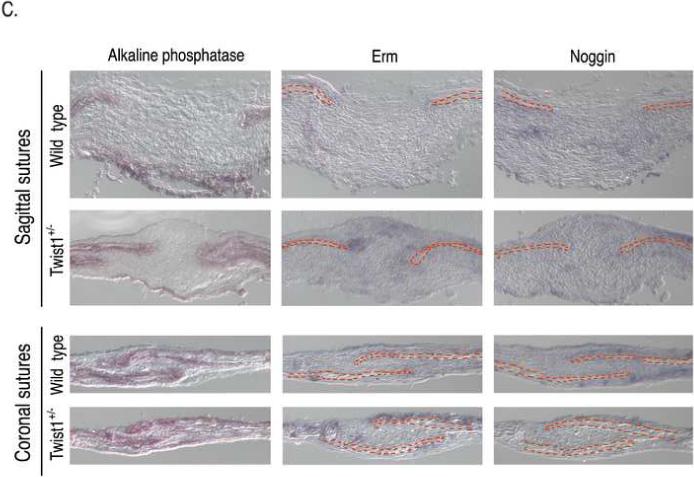
Increased FGF and BMP signaling in the sutures of Twist1+/− mice. (A) Immunohistochemistry for FGFR2, phospho-ERK1/2, phospho-smad1/5/8, and Id1 proteins in the sagittal and coronal sutures of wild type and Twist1+/− P1 mice. Arrow heads indicate the osteogenic fronts. Analysis of at least 3 mice of each genotype was performed, and representative data is shown. Note the extended staining for all of these throughout the sutures of Twist1+/− mice, while they are primarily restricted to the osteogenic fronts in wild type mice. (B) Western blot analysis of phopho-Erk1/2 in wild type and Twist1+/− sutures. Protein extracts from dissected frontal, coronal, and sagittal sutures from two P1 wild type or Twist1+/− mice were pooled and analyzed by western blot analysis for phopho-Erk1/2 and β-actin. The bar graph shows the fold change in phospho-Erk1/2 for each suture between wild type and Twist1+/− mice when corrected for β-actin expression. (C) in situ hybridization analysis of erm and noggin expression in the sagittal and coronal sutures of wild type and Twist1+/− P1 mice. The red dotted lines outline the calvaria bones.
Increased BMP signaling in Twist1+/− sutures
Our model predicts that there would be expanded BMP signaling in the sutures of Twist1+/− mice as well as increased FGF signaling. T/E dimers inhibit BMP signaling (Hayashi et al., 2007), and therefore we predicted that Twist1+/− sutures would have increased BMP signaling due to a decrease in T/E dimers. The BMP inhibitor noggin is also expressed in the suture mesenchyme, and its expression is repressed by FGF signaling (Warren et al., 2003). Therefore, an increase in FGF signaling in the Twist1+/− sutures would be expected to decrease noggin expression. The loss of T/E dimers and noggin expression would be predicted to result in an expansion of BMP signaling in these mice. To test this hypothesis, we examined the expression of noggin and the activation of the BMP pathway in the sutures of wild type and Twist1+/− mice. We analyzed noggin expression by in situ hybridization and found it to be expressed at fairly low levels throughout the suture mesenchyme of wild type mice and slightly decreased in Twist1+/− mice (Fig. 2C). The expression of noggin was originally determined using the expression of β-galactosidase from the LacZ gene that was knocked into the noggin locus, and therefore the levels of endogenous noggin were not determined (Warren et al., 2003). Our data suggest that noggin is not expressed at high levels in the sutures.
BMPs signal through the Smad proteins. Ligand activation of the BMP receptors results in the phosphorylation of Smads 1, 5, and 8, which then complex with Smad 4 to transport to the nucleus to regulate gene transcription. One of the genes that is regulated by BMP signaling is Id1 (Rice et al., 2000), and therefore we monitored BMP activity using an antibody specific for phospho-Smad 1/5/8, and by monitoring Id1 expression. We found that phospho-Smad 1/5/8 was primarily restricted to the osteogenic fronts in wild type mice, and increased and expanded to the mid-suture of Twist1+/− sutures as predicted (Fig. 2A). Id1 expression correlated closely with where Smad1/5/8 was activated. Id1 was expressed in the osteogenic fronts of wild type mice and expanded throughout the sutures in Twist1+/− mice. As with the effect on FGF signaling, the coronal sutures showed a more significant increase than the sagittal sutures. Therefore, Twist1 haploinsufficiency results in expanded activation of both the FGF and BMP signaling pathways, leading to enhanced Id1 expression, as our model predicts.
Twist1+/− sutures are more responsive to FGF2 than wild type controls
The restriction of FGFR2 expression to the osteogenic fronts of wild type mice correlates with a differential response of these cells to FGF compared with the cells of the mid-suture. FGF2-soaked beads induce suture closure when placed on the osteogenic fronts of explanted wild type E16.5 calvaria, but not when placed in the middle of the sagittal suture (Kim et al., 1998). To determine if the expanded FGFR2 expression in Twist1+/− mice altered the response of the mid-suture cells to FGF2, we used the calvaria explant assay. At E16.5, the calvaria of wild type and Twist1+/− mice are phenotypically very similar (Fig. 3A). Whole calvaria from E16.5 mice were explanted, and beads soaked in 25μg/ml BSA or FGF2 were placed on either the osteogenic fronts or on the mid-suture, as indicated in Fig. 3A. After 7 days of culture, the explants were stained with alcian blue and alizarin red to determine the extent of suture closure. We defined suture closure as when the two parietal bones touched or overlapped at any point. BSA beads did not induce suture closure in any cases. When FGF2-beads were placed on the osteogenic fronts, the sutures grew together in both wild type and Twist1+/− explants. However, when beads were placed on the mid-suture, suture closure was only seen in the Twist1+/− calvaria (Fig. 3B). Therefore, the expanded FGFR2 expression in Twist1+/− sutures leads to increased FGF-responsiveness.
