

Abstract
Heterotrimeric G-protein Gα subunits and GoLoco motif proteins are key members of a conserved set of regulatory proteins that influence invertebrate asymmetric cell division and vertebrate neuroepithelium and epithelial progenitor differentiation. GoLoco motif proteins bind selectively to the inhibitory subclass (Gαi) of Gα subunits, and thus it is assumed that a Gαi·GoLoco motif protein complex plays a direct functional role in microtubule dynamics underlying spindle orientation and metaphase chromosomal segregation during cell division. To address this hypothesis directly, we rationally identified a point mutation to Gαi subunits that renders a selective loss-of-function for GoLoco motif binding, namely an asparagine-to-isoleucine substitution in the αD-αE loop of the Gα helical domain. This GoLoco-insensitivity (“GLi”) mutation prevented Gαi1 association with all human GoLoco motif proteins and abrogated interaction between the Caenorhabditis elegans Gα subunit GOA-1 and the GPR-1 GoLoco motif. In contrast, the GLi mutation did not perturb any other biochemical or signaling properties of Gαi subunits, including nucleotide binding, intrinsic and RGS protein-accelerated GTP hydrolysis, and interactions with Gβγ dimers, adenylyl cyclase, and seven transmembrane-domain receptors. GoLoco insensitivity rendered Gαi subunits unable to recruit GoLoco motif proteins such as GPSM2/LGN and GPSM3 to the plasma membrane, and abrogated the exaggerated mitotic spindle rocking normally seen upon ectopic expression of wild type Gαi subunits in kidney epithelial cells. This GLi mutation should prove valuable in establishing the physiological roles of Gαi·GoLoco motif protein complexes in microtubule dynamics and spindle function during cell division as well as to delineate potential roles for GoLoco motifs in receptor-mediated signal transduction.
Seven transmembrane-domain receptors (7TMRs)2 mediate the actions of various extracellular sensory, hormonal, and metabolic stimuli (1). Among the signaling components coupled to the intracytosolic side of 7TMRs are the heterotrimeric G-proteins: molecular switches composed of a guanine nucleotide-binding Gα subunit and a Gβγ dimer that transduce 7TMR activation into intracellular modulation of multiple different effectors, including adenylyl cyclases, ion channels, cyclic nucleotide phosphodiesterases, and phospholipase C isoforms (2, 3). 7TMR-promoted activation of Gαβγ causes Gα to exchange the more abundant GTP for bound GDP, which in turn causes Gα·GTP and Gβγ to dissociate. Gα·GTP and Gβγ are then free to regulate effector systems that alter cell physiology (4, 5). This classical 7TMR-initiated G-protein nucleotide cycle is reset by intrinsic GTP hydrolysis activity possessed by the Gα subunit.
An evolutionarily conserved role for Gα subunits of the adenylyl cyclase inhibitory (Gαi) subfamily has recently been identified in the control of mitotic spindle orientation in cell divisions that generate cellular diversity during organismal development (6, 7). Studies of asymmetric cell division in Caenorhabditis elegans embryos and Drosophila melanogaster embryonic neuroblasts have identified initial steps of this process as generation of cell polarity and segregation of various cell fate determinants to different sides of the polarized cell (8); the mitotic spindle is then positioned to facilitate appropriate distribution of determinants to daughter cells during chromosomal segregation and cytokinesis. An integral part of the cellular machinery underlying accurate spindle positioning is the involvement of heterotrimeric G-protein Gα and Gβγ subunits in a manner considered independent of 7TMR activation and instead involving RIC-8 (a cytosolic guanine nucleotide exchange factor), GoLoco motif3 proteins (such as GPSM2/LGN, Pins, and GPR-1/2 that act as GDP dissociation inhibitors), and GTPase-accelerating proteins (“GAPs”; i.e. RGS proteins) (6-13). Vertebrate neuroepithelial progenitors use the same cellular machinery to modulate mitotic spindle orientation controlling the balance between asymmetric cell divisions that drive differentiation and planar divisions that favor maintenance and expansion of the neuroepithelial architecture (14-16). Similarly, an analogous mechanism appears to operate in the stratification and differentiation of mammalian skin (17).
An essential feature of the various emerging models of G-protein nucleotide cycling in mitotic spindle positioning is the requirement for a Gαi·GoLoco motif complex. For example, in our working model of C. elegans asymmetric cell division controlled by the Gα subunits GOA-1 and GPA-16 (18, 19), it is the Gα·GDP/GPR-1/2 complex that activates the generation of astral microtubule (MT) force on mitotic spindle poles, whereas in a competing model (3, 12, 20), the Gα·GDP/GoLoco motif complex is required for the nucleotide exchange (“GEF”) activity for RIC-8, thereby generating Gα·GTP as the presumed active form of the G-protein (12, 21, 22). However, it has not been formally established that the Gα/GoLoco motif interaction is required per se for the function of Gα subunits and GoLoco motif proteins in mitotic spindle positioning. For example, both models of C. elegans asymmetric cell division have been generated primarily by correlating various genetic phenotype data, including loss of pulling forces upon RNA interference-mediated knockdown of goa-1/gpa-16 or gpr-1/2 expression (9-11, 18, 19). These phenotypic results, although suggestive of a critical function for a Gα·GoLoco protein complex, might alternatively reflect separate and distinct functions of Gα subunits and the multidomain GPR-1/2 proteins in parallel pathways culminating in MT force generation, given that both classes of proteins have other binding partners and established functions. Furthermore, it remains unresolved as to whether Gβγ is an independent signaling entity in this system or merely a buffer of free Gα·GDP levels (14, 23, 24).
To provide a tool to address these questions, we sought to design a variant Gα subunit that will not interact with GoLoco motifs and yet retain wild type interactions with guanine nucleotides, 7TMRs, Gβγ subunits, Gα effectors, and RGS proteins. Here we describe and validate a single point mutation that renders Gαi subunits unable to bind GoLoco motif proteins, yet preserves all other aspects of Gα function. Furthermore, we use this GoLoco-insensitivity (“GLi”) mutation to demonstrate that direct Gα/GoLoco motif interaction is required for the Gα-dependent modulation of MT dynamics during mitotic spindle positioning.
EXPERIMENTAL PROCEDURES
Materials—All peptides were synthesized using Fmoc (N-(9-fluorenyl)methoxycarbonyl) group protection, high pressure liquid chromatography-purified, and validated by mass spectrometry at the Tufts University Core Facility (Medford, MA). Fluorescent guanine nucleotides were from Invitrogen. Anti-KT3 antibody MMS-125P was from Covance (Berkeley, CA). Unless elsewhere specified, all additional reagents were of the highest quality obtainable from Sigma or Fisher.