Figure 3.

Twist1+/− sutures are more responsive to FGF. (A) E16.5 calvarias stained with Alcian Blue and Alizarin Red. Note that wild type and Twist1+/− calvaria are phenotypically the same at this stage. The drawing in the middle shows the location of beads (red dots) that were placed on the osteogenic fronts (OF) or on the mid-sutures (MS). (B) BSA or FGF2-soaked beads were placed on the osteogenic fronts or mid-sutures of wild type and Twist1+/− E16.5 calvaria, and the explants were cultured for 7 days. FGF2 beads placed on the osteogenic fronts induced suture closure in both wild type and Twist1+/− explants, while only the Twist1+/− sutures closed when FGF2 beads were placed on the mid-sutures. (C) Table showing the results of beads placed either on osteogenic fronts or mid-suture of calvaria explants of wild type versus Twist1+/− mice. Suture closure was defined as when the two parietal bones came in contact.
Inhibition of FGFR rescues suture fusion in Twist1+/− mice
Since the sutures of Twist1+/− skulls had increased FGF-responsiveness, we next addressed whether FGF signaling was required for suture fusion in Twist1+/− mice. Unlike in humans where suture fusion due to TWIST1 haploinsufficiency occurs at or before birth, suture fusion in Twist1+/− mice is delayed, and the sutures are relatively normal at birth and do not show signs of craniosynostosis until 3 weeks of age (Carver et al., 2002). We took advantage of this developmental delay to try and prevent suture fusion by inhibiting FGF signaling in the sutures after birth, when the sutures are accessible to manipulation. To inhibit FGF signaling, we used transgenic mice that conditionally express the receptor tyrosine kinase inhibitor Sprouty1 (Spry1) after Cre recombinase-induced recombination (Fig. S1). We utilized an estrogen receptor/Cre recombinase (CreERT2) fusion protein driven by the β-actin promoter and CMV enhancer (CAG) to enable temporal control of Cre recombination (Hayashi and McMahon, 2002). The activation of the CreERT2 protein was induced by injection of tamoxifen into the sutures of P3 mice. The extent of activation of CreERT2 was tested by injection of tamoxifen into the sutures of R26R;CAG-CreERT2 mice, which express the LacZ gene following Cre recombination (Jiang et al., 2002). Analysis of β-galactosidase expression in the coronal sutures 3 days after injection indicated that there was efficient recombination throughout the suture after a single injection of tamoxifen at P3 (Fig. 4A). P3 pups from a cross of Twist1+/−;CAG-CreERT2 with CG-Spry1 mice were injected with tamoxifen, then harvested 5 weeks after birth and analyzed for suture fusion. Activation of Spry1 expression in Twist1+/− mice resulted in an almost complete inhibition of suture fusion (Fig. 4B). The percentage of mice showing any coronal suture fusion decreased from 81.8% of all mice with a Twist1+/− genotype without Spry1 expression to 6.06% of Twist1+/−;CG-Spry1;CAG-CreERT2 mice. To determine the degree to which craniosynostosis was rescued on the different genetic backgrounds, we used a craniosynostosis index (CI) (Connerney et al., 2006). Left and right coronal sutures were assessed individually and assigned a number between 0 (completely unfused) to 3 (completely fused). The CI given in Fig. 4C indicates the average degree of craniosynostosis for a single coronal suture. None of the mice that were wild type for Twist1 showed any signs of suture fusion, and therefore were combined to compute the CI of wild type mice. The expression of Spry1 in the sutures of Twist1+/− mice significantly inhibited the degree of suture fusion, decreasing the CI from 1.2 in Twist1+/− mice to 0.12 in Twist1+/−;CG-Spry1;CAG-CreERT2 mice. Mice expressing Spry1 on a wild type background were similar to wild type and had a CI of 0. Suture patency was confirmed by sectioning through the coronal sutures. This demonstrated that expression of Spry1 on all backgrounds resulted in slightly thinner calvaria bones (Fig. 4B), indicating a requirement of FGF signaling for normal calvaria growth. Cre expression alone had no distinguishable affect on calvaria growth (data not shown). Therefore, inhibition of FGF signaling can prevent craniosynostosis in Twist1+/− mice.
Figure 4.
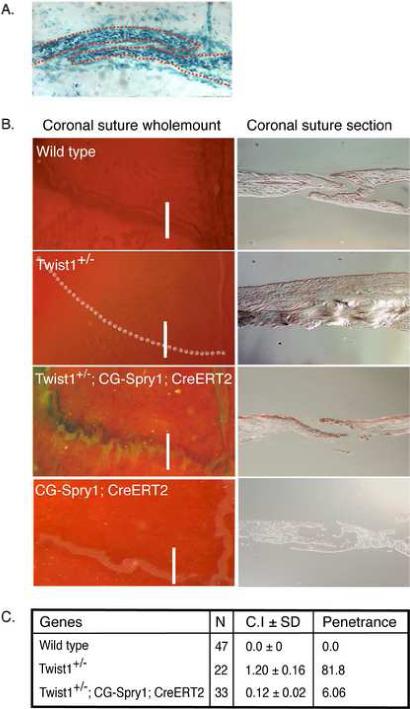
FGF signaling is required for craniosynostosis in Twist1+/− mice. (A) Efficient tamoxifen-induced cre recombination in the sutures. The sutures of 3-day-old pups from R26R reporter mice crossed with CAG-CreERT mice were injected with tamoxifen to induce expression of β-galactosidase from the R26R locus. The mice were then harvested at P7 and the coronal sutures were analyzed for β-galactosidase expression. Note expression throughout the coronal suture. (B) Expression of Spry1 in the sutures of Twist1+/− mice prevents craniosynostosis. Spry1 expression was induced from the CG-Spry1 transgene in CG-Spry1;CAG-CreERT2 mice on a wild type or Twist1+/− background by injection of tamoxifen into the sutures of P3 pups, and the coronal sutures were analyzed at 5 weeks. The skulls were stained with alcian blue and alizarin red. The left panels are wholemount analyses of the coronal sutures. The right panels are cross sections through the coronal sutures indicated by the white lines in the left panels. Representative examples are shown. (C) Table indicating the penetrance and degree of suture fusion. Skulls were assessed using a scoring system in which a suture was assigned a value from 0 to 3: 0 is completely unfused; 1 is 30% fused; 2 is 60% fused; and 3 is 100% fused. Left and right coronal sutures were scored individually for each skull. For each genotype, the scoring mean SEM was determined and was termed the craniosynostosis index (CI). Statistical analysis was performed by one-way analysis of variance. Cre expression alone did not affect the Twist1+/− phenotype, and therefore all of the mice that were Twist1+/− but did not have both the CAG-CreERT2 and CG-Spry1 transgenes, and therefore did not express Spry1, were designated as being Twist1+/−. For the same reason, all mice that were wild type for Twist1 were all considered wild type in computing the CI. The difference between Twist1+/− and Twist1+/−;CG-Spry1;CAG-CreERT2 mice, with activated Spry1 expression, was highly significant (p =0.001). Penetrance was calculated as the percentage of animals that exhibited any coronal suture fusion.
Twist1 dimers have opposing effects on osteoblast differentiation and FGF-responsiveness
Our previous data suggested that the balance between T/T and T/E is altered in Twist1+/− mice and that restoration of this balance prevents suture closure (Connerney et al., 2006). However, these experiments did not distinguish if suture closure in these mice occurs due to the relative increase in T/T formation, or to the decrease in T/E dimers, or both. Twist1 has been implicated in the inhibition of osteoblast differentiation (Bialek et al., 2004; Funato et al., 2001; Hayashi et al., 2007; Lee et al., 1999), and we found that this was mediated by T/E dimers (Connerney et al., 2006). However, since T/T dimers regulate FGFR2 expression (Connerney et al., 2006), which is expanded to the mid-suture in Twist1+/− mice (Fig. 2) (Connerney et al., 2006; Rice et al., 2000), T/T dimers may play an active role in promoting suture fusion. To further test this hypothesis, we asked if the expression of TT forced dimers, in which the Twist1 monomers are linked by a flexible glycine-serine polylinker (Connerney et al., 2006), in otherwise wild type sutures would result in a similar phenotype as that due to Twist1 haploinsufficiency. Conversely, we also determined if increasing the levels of TE would inhibit osteoblast differentiation in vivo, and lead to open sutures. We achieved this by using a conditional transgenic system similar to the CG-Spry1 transgenic mice explained above. After Cre-induced recombination, the CC-TT or CC-TE mice constitutively express a “tethered” Twist1 homodimer (TT) or heterodimer (TE) construct, respectively (Fig. S1). CC-TT and CC-TE mice were crossed with CAG-CreERT2 mice so that Cre recombination could be controlled in a temporal manner.
To determine if increased TT or TE expression would affect calvaria differentiation or FGFR2 expression, we activated CC-TT and CC-TE transgene expression by injecting tamoxifen into the sutures of P3 pups. The sutures were then analyzed for the osteoblast differentiation marker alkaline phosphatase and FGFR2 expression at P7 when there is active calvaria growth. TE expression dramatically reduced alkaline phosphatase expression, while TT expression may have even enhanced calvaria differentiation, as these mice had increased alkaline phosphatase staining (Fig. 5A). Alkaline phosphatase staining of Twist1+/− sutures was slightly expanded compared to wild type sutures (see also Fig 2). FGFR2 expression was expanded throughout the sagittal sutures of both Twist1+/− and TT-expressing mice. TE expression seemed to decrease FGFR2 expression, similar to what was observed in vitro (Connerney et al., 2006). Therefore, TE expression inhibited osteoblast differentiation and FGFR2 expression while TT expression enhanced them.
Figure 5.
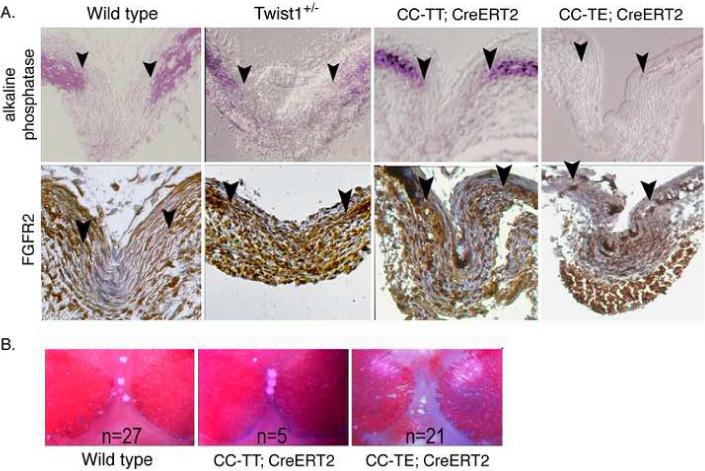
Expression of TT leads to a similar increase in FGFR2 expression and responsiveness as Twist1+/− mice. (A) Twist1 homodimers (CC-TT) promote FGFR2 expression and osteoblast differentiation, while Twist1 heterodimers (CC-TE) inhibit these. All pups were injected with tamoxifen at P3 to induce transgene expression, and the sutures were analyzed at P7. Sections through the sagittal sutures of P7 skulls from wild type, Twist1+/−, CC-TT;CAG-CreERT2, and CC-TE;CAG-CreERT2 mice were analyzed for alkaline phosphatase and FGFR2 protein expression. Arrowheads indicate the osteogenic fronts. Analysis of at least 3 mice of each genotype was performed, and representative data is shown. Note that FGFR2 expression was limited to the osteogenic fronts in wild type sutures, while Twist1+/− and CC-TT;CAG-CreERT2 sutures had an expansion of this expression to the middle of the suture. Mice expressing CC-TE had even lower FGFR2 expression than wild type. Alkaline phosphatase staining was slightly expanded from the osteogenic fronts in Twist1+/− sutures, but was significantly enhanced in CC-TT;CAG-CreERT2 mice and almost absent in CC-TE;CAG-CreERT2 mice. (B) FGF responsiveness is enhanced by TT and decreased by TE expression. Pregnant females were injected with tamoxifen at E15 and then at E16.5 calvaria explants of wild type, CC-TT;CAG-CreERT2, and CC-TE;CAG-CreERT2 were incubated for 5 days in differentiation media with FGF2 soaked beads placed on the mid-suture. FGF beads placed on the mid-suture of CC-TT-expressing calvaria promoted suture closure in all of the explanted calvaria, which did not occur in any of the control or CC-TE-expressing explants.