Molecular Biology—The expression vectors pcDNA3.1 human Gαi1 (Missouri Science and Technology cDNA Resource Center), pPROEXHTb human Gαi1 (25), pcDNA3.1 human Gαi1-KT3 (26), pCI rat Gαi1(C352G) (27), pCI rat Gαi2(C353G) (27), pCI rat Gαi3(C352G) (27), and pPROEXHTb GOA-1 (encoding aa 28-351) (9) were each subjected to site-directed mutagenesis to create N149I or N150I variants. All mutagenesis was performed using the QuikChange system (Stratagene, La Jolla, CA). The mammalian expression vectors pCI bovine Gβ1 and pCI bovine Gγ2 are described in Ref. 28, and pCI rat mGluR2 is described in Ref. 29. pK mammalian expression vectors and derivatives thereof (including venus yellow fluorescent protein (YFP) fusion (30) (pK-VENUS), monomeric red fluorescent protein (mRFP) fusion (31) (pK-mRFP), and 3× HA tag fusion (pK-HA3)), originated from the Macara laboratory (University of Virginia, VA) and are derived from pRK5 (BD Biosciences). pK-YFP-GPSM2 and pK-Gαi1-YFP are described in Ref. 32. Wild type and N149I pK-Gαi1-YFP, pK-Gαi1-mRFP, and pK-Gαi1-HA3 were made by PCR amplification of pcDNA3.1(Gαi1, wild type and N149I) and subcloning into the XbaI sites of pK-VENUS, pK-mRFP, and pK-HA3 respectively. To construct pK-GPSM1-YFP, mouse GPSM1 cDNA was PCR-amplified and subcloned into the BamHI/EcoRI sites of pK-VENUS. A pFLAG expression construct encoding the adenosine A2A receptor fused to venus-enhanced YFP is described in Ref. 33. C. elegans RGS-7 in pBluescript was provided by Pierre Gonczy (ISREC, Lausanne, Switzerland). DNA encoding the predicted minimal RGS domain of RGS-7 (aa 667-808 of RGS-7A (12)) was cloned into pPROEXHTb using heterostagger PCR (34). All DNA constructs were verified by DNA sequencing.
Protein Purification—GST fusion proteins were purified to homogeneity using standard methods (34, 35). The GST-GoLoco motif fusion proteins purified were rat GPSM1(GL1234, aa 361-650 (36)), human GPSM2 (GL1234, aa 481-657 (37)), human GPSM3/G18 (GL123, aa 61-160 (26)), human PCP-2/GPSM4 (GL12, full-length (38)), rat RGS12 (aa 1184-1228 (25)), rat RGS14 (aa 496-531 (39)), Rap1GAP1a (aa 1-34 (40)), and Rap1GAP1b (aa 25-65 (40)) (see also supplemental Fig. S1 for a graphical representation). Gα subunits were purified to homogeneity using previously described methods, including the removal of His6 tags by tobacco etch virus protease cleavage (25, 34, 35). The specific activities of wild type and N149I Gαi1 were determined using [35S]GTPγS binding (mean ± S.E. of mol of GTPγS bound per mol of Gαi1) as follows: wild type, 0.93 ± 0.02; N149I 0.93 ± 0.02. C. elegans RGS-7, also with its His6 tag removed, was purified to homogeneity using methods standard for other RGS domains (41).
Surface Plasmon Resonance—Surface plasmon resonance analysis of GoLoco motif/Gα interactions was conducted as described in Refs. 25, 38.
Fluorescence Anisotropy—Fluorescence anisotropic assays of Gα binding to FITC-labeled GoLoco motif peptides was conducted as described in Ref. 42 for Fig. 2 and Fig. 7 and as described in Ref. 40 for Fig. 3. A minor modification was the use of a 5 nm final concentration of the FITC-RGS14, FITC-RGS12, FITC-GPSM2(GL2), and FITC-KB-1753 peptides. FITC-RGS12 is described in Ref. 42. FITC-GPSM2(GL2) is described in Ref. 40. FITC-KB-1753 is described in Ref. 43. The FITC-RGS14 peptide included amino acids 496-531 of rat RGS14 (FITC-β-alanine-S-DIEGLVELLNRVQSSGAHDQRGLLRKEDLVLPEFLQ-NH2). Anisotropy data are presented as millipolarization units (mP) following data analysis as described in Ref. 42.
FIGURE 2.

Asparagine 149 is an evolutionarily conserved
Gαi/o class-specific amino acid that is crucial for
GoLoco motif interaction. A, multiple sequence alignment of the
αD/αE loops within the all α-helical domains of human
(Hs)Gα subunits. Sequences are grouped into the four classical
Gα subclasses (s, i/o, q, and 12/13). C. elegans (Ce)
and D. melanogaster (Dm)Gα subunits known to interact
with GoLoco motifs are also included in the alignment. SwissProt or
GenBank™ accession numbers are as follows: human Gαs,
P63092; human Gαolf, P38405; human GαT1,
P11488; human GαT2, P19087; human GαT3,
NP_001095856; human GαZ, P19086; C. elegans GOA-1,
P51875; D. melanogaster Gαo, P16378; human
Gαi1, P63096; human Gαi2, P04899; human
Gαi3, P08754; C. elegans GPA-16, Q60XS3; D.
melanogaster Gαi, P20353; human Gαo, P59215; human
Gαq, P50148; human Gα14, O95837; human
Gα15, P30679; human Gα12, Q03113; human
Gα13, Q14344. B, surface plasmon resonance was used
to measure interactions between antibody-immobilized RGS14 GoLoco motif GST
fusion protein and either GDP-bound or
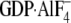 bound Gα
subunits. Injections of either 1 μm Gαi1(wild
type) or 10 μm Gαi1(N149I) were used. Binding
curves were obtained by subtracting nonspecific binding to GST alone surfaces.
C, affinity of wild type and N149I Gαi1 proteins for
the RGS14 GoLoco motif was measured using fluorescence anisotropy. 5
nm FITC-RGS14(GoLoco motif) peptide was mixed with increasing
amounts of Gαi1 proteins in the presence of either GDP or
bound Gα
subunits. Injections of either 1 μm Gαi1(wild
type) or 10 μm Gαi1(N149I) were used. Binding
curves were obtained by subtracting nonspecific binding to GST alone surfaces.