We next asked whether the differential expression of FGFR2 would affect the FGF-responsiveness of the sutures in the CC-TT and CC-TE mice as it did in the Twist1+/− mice. Pregnant CC-TT or CC-TE females that had been crossed with CAG-CreERT2 males were injected with tamoxifen at E15.5 to activate transgene expression, and embryos were then harvested and the calvaria explanted at E16.5. Activation of either CC-TT or CC-TE had no discernable phenotypic effect at the time of explant (data not shown). FGF2 or BSA beads were placed in the mid-suture as in Fig. 3, and the explants were cultured for 5 days. FGF beads placed in the mid-suture of TT-expressing calvaria promoted suture closure in all of the explanted calvaria, which did not occur in any of the control or TE-expressing explants (Fig. 5B). Therefore, increased expression of Twist1 homodimers enhanced FGFR2 expression and FGF-responsiveness consistent with our model in Twist1+/− mice.
Twist1 homodimers promote suture overgrowth and craniosynostosis
Since TT expression enhanced suture closure in explant cultures, we next asked if TT expression would affect the post-natal development of the coronal sutures in otherwise wild type mice. For these experiments, CC-TT;CAG-CreERT2 or CC-TE;CAG-CreERT2 and control littermates were injected with tamoxifen in the sutures at P3 to activate transgene expression. The mice were then sacrificed at 5 weeks of age and the skulls were analyzed for suture closure as above. In 6 of 8 CC-TT;CAG-CreERT2 mice, the coronal sutures were distinctly different from wild type (Fig. 6A) and uninjected control sutures. In these mice, the coronal sutures had an undulating or serrate phenotype (Fig. 6D,E). The coronal sutures of CC-TE;CAG-CreERT2 mice were slightly more open than controls, but otherwise looked fairly normal (Fig. 6C). Sections through the coronal sutures further emphasized the different suture phenotypes. The coronal sutures from wild type mice had a defined opening between the parietal and frontal bones (Fig. 6F), which was clearly absent in the Twist1+/− mice where the parietal and frontal bones were fused, and in comparison with the wild type, these bones appeared thicker (Fig. 6G). TE expression resulted in a slightly more open suture than wild type, with thinner tips of the bones (Fig. 6H). In contrast, CC-TT;CAG-CreERT2 mice had dramatically extended sutures, with the overlap of the parietal and frontal bones being more than 5 times that of wild type mice. Sections through the sutures of 2 different mice with either undulate or serrate phenotypes are shown at both 4× and 10× magnification (Fig. 6I-L). Interestingly, while TT expression caused overgrowth of the calvaria bones, no actual fusion between the frontal and parietal bones was observed, perhaps indicating that constitutive TT expression inhibits some activity that is required for bone fusion.
Figure 6.
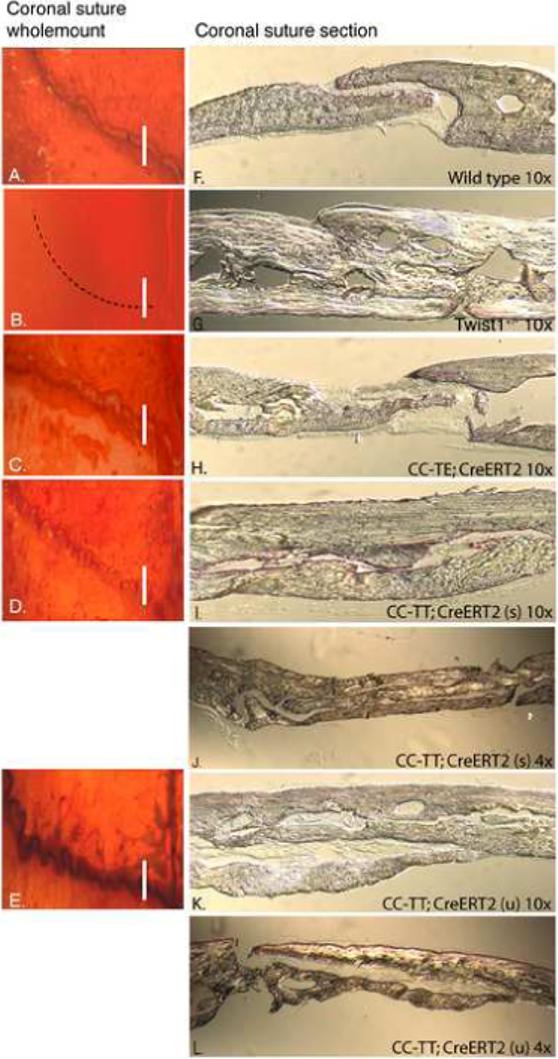
Expression of TT promotes overgrowth of the calvaria bones. (A-L) Mice were treated at P3 as in Figure 4 and were then harvested for analysis at 5 weeks of age. Skulls were stained with alcian blue and alizarin red and analyzed by wholemount (A-E) and sectioning (F-L). The number of mice treated was wild type (n=9), CC-TE (n=19), and CC-TT (n=8). Coronal sutures appeared patent in wild type mice (A), while Twist1+/− sutures were fused (B). TT expression resulted in varied degrees of two phenotypes: CC-TT (u) presented with an undulate phenotype (D), and CC-TT (s) had a serrate phenotype, which appeared to be tightly closed (E). The TE-expressing sutures were patent and showed greater separation between the bones (C). Cross-sections of coronal sutures confirmed the difference between the sutures: (F) coronal sutures from wild type adult mice showed a defined opening between the parietal and frontal bones; (G) Twist1+/− coronal sutures were clearly fused and the resulting bone was thicker than wild type; (H) CC-TE;CAG-CreERT2 sutures showed an increased mid-suture space with thinner bone ends compared to wild type; (I-L) CC-TT;CAG-CreERT2 expressing sutures of both serrate (s) (I,J) and undulate (u) (K,L) phenotypes were significantly expanded with severely overgrown calvaria bones that resembled the thickness of the Twist1+/− calvaria. These sutures are also shown using 4× magnification objectives to be able to view the entire sutures (J,L).
Discussion
About 25% of craniosynostosis cases can be classified into one of about 100 different craniosynostotic syndromes, with most being due to activating mutations in the FGFR1, FGFR2, or FGFR3 genes, or inactivating mutations in the TWIST1 gene resulting in haploinsufficiency (Cunningham et al., 2007; Lenton et al., 2005; Rice, 2005). The mechanisms linking these two pathways, however, are unclear. We previously proposed a model where Twist1 homodimers and heterodimers differentially regulate cranial suture patency (Connerney et al., 2006). TWIST1 haploinsufficiency results in an imbalance between these dimers due to increased competition with Id1 proteins for dimerization with E proteins. This results in a higher T/T to T/E ratio throughout the sutures leading to an expansion of the osteogenic fronts and suture closure. Our data supported this model, and importantly we were able to show that restoration of the normal balance between T/T and T/E, by modulating the expression of E proteins or Id1, prevented craniosynostosis in Twist1+/− mice (Connerney et al., 2006). The data we present here further supports this model and extends it to integrate the Twist1 dimers with the FGF and BMP pathways, which are known to regulate cranial suture patency (Fig. 1A). We show that the expression of T/T and T/E differentially affect the FGF responsiveness of the sutures to FGF, and that inhibition of FGF signaling prevents craniosynostosis in Twist1+/− mice, creating a more dynamic model of the regulation of cranial suture patency (Fig. 1B).
Integration of Twist1 dimer formation and activity with the FGF and BMP pathways
In several different organisms, changes in Twist expression alter FGF receptor expression, however it has been unclear how direct this regulation is, and whether the regulation is positive or negative. Our data indicate that T/T homodimers increase, and T/E heterodimers decrease FGFR2 expression. Over-expression of TT homodimers on a wild type background resulted in a similar phenotype as Twist1 haploinsufficiency, giving strong support for our model that Twist1 haploinsufficiency leads to a high T/T to T/E ratio throughout the sutures. This also indicates that the T/T to T/E ratio in a cell likely determines the functional output of Twist expression. This can be difficult to quantify because it is determined by the relative ratio of Twist to other HLH proteins in a cell. Because there are 2 vertebrate Twist proteins, Twist1 and Twist2 (also known as Dermo1), 4 Id proteins and 4 E proteins, measurements can be complex. Furthermore, other bHLH proteins compete with Twist1 for dimerization with E proteins (Spicer et al., 1996), and Twist1 can dimerize with some other bHLH proteins such as the Hand proteins (Firulli et al., 2005; Firulli et al., 2007), further increasing the complexity.
In the cranial sutures, Twist1, Id1, and E proteins are all expressed. E2A and HEB E proteins are expressed throughout the sutures and osteogenic fronts, as well as in the differentiating osteoblasts (Funato et al., 2001). Twist1 is expressed in the osteogenic fronts and suture mesenchyme, while Id1 proteins are restricted to the osteogenic fronts (Connerney et al., 2006; Johnson et al., 2000; Rice et al., 2000). Because we previously found that Twist1 and Id1 compete for dimerization with E proteins, and the relative level of Twist1 to Id1 determines the T/T to T/E ratio within a cell (Connerney et al., 2006), we proposed that these expression patterns within the sutures would result in primarily T/T dimers forming in the osteogenic fronts where Twist1 and Id1 are co-expressed, and a predominance of T/E dimers in the mid-sutures where only Twist1 is expressed. This was supported by the expression patterns of genes that are differentially regulated by T/T and T/E dimers (Connerney et al., 2006). The expression domains of Twist1 and Id1 are largely regulated by FGF and BMP signaling, which integrate with TGFβ to regulate suture patency (Cunningham et al., 2007; Lenton et al., 2005; Rice, 2005). Twist1 expression is at least in part regulated by FGF signaling, while Id1 expression is induced by BMP signaling (Isaac et al., 2000; Rice et al., 2000). Conversely, Twist1 dimers differentially affect the response of cells to FGF and BMP, and may affect TGFβ-responsiveness as well. Our data demonstrate that T/T and T/E dimers have opposing effects on the expression of FGFR2, resulting in a differential response of cells in the osteogenic fronts and mid-suture to FGF. We also show that BMP signaling is restricted to the osteogenic fronts of wild type sutures, even though BMP4 is expressed throughout the sutures (Warren et al., 2003). This restriction may be due to T/E dimers in the mid-suture, since they have been shown to inhibit BMP signaling (Hayashi et al., 2007). The BMP inhibitor noggin is also expressed in the mid-suture mesenchyme (Warren et al., 2003), and may cooperate with the T/E dimers to restrict BMP signaling to the osteogenic fronts, however we found noggin expression to be quite low in the sutures. The role of TGFβ in regulating suture patency is complex, but may also be affected by Twist1 dimer formation. TGFβ2 promotes suture closure, while TGFβ3 inhibits closure (Opperman et al., 1999). Interestingly, we found that T/E heterodimers regulate the expression of TSP1 in the mid-suture mesenchyme (Connerney et al., 2006). One of the functions of TSP1 is to activate latent TGFβ (Annes et al., 2003). The mechanism of TGFβ activation in the cranial sutures has not been studied, and perhaps TSP1 plays a role in this.