C, affinity of wild type and N149I Gαi1 proteins for
the RGS14 GoLoco motif was measured using fluorescence anisotropy. 5
nm FITC-RGS14(GoLoco motif) peptide was mixed with increasing
amounts of Gαi1 proteins in the presence of either GDP or
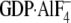 , and equilibrium
fluorescence anisotropy was measured (expressed as millipolarization units
(mP) as described in Ref.
42). Dissociation constants
were determined by nonlinear regression: wild type
Gαi1·GDP (14 ± 1 nm). Dissociation
constants for wild type
Gαi1·
, and equilibrium
fluorescence anisotropy was measured (expressed as millipolarization units
(mP) as described in Ref.
42). Dissociation constants
were determined by nonlinear regression: wild type
Gαi1·GDP (14 ± 1 nm). Dissociation
constants for wild type
Gαi1·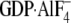 ,
N149I Gαi1·GDP, and N149I
Gαi1·
,
N149I Gαi1·GDP, and N149I
Gαi1·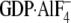 could not be accurately determined as calculated values were <50% of S.E.M.
D, GDI effect of RGS14 GoLoco motif binding on
Gαi1·GDP was quantified using fluorescence
spectroscopy. The fluorescence of 1 μm BODIPYFL-GTPγS was
measured alone (blue trace) or in the presence of 200 nm
Gαi1(N149I) (red trace), 200 nm
Gαi1(N149I) + 1 μm RGS14 (black
trace), or 200 nm Gαi1(N149) + 5
μm RGS14 (green trace). E, concentration
dependence of RGS14 GoLoco motif GDI activity was measured by quantifying the
initial rates of BODIPYFL-GTPγS binding to 200 nm wild type
Gαi1(blue) or Gαi1(N149I)
(red) in the presence of increasing amounts of RGS14 GoLoco motif.
IC50 values were determined by nonlinear regression (95% confidence
intervals in parentheses): wild type Gαi1, 243
(190-310) nm; N149I Gαi1, 490 (220-1000)
μm. RU, resonance units.
could not be accurately determined as calculated values were <50% of S.E.M.
D, GDI effect of RGS14 GoLoco motif binding on
Gαi1·GDP was quantified using fluorescence
spectroscopy. The fluorescence of 1 μm BODIPYFL-GTPγS was
measured alone (blue trace) or in the presence of 200 nm
Gαi1(N149I) (red trace), 200 nm
Gαi1(N149I) + 1 μm RGS14 (black
trace), or 200 nm Gαi1(N149) + 5
μm RGS14 (green trace). E, concentration
dependence of RGS14 GoLoco motif GDI activity was measured by quantifying the
initial rates of BODIPYFL-GTPγS binding to 200 nm wild type
Gαi1(blue) or Gαi1(N149I)
(red) in the presence of increasing amounts of RGS14 GoLoco motif.
IC50 values were determined by nonlinear regression (95% confidence
intervals in parentheses): wild type Gαi1, 243
(190-310) nm; N149I Gαi1, 490 (220-1000)
μm. RU, resonance units.
FIGURE 7.

GoLoco-insensitive Gαil has normal interactions
with the effector adenylyl cyclase and the effector-mimetic peptide
KB-1753. A, affinity of wild type and N149I Gαi1
proteins for the Gα-effector mimetic peptide KB-1753
(43) was measured using
fluorescence anisotropy. 5 nm FITC-KB-1753 peptide was mixed with
increasing amounts of Gαi1 proteins, and equilibrium
fluorescence anisotropy was measured. Data are presented as the mean ±
S.E.M. of triplicate determinations. Dissociation constants were determined by
nonlinear regression: wild type Gαi1·GDP (24.1
± 4 μm), wild type
Gαi1·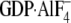 (294 ± 40 nm), N149I Gαi1·GDP (13.0
± 2 μm), N149I
Gαi1·
(294 ± 40 nm), N149I Gαi1·GDP (13.0
± 2 μm), N149I
Gαi1·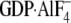 (311 ± 40 nm). B, cells were transiently
transfected with cDNA encoding Gαi1(Q204L,C352G),
Gαi1(N149I,Q204L,C352G), or pcDNA3.1(+) as a vector control,
with the adenosine A2A receptor. Cyclic AMP accumulation was
stimulated with 1 μm MECA for 15 min at 37 °C. Data
represent the mean ± S.E.M. of four independent experiments in
duplicate. *, p < 0.05; **, p <
0.01 compared with A2A-R + empty vector transfection under matched
stimulation (basal or MECA), one-way analysis of variance followed by
Dunnett's post hoc test.
(311 ± 40 nm). B, cells were transiently
transfected with cDNA encoding Gαi1(Q204L,C352G),
Gαi1(N149I,Q204L,C352G), or pcDNA3.1(+) as a vector control,
with the adenosine A2A receptor. Cyclic AMP accumulation was
stimulated with 1 μm MECA for 15 min at 37 °C. Data
represent the mean ± S.E.M. of four independent experiments in
duplicate. *, p < 0.05; **, p <
0.01 compared with A2A-R + empty vector transfection under matched
stimulation (basal or MECA), one-way analysis of variance followed by
Dunnett's post hoc test.
FIGURE 3.

N149I substitution is a loss-of-function Gα mutation for
all mammalian GoLoco motifs. The universality of the Gαi1
N149I mutation was analyzed by surface plasmon resonance. GST fusion proteins
of the GoLoco motifs of GPSM1(GL1,2,3,4) (A); GPSM2(GL1,2,3,4)
(B); GPSM3(GL1,2,3) (C); PCP-2(GL1,2) (D); RGS12
(E); Rap1GAP1a (F); and Rap1GAP1b (G) were
immobilized on SPR biosensor surfaces. 1 μm wild type
Gαi1·GDP (blue), 1 μm wild type
Gαi1·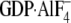 (red), 10 μm N149I Gαi1·GDP
(green), and 10 μm N149I
Gαi1·
(red), 10 μm N149I Gαi1·GDP
(green), and 10 μm N149I
Gαi1·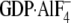 (black) were separately injected over biosensor surfaces. Binding
curves were obtained by subtracting nonspecific binding to GST alone.
H, affinity of wild type and N149I Gαi1 proteins for
GPSM2(GL2) was measured using fluorescence anisotropy. 5 nm
FITC-GPSM(GL2) peptide was mixed with increasing amounts of
Gαi1 proteins, and equilibrium fluorescence anisotropy was
measured. Data are expressed as millipolarization units as described in Ref.