Our model (Fig. 1) indicates how relatively small changes in the dosage of Twist1 can dramatically alter the regulation of suture patency by affecting FGF, BMP, and potentially TGFβ signaling, and these pathways in turn can affect the balance between T/T and T/E by modulating the expression of Twist1 and Id1. Because Twist1 and Id1 compete for dimerization with E proteins (Connerney et al., 2006), we hypothesize that the decrease in Twist1 expression due to Twist1 haploinsufficiency results in a higher T/T to T/E ratio extending beyond the osteogenic fronts due to Id1 levels out-competing Twist1 for dimerization with E proteins. This results in an expansion of FGFR2 expression and an increase in FGF responsiveness beyond the osteogenic fronts. The decrease in T/E formation, perhaps combined with a decrease in noggin expression due to the increased FGF signaling (Warren et al., 2003), would lead to an increase in BMP signaling. The BMP signaling would increase Id1 expression, further enhancing T/T formation. This positive feedback loop would continue to promote the expansion of FGF and BMP signaling throughout the sutures, resulting in their premature closure and craniosynostosis. Our data support this model, showing increased FGF and BMP signaling, and Id1 expression throughout the sutures of Twist1+/− mice.
T/T and T/E dimers differentially regulate osteoblast differentiation
Twist1 inhibits osteoblast differentiation (Bialek et al., 2004; Funato et al., 2001; Hayashi et al., 2007; Lee et al., 1999), but our data indicate that this activity is restricted to T/E dimers because Twist1 required heterodimerization with E proteins to inhibit the differentiation of primary calvaria osteoblasts (Connerney et al., 2006). Furthermore, T/E heterodimerization was shown to be required to inhibit BMP-induced osteoblast differentiation (Hayashi et al., 2007). Interestingly, this inhibition was relieved by the expression of Id1, which would be expected to promote T/T formation (Hayashi et al., 2007). In the present study, we found that TT expression resulted in increased alkaline phosphatase activity and dramatic overgrowth of the calvaria bones, which suggest that T/T dimers may even promote osteoblast differentiation. TE expression resulted in significant inhibition of alkaline phosphatase expression in the osteogenic fronts at P7, however at 5 weeks the sutures were only slightly more open than controls with thinner tips of the bones. This suggests that osteoblast differentiation is only delayed by T/E dimers, as compared to myoblast differentiation, which is completely inhibited due to T/E expression (Hamamori et al., 1997; Hebrok et al., 1994; Leshem et al., 2000; Spicer et al., 1996). A lack of severe inhibition by TE dimers was also recently observed by expression of TE forced dimers during limb development, where the bones ended up being slightly shorter, but there was clearly substantial osteogenesis occurring (Firulli et al., 2007). We have also observed that the initial inhibition of osteoblast differentiation by Twist1 in vitro is much less significant as differentiation proceeds, which is not true for the inhibition of myogenesis by Twist1 (data not shown). We believe that this, coupled with different culture conditions affecting which Twist1 dimer is favored, can lead to confusing results. For instance, three studies on the differentiation properties of TWIST1 haploinsufficient osteoblasts came to three different conclusions about whether the cells differentiated faster, slower, or the same as controls (Guenou et al., 2005; Ratisoontorn et al., 2005; Yousfi et al., 2001). Since the expression of the Id1 gene is highly regulated by culture conditions, small changes in confluency, or even the type of serum used, or when it was added could easily affect the T/T to T/E ratio in the cells and alter the effect on differentiation.
T/T dimers play an active role in promoting craniosynostosis
Expression of TT dimers in the sutures resulted in a phenotype that closely resembled that of Twist1 haploinsufficiency. The expression of TT caused a similar expansion of FGFR2 expression and increased FGF-responsiveness of the sutures. Furthermore, the TT over-expressing sutures had a serrate and undulate phenotype sometimes observed in Twist1+/− sutures. Histological analysis of calvarial bones of patients with activating mutations of FGFR2 show that these samples have increased alkaline phosphatase activity, accelerated ossification, and increased thickness of the calvaria bones compared with normal controls (Lomri et al., 1998). Interestingly, the calvarial bones around the coronal suture in our TT over-expressing transgenic mice also had increased alkaline phosphatase activity, accelerated ossification, and increased thickness of the calvarial bone. These data strongly support an active role for T/T dimers in promoting craniosynostosis in TWIST1 haploinsufficiency.
Our data indicate that TT expression affects the osteogenic fronts as well as the mid-suture mesenchyme, and these changes closely reflect the changes seen in the sutures of Twist1+/− mice. While the sutures were not significantly morphologically different than wild type sutures up to P7, there was increased alkaline phosphatase in the osteogenic fronts and changes in gene expression and increased FGF and BMP signaling throughout the sutures. These changes in gene expression significantly altered the response of the mid-suture cells to FGF and promoted suture closure in our explant studies. Our experiments cannot distinguish whether TT expression in the osteogenic fronts, mid-suture mesenchyme, or both was important for promoting the calvaria outgrowth seen at 5 weeks, however our data from the calvaria explants suggest that it may be due to an effect on both cell populations. A recent study using cell transplants from different regions of the sagittal suture demonstrated that in wild type mice cells just outside of the osteogenic fronts are able to participate in the formation of the calvaria bones, however the cells in the mid-suture are not (Lana-Elola et al., 2007). It would be interesting to do similar assays with Twist1+/− and TT-expressing sutures to see if the fate, or potency of these mid-suture cells has changed.