42. Dissociation constants
were determined by nonlinear regression as follows: wild type
Gαi1·GDP (150 ± 20 nm) and N149I
Gαi1·GDP (>99 μm). I,
affinity of wild type and N149I Gαi1 proteins for the RGS12
GoLoco motif was measured using fluorescence anisotropy. 5 nm
FITC-RGS12 peptide was mixed with increasing amounts of Gαi1
proteins and equilibrium fluorescence anisotropy was measured. Data are
expressed as millipolarization units as described in Ref.
42. Dissociation constants
were determined by nonlinear regression: wild type
Gαi1·GDP (44 ± 6 nm), N149I
Gαi1·GDP (9.3 ± 0.5 μm).
RU, resonance units.
(black) were separately injected over biosensor surfaces. Binding
curves were obtained by subtracting nonspecific binding to GST alone.
H, affinity of wild type and N149I Gαi1 proteins for
GPSM2(GL2) was measured using fluorescence anisotropy. 5 nm
FITC-GPSM(GL2) peptide was mixed with increasing amounts of
Gαi1 proteins, and equilibrium fluorescence anisotropy was
measured. Data are expressed as millipolarization units as described in Ref.
42. Dissociation constants
were determined by nonlinear regression as follows: wild type
Gαi1·GDP (150 ± 20 nm) and N149I
Gαi1·GDP (>99 μm). I,
affinity of wild type and N149I Gαi1 proteins for the RGS12
GoLoco motif was measured using fluorescence anisotropy. 5 nm
FITC-RGS12 peptide was mixed with increasing amounts of Gαi1
proteins and equilibrium fluorescence anisotropy was measured. Data are
expressed as millipolarization units as described in Ref.
42. Dissociation constants
were determined by nonlinear regression: wild type
Gαi1·GDP (44 ± 6 nm), N149I
Gαi1·GDP (9.3 ± 0.5 μm).
RU, resonance units.
Nucleotide Binding and Hydrolysis Assays—[35S]GTPγS binding and [γ-32P]GTP hydrolysis assays were conducted as described in Ref. 9, 44. [35S]GTPγS binding was used to measure GPR-1/2-mediated GDI activity on GOA-1 as described in Refs. 40, 44. The GPR-1/2 peptide (aa 423-461) is described in Refs. 9, 42. BODIPYFL-GTPγS binding assays were used to quantify GoLoco motif-promoted Gαi1 GDI activity, as described previously (38). RGS domain-mediated acceleration of GTP hydrolysis by 200 nm GOA-1 was measured using 100 nm BODIPYFL-GTP as described (34).
Dissociation of Superior Cervical Ganglion and cDNA Microinjection—Detailed methods of preparing rat superior cervical ganglion (SCG) neurons and cDNA microinjection were described previously (27). In brief, adult male Wistar rats were anesthetized by CO2 inhalation and decapitated as approved by the Institutional Animal Care and Use Committee. Superior cervical ganglia were digested within modified Earle's balanced salt solution containing 0.6 mg/ml collagenase, 0.3 mg/ml trypsin, and 0.05 mg/ml DNase I for 1 h in a shaking water bath at 36 °C under an atmosphere of 5% CO2, 95% O2. The dissociated cells were then washed and plated on poly-l-lysine-coated tissue culture dishes containing minimum essential medium and 10% (v/v) fetal calf serum. After cDNA injection, the neurons were incubated overnight at 37 °C, and electrophysiology experiments were performed the next day. For some experiments, neurons were incubated overnight with 500 ng/ml Bordetella pertussis toxin (PTX, List Biological Laboratories, Campbell, CA). Microinjection of cDNA was performed with an Eppendorf FemtoJet microjector and 5171 micromanipulator (Eppendorf, Madison, WI) using custom designed software. Constructs containing inserts coding for mGluR2, Gαi1(N149I, C352G), Gαi2(N150I, C353G), Gαi3(N149I, C352G), Gαi3(C352G), Gβ1, and Gγ2 were stored at -20 °C as 0.6-1 μg/μl stock solutions in TE buffer (10 mmTris, 1 mm EDTA, pH 8) and injected at a final concentration of 0.1 μg/μl.
Electrophysiolgical Studies—The method of recording whole cell Ca2+ currents from rat SCG neurons with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) was described in detail previously (27, 45). Patch electrodes were fire-polished to final resistances of ∼2 megohms when filled with internal solution. Uncompensated series resistance was <5 megohms and electronically compensated ∼80%. Voltage protocol generation and data acquisition were performed using the custom-designed software S5. Current traces were filtered at 2 kHz and digitized at 10 kHz. All recordings were performed at room temperature (21-24 °C).
Electrophysiology Solutions and Chemicals—The external solution consisted of the following (in mm): 140 methanesulfonic acid, 145 tetraethylammonium hydroxide (TEA-OH), 10 HEPES, 10 glucose, 10 CaCl2, and 0.0003 tetrodotoxin, pH 7.4, with TEA-OH. The internal solution contained the following (in mm): 120 N-methyl-d-glucamine, 20 TEA-OH, 11 EGTA, 10 HEPES, 10 sucrose, 1 CaCl2, 4 MgATP, 0.3 Na2GTP, and 14 Tris creatine phosphate, pH 7.2, with methanesulfonic acid. The osmolalities of the external and internal solutions were adjusted with sucrose to 325 and 300 mosmol/kg, respectively. All drug and control solutions were applied to neurons via a custom-designed gravity-driven perfusion system as described previously (45).
Cyclic AMP Accumulation Assay—HEK 293 cells stably expressing the rat D2L dopamine receptor (46) were propagated in Dulbecco's modified Eagle's medium supplemented with 5% (v/v) bovine calf serum, 5% (v/v) FetalClone 1 serum (Thermo Fisher, Waltham, MA), 1 unit/ml penicillin, 1 μg/ml streptomycin, 2.5 ng/ml amphotericin B, and 2 μg/ml puromycin and maintained in a humidified incubator at 37 °C and 6% CO2. Cells were seeded into 24-well cluster plates and, upon reaching ∼80% confluence, were transiently transfected with 200 ng of pcDNA3.1(+), pCI rat Gαi1(Q204L, C352G), or Gαi1(N149I, Q204L, C352G) together with 50 ng of pFLAG-YFP-A2A using Lipofectamine 2000 reagent (Invitrogen, 1 μl/well). At 24 h post-transfection, cAMP accumulation assays were carried out on ice in Earle's balanced salt solution containing 15 mm Na+-HEPES, 2% bovine calf serum, and 0.02% ascorbic acid following a 5-min preincubation in assay buffer. Cyclic AMP was stimulated by activation of the adenosine A2A receptor with the agonist 5-N-methylcarboxamidoadenosine (MECA, 1 μm) at 37 °C for 15 min in the presence of the phosphodiesterase inhibitor 4-(3-butoxy-4-methoxybenzyl)imidazolidin-2-one (Ro-20-1724, 100 μm). The stimulation medium was decanted, and the reaction was terminated by addition of ice-cold 3% trichloroacetic acid. The plate was stored at 4 °C for at least 1 h before cAMP quantification. Cyclic AMP was quantified using a competitive binding assay (47).
GPSM3 Membrane Recruitment Experiments—Similar to the transmembrane domain-anchored Gα subunits described in Ref. 48, a pcDNA3.1-based mammalian expression vector was generated to encode CFP-TM-Gαoi subunits consisting of (starting at the N terminus) a signal peptide, enhanced CFP (49), the N-terminal 103 amino acids of the rat μ-opioid receptor, the N-terminal 33 amino acids of human GαoA, and amino acids 34-354 of human Gαi1. A pcDNA3.1-based mammalian expression vector was generated to encode YFP-GPSM3 consisting of the venus variant of enhanced YFP (30), a c-Myc epitope tag, a hexahistidine tag, and human GPSM3 fused inframe (derived from pcDNA3.1mycHis human GPSM3 (26)).
Human embryonic kidney 293 cells (ATCC; Manassas, VA) were propagated in plastic flasks and seeded onto polylysine-coated glass coverslips according to the supplier's protocol. Cells were transfected using polyethyleneimine and were used for experiments 12-48 h later. Coverslips bearing transfected cells were imaged using a Leica (Bannockburn, IL) SP2 scanning confocal microscope and a 63×, 1.4 NA objective; cells were excited using 458 nm (for CFP) or 514 nm (for YFP) laser lines. Images were acquired and analyzed by an experimenter who was blinded to the transfection condition. A 5-μm profile drawn normal to, and centered on, the plasma membrane was obtained for each cell.
MDCK Cell and Spindle Rocking Experiments—MDCK II cells were cultured, transfected, and processed for imaging as described (32). MDCK cells were transfected with either wild type or N149I Gαi1-YFP. Time-lapse images of Hoechst 33342-stained chromosomal DNA condensation and segregation were recorded as described (32). The angles of the long axis of the metaphase chromosomal array in each frame were measured using Metamorph software (Molecular Devices, Sunnyvale, CA). The absolute angle changes of at least 100 sets of adjacent frames were binned into <5°, 5-10°, and >10° groups.
Co-immunoprecipitation—COS-7 cell culture, transfection, and immunoprecipitation was performed as described (32).
Statistics and Curve Fitting—Unless otherwise indicated, data analysis and curve fitting were performed using PRISM version 4.0 (GraphPad; San Diego). All data are representative of three or more independent experiments.
Structural Analysis of the GoLoco Motif/Gα Interaction—The crystallographic structure of Gαi1·GDP bound to the GoLoco motif of RGS14 has been determined at 2.7 Å resolution (PDB code 1KJY (39)), and more recently at 2.2 Å resolution (PDB code 2OM2 (50)). The two structures share the same overall global architecture; however, there are appreciable differences between the two structural models (50). In this study, we confined our analysis to the 2OM2 structure, as it has higher overall resolution and better refinement statistics. Similarly, subtle but discrete differences exist between the two asymmetric units in both 2OM2 and 1KJY structures; for this reason, we have generally confined our analysis to the A and B chains of 2OM2 as the refinement of this asymmetric unit appeared superior. PyMol (DeLano Scientific; Palo Alto, CA) was used for analysis of structures and the generation of images. Amino acid interaction data were derived using SPACE(CMA) (51) and plotted using MATLAB (The MathWorks, Natick, MA).
RESULTS AND DISCUSSION
Rational Design of a Loss-of-Function Point Mutation in Gαi to Prevent GoLoco Motif Interaction—A major feature of the Gαi1·GDP/RGS14 GoLoco motif complex (39, 50) consists of the GoLoco motif N terminus forming an α-helix that binds in the pocket formed by the α2 helix (“switch II”) and the α3 helix of the Ras-like domain of Gαi1 (Fig. 1A). The invariant glutamine residue (Gln515) of the GoLoco motif (D/E)QR triad terminates this helical portion of the GoLoco motif. The GoLoco motif peptide continues to transit across the surface of Gαi1 to contact the all α-helical domain of Gαi1. The conserved Asp514-Gln515-Arg516 triad is responsible for turning the GoLoco motif peptide and positioning Arg516 into the nucleotide binding pocket of Gαi1 so that the Arg516 side chain is able to make direct contact with the α- and β-phosphates of GDP, thus stabilizing the bound nucleotide and conferring GDI activity (39). The C-terminal segment of the GoLoco motif makes a sharp hydrogen-bonded turn (Lys521-Glu522-Asp523-Leu524) as it enters the central grove between the αA- and αB-helices of the G-protein helical domain where it makes an extensive network of contacts. The RGS14 GoLoco motif culminates in a short 310 helix at amino acids Glu528-Phe529-Leu530. Thus, the main points of contact between Gα and the GoLoco motif are in the switch II/α3 helix pocket, switch I, the helical domain (αA and αB helices), the phosphate binding “P-loop,” and in the αD/αE loop of the helical domain. A comprehensive analysis of these Gα/RGS14 GoLoco motif interactions is reported in a contact map in supplemental Fig. S2.
FIGURE 1.

Structural analysis of the function of Gαil residue asparagine 149 in mediating the interaction between Gαi1·GDP and the GoLoco motif of RGS14. Ribbon diagram of the RGS14 GoLoco motif (blue) bound to Gαi1·GDP (yellow). Gα residues are annotated in red; GoLoco motif residues are annotated in blue, and bound GDP is colored magenta. A, overall view of the complex highlighting the position of the αD/αE helix. Asn149 is highlighted in stick format (red). B, enlarged view of the Gα/GoLoco motif interaction interface. The side chain oxygen of Gln515 forms a hydrogen bonding network with the side chain and backbone amine of Asn149. C, enlarged view of intramolecular interactions of Asn149. The side chain amine of Asn149 forms a hydrogen bond with the side chain oxygen of Ser80. Asn149 also makes several stabilizing contacts with Arg178, predominantly between the α-carbon of Asn149 and the ζ-carbon of Arg178.