Interestingly, TT expression resulted in overgrowth of the calvaria bones, but no overt fusion was observed. The mechanisms regulating fusion of the calvaria bones are really unknown. The transgenic system that we used results in constitutive expression of TT, or TE, throughout the suture and osteogenic fronts after recombination. Twist1 is normally expressed in these tissues, but its expression declines as the cells differentiate. There is the possibility that maintenance of TT expression in these cells may block some aspect that is required for the two bones to adhere and fuse with each other. Alternatively, actual fusion may require an absence of T/E dimers, which may still be present in the sutures of the TT-expressing mice, but not in Twist1+/− mice. Increased proliferation and differentiation has been proposed to be the mechanism that drives craniosynostosis due to activated FGFR2 mutations (Ornitz and Marie, 2002), however our data indicate that simply overgrowth of the calvaria is not sufficient to cause suture fusion.
In conclusion, we propose that the balance between T/T and T/E dimers plays a central role in integrating FGF, BMP, and perhaps TGFβ signaling to regulate cranial suture growth and patency. Clearly, there are other factors that need to be incorporated into this model, but this type of complex feedback regulation can help explain why there is such phenotypic variability in many craniosynostosis syndromes.
Supplementary Material
Figure S1. Diagram of transgenes used. (A) CG-Spry1 transgene before and after Cre-induced excision of the GFP open reading frame (ORF). The CAG promoter is followed by the GFP ORF and transcription termination sequences, which are flanked by loxP1 sites (◆). The Spry1 ORF is downstream of the second loxP1 site. Before excision, GFP is expressed and transcription is stopped by termination sequences after the GFP ORF. After excision, Spry1 is immediately downstream of the CAG promoter and is expressed. (B) CC-TT and CC-TE transgenes before and after excision. These transgenes are similar to the CG-Spry1 transgene with the following differences. The GFP ORF in CG-Spry1 is replaced with the chlorumphenicol acelyl transferase (CAT) gene, and there is an IRES-GFP cassette downstream of the TT and TE ORFs.
Acknowledgements
We would like to thank members of the Spicer lab and MMCRI for helpful comments and discussions. This research was supported by grants from NIH to D.B.S. (P20 RR15555 and R01 DE015329) and R.E.F. (P20 RR15555 and R01 DK73871).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annes JP, et al. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Araki K, et al. Site-specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A. 1995;92:160–4. doi: 10.1073/pnas.92.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialek P, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–35. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- Brent AE, Tabin CJ. FGF acts directly on the somitic tendon progenitors through the Ets transcription factors Pea3 and Erm to regulate scleraxis expression. Development. 2004;131:3885–96. doi: 10.1242/dev.01275. [DOI] [PubMed] [Google Scholar]
- Carver EA, et al. Craniosynostosis in Twist heterozygous mice: A model for Saethre-Chotzen syndrome. Anat Rec. 2002;268:90–2. doi: 10.1002/ar.10124. [DOI] [PubMed] [Google Scholar]
- Castanon I, Baylies MK. A Twist in fate: evolutionary comparison of Twist structure and function. Gene. 2002;287:11–22. doi: 10.1016/s0378-1119(01)00893-9. [DOI] [PubMed] [Google Scholar]
- Chun K, et al. Genetic analysis of patients with the Saethre-Chotzen phenotype. Am J Med Genet. 2002;110:136–43. doi: 10.1002/ajmg.10400. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr. Merging the old skeletal biology with the new. I. Intramembranous ossification, endochondral ossification, ectopic bone, secondary cartilage, and pathologic considerations. J Craniofac Genet Dev Biol. 2000;20:84–93. [PubMed] [Google Scholar]
- Connerney J, et al. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dyn. 2006;235:1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- Corson LB, et al. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–37. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- Cunningham ML, et al. Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res. 2007;10:67–81. doi: 10.1111/j.1601-6343.2007.00389.x. [DOI] [PubMed] [Google Scholar]
- Dupont J, et al. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J Biol Chem. 2001;276:26699–707. doi: 10.1074/jbc.M102664200. [DOI] [PubMed] [Google Scholar]
- el Ghouzzi V, et al. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–6. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- Fang MA, et al. Transcriptional regulation of alpha 2(I) collagen gene expression by fibroblast growth factor-2 in MC3T3-E1 osteoblast-like cells. J Cell Biochem. 2001;80:550–9. [PubMed] [Google Scholar]
- Firnberg N, Neubuser A. FGF signaling regulates expression of Tbx2, Erm, Pea3, and Pax3 in the early nasal region. Dev Biol. 2002;247:237–50. doi: 10.1006/dbio.2002.0696. [DOI] [PubMed] [Google Scholar]
- Firulli BA, et al. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nat Genet. 2005;37:373–81. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli BA, et al. Mutations within helix I of twist1 result in distinct limb defects and variation of DNA-binding affinities. J Biol Chem. 2007 doi: 10.1074/jbc.M702613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato N, et al. Common regulation of growth arrest and differentiation of osteoblasts by helix-loop-helix factors. Mol Cell Biol. 2001;21:7416–28. doi: 10.1128/MCB.21.21.7416-7428.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenou H, et al. A role for fibroblast growth factor receptor-2 in the altered osteoblast phenotype induced by Twist haploinsufficiency in the Saethre-Chotzen syndrome. Hum Mol Genet. 2005;14:1429–39. doi: 10.1093/hmg/ddi152. [DOI] [PubMed] [Google Scholar]