Based on these structural data (39, 50), it is not intuitively obvious how to create a loss-of-function point mutation(s) in a Gαi subunit to abrogate GoLoco motif binding yet retain wild type nucleotide binding and hydrolysis, receptor coupling, and effector activation. Many of the GoLoco motif contact sites on Gα are also used by other regulatory proteins (2, 39); this is especially true for the main sites of GoLoco motif contact, e.g. switch-II/α3 helix, which is essential for Gβγ, effector, and RGS domain interactions (2). Initial structure/function studies on the molecular determinants of the Gα/GoLoco interaction indicated that the αD/αE loop of the Gαi1 helical domain was essential for binding (52). In testing various Gαi1/Gαs chimeric proteins, amino acids 144-151 in the αD-αE loop were identified by Natochin et al. (52) as one determinant responsible for the inability of Gαs to interact with GoLoco motifs. The three residues that differ between Gαi1 and Gαs in the αD-αE loop are Arg144 to Asn, Asn149 to Ile, and Ser151 to Cys (illustrated in Fig. 2A). Examination of the Gαi1·GDP/RGS14 GoLoco motif structure indicates that, of these three residues, only Asn149 of Gαi1 makes contact with the GoLoco motif (Fig. 1B and supplemental Fig. S1). Asn149 isaGαi/o class-specific residue (Fig. 2A) and therefore also a good candidate for a GoLoco motif interaction loss-of-function mutation. We hypothesized that a single point mutation of N149I would be sufficient to selectively abrogate GoLoco motif binding to Gαi1 and create a GoLoco-insensitive (GLi) Gα subunit. We tested this hypothesis using multiple independent techniques.
Asn149 to Ile Mutation in Gαil Prevents Interaction with GoLoco Motifs in Vitro—Mutation of the Gαi1 amino acid Asn149 to isoleucine dramatically attenuated GDP-dependent binding of Gαi1 to the affinity of Gαi1·GDP for the RGS14 GoLoco motif by 300-fold (Fig. 2C). The signature biochemical activity of GoLoco motifs is GDI activity (7). We therefore also tested GDI activity of the RGS14 GoLoco motif on wild type and N149I Gαi1 (Fig. 2, D and E). We were unable to observe significant GoLoco motif-mediated GDI activity using Gαi1(N149I), whereas wild type Gαi1 was a substrate for RGS14 GoLoco motif GDI activity in the nanomolar range (Fig. 2, D and E), as observed previously (25).
To test the universality of this GoLoco-insensitivity point mutation, we analyzed the binding of N149I Gαi1 to all human GoLoco motifs using SPR. A graphical representation of all known GoLoco motif proteins, as well as the purified protein constructs used in these SPR analyses, is presented in supplemental Fig. S2.4 Wild type Gαi1 (at 1 μm) exhibited robust, GDP-selective binding to all known GoLoco motifs (Fig. 3, A-G); at a 10-fold higher concentration, N149I Gαi1 did not demonstrate any binding to GPSM2 (Fig. 3B) nor to PCP-2 (Fig. 3D). Low but measurable levels of N149I Gαi1 binding were observed on immobilized surfaces of GPSM1 (Fig. 3A), GPSM3 (Fig. 3C), and the GoLoco motifs of RGS12 (Fig. 3E), Rap1GAP1a (Fig. 3F), and Rap1GAP1b (Fig. 3G). We used fluorescence anisotropy to further quantify the binding of GPSM2(GL2) and RGS12GL to Gαi1(N149I). Calculated KD values for Gαi1(N149I) versus wild type suggest that the GLi mutation reduces affinity for GoLoco motifs by at least 700- and 200-fold, respectively (Fig. 3, H and I).
In Vitro Biochemical Properties of GoLoco-insensitive Gα Subunits—Mutations in Gα subunits can alter nucleotide binding, nucleotide hydrolysis, and interaction with regulatory proteins (53). We therefore wanted to test whether the GLi mutation may have also altered the nucleotide binding and/or hydrolysis properties of Gα subunits. Using [35S]GTPγS binding, we observed that wild type and N149I Gαi1 have equivalent nucleotide exchange rates (Fig. 4A). The nucleotide exchange rate as assayed by [35S]GTPγS binding is an indirect measure of spontaneous GDP release, and thus an index of Gα affinity for GDP (54). Similarly, we used single turnover [32P]GTP hydrolysis assays to measure the catalytic rate of GTPase activity for wild type and N149I Gαi1 (Fig. 4B). We observed no significant difference in the ability of wild type or N149I Gαi1 to hydrolyze GTP. Finally, we constructed the GLi mutation (N150I) in the C. elegans Gαo-like G-protein GOA-1, known to functionally interact with GoLoco motifs to regulate asymmetric cell division in the one-cell embryo (9). We measured RGS-75-mediated acceleration of GTP hydrolysis by GOA-1. We observed that the N150I mutation had no appreciable effect on the ability of RGS-7 to stimulate the GTPase activity of GOA-1 in a dose-dependent fashion (Fig. 4C). Additionally, we verified that GOA-1(N150I) is indeed resistant to GoLoco motif-mediated GDI activity (Fig. 4D), suggesting that the Asn to Ile mutation can be transferred across species, consistent with the conserved evolutionary relationships among metazoan G-proteins and GoLoco motif proteins (6, 7).
FIGURE 4.

Biochemical properties of GoLoco-insensitive Gα subunits. A, spontaneous nucleotide exchange rates (kex) of wild type (black) and N149I (gray)Gαi1·GDP were measured. A time course of specific binding of 100 nm Gα subunit to 1 μm GTPγS was determined using an [35S]GTPγS filter-binding assay. Data were fit to single exponential functions with rate constants as follows: wild type Gαi1 0.013 ± 0.002 min-1 and N149I Gαi1 0.009 ± 0.0004 min-1. B, spontaneous GTP hydrolysis rates (kcat) of wild type (black) and N149I (gray)Gαi1 were measured using [γ-32P]GTP hydrolysis assays. A time course of 32Pi (inorganic phosphate) production was determined using activated charcoal filtration. Data were fit to single exponential functions with rate constants as follows: wild type Gαi1 0.40 ± 0.003 min-1 and N149I Gαi1 0.30 ± 0.003 min-1. C, GTPase-accelerating protein (GAP) activity of C. elegans RGS7 on 200 nm wild type and N150I-mutated C. elegans GOA-1 was measured using 100 nm BODIPYFL-GTP and fluorescence spectroscopy. Data were fit to the four parameter logistic equation to determine EC50 values of RGS-7 GAP activity (95% confidence intervals in parentheses) as follows: wild type GOA-1, 830 (570-1300) nm; N149I GOA-1, 670 (580-790) nm. D, GDI effect of the C. elegans GPR-1/2 GoLoco motif on C. elegans GOA-1 was quantified using [35S]GTPγS filter binding. Time courses were obtained by preincubating 100 nm GOA-1 (wild type or N150I) with either buffer or 10 μm GPR-1/2 GoLoco motif peptide for 5 min. Samples were then added to 1 μm GTPγS, and specific [35S]GTPγS binding was quantified by filtration and scintillation counting. Data were fit to exponential association functions (95% confidence intervals in parentheses) as follows: wild type GOA-1 alone, 0.202 (0.160-0.240) min-1; wild type GOA-1 + GoLoco peptide, 0.068 (0.055-0.081) min-1; N150I GOA-1 alone, 0.178 (0.150-0.210) min-1; N150I GOA-1 + GoLoco peptide, 0.194 (0.140-0.250) min-1.
Functional Properties of the GoLoco-insensitive Gα—Ca2+ channel modulation in sympathetic neurons was used to examine the ability of GoLoco-insensitive Gα mutants to complex with endogenous Gβγ subunits. N-type Ca2+ channels respond to both tonic and 7TMR-mediated G-protein activation with a characteristic voltage-dependent modulation mediated by Gβγ subunits (55). Ca2+ channel currents in whole-cell voltage-clamped neurons were evoked with a double-pulse voltage protocol consisting of two 25-ms test pulses to +10 mV separated by a depolarizing conditioning pulse to +80 mV (56). In control neurons under basal conditions (Fig. 5A, open circle), the amplitude during the first test pulse (prepulse) is slightly smaller than that evoked by the second test pulse (postpulse) resulting in a mean facilitation ratio (postpulse/prepulse amplitude) greater than 1 (Fig. 5F, open bar). Basal (in the absence of agonist) facilitation has been shown to arise from tonic modulation by Gβγ subunits (28, 57). Application of norepinephrine (NE, 10 μm) activates endogenous α2-adrenergic receptors, resulting in a large inhibition of prepulse amplitude (Fig. 5A, filled circle; Fig. 5E, open bar) and changes in current kinetics (slowing) and facilitation ratio characteristic of Gβγ modulation (55). Heterologous expression of GLi Gα subunits (Fig. 5, B-D) abolished both tonic and agonist-mediated modulation as indicated by decreases in mean basal facilitation (Fig. 5F) and agonist-mediated inhibition of the prepulse amplitude (Fig. 5E). The decreases were comparable with those produced by heterologous expression of Gαi3(C352G) (i.e. lacking the GLi mutation). These results indicate that GLi Gαi subunits are capable of binding constitutive Gβγ subunits and buffering Gβγ “released” from heterotrimers activated by receptor stimulation (58).
FIGURE 5.

GoLoco-insensitivity mutation does not alter the ability of Gαi to buffer free Gβγ subunits. A-D, superimposed Ca2+ current traces evoked with a double-pulse voltage protocol in the absence (open circle) or presence of 10 μm NE (filled circle) from control (A), Gαi1(N149I, C352G) (B), Gαi2(N150I, C353G) (C), and Gαi3(N149I, C352G) (D) expressing SCG neurons. Currents were evoked every 10 s. The dashed lines indicate the zero current level. E, summary graph of Ca2+ current inhibition by 10 μm NE from control neurons and neurons expressing Gαi1(N149I,C352G), Gαi2(N150I,C353G), or Gαi3 (N149I,C352G). Ca2+ current inhibition was measured 10 ms after initiation of the test pulse (+10 mV) in the absence or presence of 10 μm NE. F, basal facilitation from control neurons or neurons expressing Gαi1(N149I,C352G), Gαi2(N150I,C353G), or Gαi3(N149I,C352G). Basal facilitation was calculated as the ratio of Ca2+ current amplitude determined from the test pulse (+10 mV) occurring after and before the +80 mV conditioning pulse. E and F, bars represent mean±S.E.M. Numbers in parentheses indicate the number of neuron tested. The mean for all experimental conditions (colored bars) was different (p < 0.05) from the control condition (open bar) as determined by one-way analysis of variance followed by Neuman-Keuls multiple comparison test. Means among experimental groups were not different.
The ability to bind Gβγ subunits does not establish whether the GLi Gα subunits are capable of forming functional heterotrimeric complexes. Thus, to examine directly the ability of GLi Gα subunits to form functional heterotrimers, a reconstitution assay was employed that utilizes a PTX-resistant mutation (C-terminal cysteine to glycine mutation or “CG”) to distinguish responses arising from endogenous (PTX-sensitive) versus heterologously expressed Gα-containing heterotrimers (27). Expression of the 7TMR metabotropic glutamate receptor (mGluR2) in sympathetic neurons renders Ca2+ channels sensitive to application of glutamate (Fig. 6A). The expressed mGluR2 receptors couple to endogenous Gi/o family heterotrimers as indicated by the near complete sensitivity of the voltage-dependent inhibition to PTX pretreatment (Fig. 6B). Co-expression of a PTX-insensitive Gαi3 mutant along with Gβ1 and Gγ2 reconstituted the response in PTX-treated neurons (Fig. 6C) (29). Similarly, the GoLoco- and PTX-resistant mutant Gαi3(N149I, C352G) was capable of reconstituting the response to glutamate following PTX treatment (Fig. 6D). Fig. 6E depicts the prepulse Ca2+ current inhibition for individual neurons as well as the median response. The magnitude of inhibition varies for the PTX-resistant mutants as the stoichiometric balance of Gα and Gβγ influences the response (59). These results demonstrate that the Gαi3(N149I,C352G) double mutant is competent to form a Gαβγ heterotrimer that couples to mGluR2 and “releases” Gβγ upon receptor activation.
FIGURE 6.