- Hamamori Y, et al. The basic domain of myogenic basic helix-loop-helix (bHLH) proteins is the novel target for direct inhibition by another bHLH protein, Twist. Mol Cell Biol. 1997;17:6563–73. doi: 10.1128/mcb.17.11.6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, et al. Comparative roles of Twist-1 and Id1 in transcriptional regulation by BMP signaling. J Cell Sci. 2007;120:1350–7. doi: 10.1242/jcs.000067. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–18. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Hebrok M, et al. M-twist is an inhibitor of muscle differentiation. Dev Biol. 1994;165:537–44. doi: 10.1006/dbio.1994.1273. [DOI] [PubMed] [Google Scholar]
- Howard TD, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- Isaac A, et al. FGF and genes encoding transcription factors in early limb specification. Mech Dev. 2000;93:41–8. doi: 10.1016/s0925-4773(00)00261-6. [DOI] [PubMed] [Google Scholar]
- Jiang X, et al. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–16. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Johnson D, et al. Expression patterns of Twist and Fgfr1, −2 and −3 in the developing mouse coronal suture suggest a key role for twist in suture initiation and biogenesis. Mech Dev. 2000;91:341–5. doi: 10.1016/s0925-4773(99)00278-6. [DOI] [PubMed] [Google Scholar]
- Kim HJ, et al. FGF-, BMP- and Shh-mediated signalling pathways in the regulation of cranial suture morphogenesis and calvarial bone development. Development. 1998;125:1241–51. doi: 10.1242/dev.125.7.1241. [DOI] [PubMed] [Google Scholar]
- Lana-Elola E, et al. Cell fate specification during calvarial bone and suture development. Dev Biol. 2007;311:335–46. doi: 10.1016/j.ydbio.2007.08.028. [DOI] [PubMed] [Google Scholar]
- LeClair RJ, et al. Cthrc1 is a novel inhibitor of transforming growth factor-beta signaling and neointimal lesion formation. Circ Res. 2007;100:826–33. doi: 10.1161/01.RES.0000260806.99307.72. [DOI] [PubMed] [Google Scholar]
- Lee MS, et al. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem. 1999;75:566–77. doi: 10.1002/(sici)1097-4644(19991215)75:4<566::aid-jcb3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Lenton KA, et al. Cranial suture biology. Curr Top Dev Biol. 2005;66:287–328. doi: 10.1016/S0070-2153(05)66009-7. [DOI] [PubMed] [Google Scholar]
- Leshem Y, et al. Hepatocyte growth factor (HGF) inhibits skeletal muscle cell differentiation: a role for the bHLH protein twist and the cdk inhibitor p27. J Cell Physiol. 2000;184:101–9. doi: 10.1002/(SICI)1097-4652(200007)184:1<101::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Lomri A, et al. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest. 1998;101:1310–7. [PMC free article] [PubMed] [Google Scholar]
- Lunn JS, et al. A spatial and temporal map of FGF/Erk1/2 activity and response repertoires in the early chick embryo. Dev Biol. 2007;302:536–52. doi: 10.1016/j.ydbio.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–40. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke MP, Tam PP. Twist functions in mouse development. Int J Dev Biol. 2002;46:401–13. [PubMed] [Google Scholar]
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219:472–85. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Opperman LA, et al. Cranial suture obliteration is induced by removal of transforming growth factor (TGF)-beta 3 activity and prevented by removal of TGF-beta 2 activity from fetal rat calvaria in vitro. J Craniofac Genet Dev Biol. 1999;19:164–73. [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–65. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Paznekas WA, et al. Genetic heterogeneity of Saethre-Chotzen syndrome, due to TWIST and FGFR mutations. Am J Hum Genet. 1998;62:1370–80. doi: 10.1086/301855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raible F, Brand M. Tight transcriptional control of the ETS domain factors Erm and Pea3 by Fgf signaling during early zebrafish development. Mech Dev. 2001;107:105–17. doi: 10.1016/s0925-4773(01)00456-7. [DOI] [PubMed] [Google Scholar]
- Ratisoontorn C, et al. In vitro differentiation profile of osteoblasts derived from patients with Saethre-Chotzen syndrome. Bone. 2005;36:627–34. doi: 10.1016/j.bone.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Rice DP. Craniofacial anomalies: from development to molecular pathogenesis. Curr Mol Med. 2005;5:699–722. doi: 10.2174/156652405774641043. [DOI] [PubMed] [Google Scholar]
- Rice DP, et al. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–55. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Spicer DB, et al. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- Warren SM, et al. The BMP antagonist noggin regulates cranial suture fusion. Nature. 2003;422:625–9. doi: 10.1038/nature01545. [DOI] [PubMed] [Google Scholar]
- Yoshida T, et al. Twist is required for establishment of the mouse coronal suture. J Anat. 2005;206:437–44. doi: 10.1111/j.1469-7580.2005.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousfi M, et al. Increased bone formation and decreased osteocalcin expression induced by reduced Twist dosage in Saethre-Chotzen syndrome. J Clin Invest. 2001;107:1153–61. doi: 10.1172/JCI11846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Diagram of transgenes used. (A) CG-Spry1 transgene before and after Cre-induced excision of the GFP open reading frame (ORF). The CAG promoter is followed by the GFP ORF and transcription termination sequences, which are flanked by loxP1 sites (◆). The Spry1 ORF is downstream of the second loxP1 site. Before excision, GFP is expressed and transcription is stopped by termination sequences after the GFP ORF. After excision, Spry1 is immediately downstream of the CAG promoter and is expressed. (B) CC-TT and CC-TE transgenes before and after excision. These transgenes are similar to the CG-Spry1 transgene with the following differences. The GFP ORF in CG-Spry1 is replaced with the chlorumphenicol acelyl transferase (CAT) gene, and there is an IRES-GFP cassette downstream of the TT and TE ORFs.