GoLoco-insensitive Gαi3 reconstitutes a functional heterotrimer that couples glutamate receptor activation to Ca2+ channel inhibition. A and B, superimposed Ca2+ current traces evoked with the double-pulse voltage protocol in the absence or presence of 100 μm glutamate from SCG neurons heterologously expressing the metabotropic glutamate receptor mGluR2 without (A) or with PTX pretreatment (B). C and D, superimposed Ca2+ current traces evoked with the same double-pulse protocol in the absence and presence of 100 μm glutamate from SCG neurons heterologously expressing mGluR2, Gαi3(C352G), Gβ1γ2 (C) or mGluR2, Gαi3(N149I,C352G), Gβ1γ2 (D) after pretreatment with PTX. E, summary graph of Ca2+ current inhibition by 100 μm glutamate from neurons expressing different combinations of constructs as described above. Solid line represents the intragroup medians. The median inhibition for the mutant Gα reconstitution conditions differed significantly (p < 0.05) from the PTX-treated condition (Kruskal-Wallis test). Numbers in parentheses indicate the number of neuron tested.
Normal Gα Effector Modulation by GoLoco-insensitive
Gα Subunits—Our demonstration that GLi Gα can
functionally couple to G-protein-coupled receptors and Gβγ
effectors does not preclude the possibility that the GLi mutation is in some
way deleterious to Gα effector interactions. To address this
possibility, we measured the interaction of Gαi1 with the
Gα effector mimetic peptide KB-1753. KB-1753 interacts selectively with
activated Gαi subunits (i.e. GTPγS and
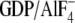 forms but not GDP-bound
Gα) in an effector-like conformation by binding to switch-II of
Gαi (43). We
measured the binding of wild type and GLi Gαi1 to KB-1753
using fluorescence anisotropy. Binding of both proteins to FITC-KB-1753 was
selective for the activated
(
forms but not GDP-bound
Gα) in an effector-like conformation by binding to switch-II of
Gαi (43). We
measured the binding of wild type and GLi Gαi1 to KB-1753
using fluorescence anisotropy. Binding of both proteins to FITC-KB-1753 was
selective for the activated
(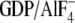 ) conformation of Gα
(Fig. 7A) and similar
in magnitude (KD values as follows: wild type, 294
± 40 nm; GLi, 311 ± 40 nm).
) conformation of Gα
(Fig. 7A) and similar
in magnitude (KD values as follows: wild type, 294
± 40 nm; GLi, 311 ± 40 nm).
The ability of the GoLoco-insensitive Gαi1 mutant to inhibit the Gα effector adenylyl cyclase was assessed by measuring the inhibition of agonist-stimulated cAMP accumulation in cells co-expressing constitutively active Gαi1(Q204L) subunits and the A2A adenosine receptor. Co-transfection of Gαi1(Q204L) inhibited MECA-stimulated cAMP accumulation by more than 50% when compared with cells co-transfected with the vector control (Fig. 7B). Cells co-expressing Gαi1(N149I,Q204L) also reduced MECA-stimulated cAMP accumulation, indicating that GLi Gαi1 retains the canonical Gαi inhibitory function on adenylyl cyclase.
Effect of the GoLoco-insensitivity Mutation on GoLoco Motif-dependent Properties of Gαi Subunits—To examine the effect of the GLi mutation on Gα regulation of GoLoco motif protein biology, we undertook multiple approaches. Gαi subunits facilitate the membrane localization of GoLoco motif proteins in various model systems, including Drosophila neuroblasts and mammalian cell lines (23, 32). Using MDCK cells, we measured the ability of exogenously expressed KT3 epitope-tagged wild type and GLi Gαi1 subunits to regulate the cellular distribution of endogenous GPSM2 (Fig. 8). We consistently observed that wild type Gαi1 expression promoted the plasma membrane recruitment of GPSM2, whereas GLi Gαi1 had no effect on GPSM2 cellular distribution. Analogous results were observed using mRFP-tagged Gαi1 and endogenous GPSM2 (supplemental Fig. S3). We also observed that exogenous expression of GPSM2 frequently resulted in the accumulation of GPSM2 in “vesicle-like” intracellular organelles. These structures were eliminated by co-transfection with wild type Gαi1, presumably by Gα-mediated recruitment of GPSM2 to the plasma membrane (supplemental Fig. S4). However, co-transfection of YFP-GPSM2 with Gαi1(N149I) did not alter the morphology of YFP-GPSM2-containing vesicular structures (supplemental Fig. S4).
FIGURE 8.

Wild type Gαil, but not GoLoco-insensitive Gαil, causes membrane localization of endogenous GPSM2. MDCK II cells were transfected with KT3-epitope tagged Gαi1(wild type) (top panel) or Gαi1(N149I) (bottom panel). Twenty four hours later, cells were fixed and stained with anti-GPSM2 antibodies, anti-KT3 antibodies, and DNA was stained with 4′,6-diamidino-2-phenylindole. Images were obtained using confocal microscopy.
We also examined the effect of the GLi mutation on interactions between Gαi1 and the triple GoLoco motif protein GPSM3 (26). We used a CFP-tagged and transmembrane domain-immobilized chimeric GαoA/i1 subunit6 (“CFP-TM-Gαoi”) to demonstrate the ability of Gα subunits to specify the membrane localization of GPSM3 (48). Expression of YFP-tagged GPSM3 in the absence of co-expressed Gα subunits is characterized by a uniform distribution of the GoLoco motif protein throughout the cell (Fig. 9, left panel). Expression of GPSM3-YFP in the presence of membrane-tethered, wild type CFP-TM-Gαoi causes a redistribution of GPSM3 to the plasma membrane (Fig. 9, middle panel). In contrast, expression of GPSM3-YFP in the presence of the N149I mutant CFP-TM-Gαoi results in a predominantly cytoplasmic distribution of GPSM3 (Fig. 9, right panel). As yet another alternative technique to monitor Gαi/GoLoco motif interaction in cells, we used co-immunoprecipitation from lysates of co-transfected COS-7 cells; both GPSM1 and GPSM2 interacted robustly with wild type but not GLi, Gαi1 (Fig. 10). In summary, these data are all consistent with the GLi mutation being a loss-of-function with respect to GoLoco motif binding in cells.
FIGURE 9.

GPSM3-YFP translocates to the plasma membrane after overexpression of transmembrane domain-tethered wild type, but not GoLoco-insensitive Gα subunits. Confocal images of HEK 293 cells transiently expressing GPSM3-YFP and either vector control (pcDNA; left panels), CFP-TM-GαoA/i1 (middle panels), or CFP-TM-GαoA/i1 N149I (right panels). CFP fluorescence micrograph is shown above the corresponding YFP fluorescence micrograph for the same cells. GPSM3-YFP fluorescence is distributed throughout the nucleus and cytoplasm in control cells and in cells expressing CFP-TM-GαoA/i1 with the GoLoco-insensitivity N149I mutation, whereas GPSM3-YFP fluorescence is enriched at the plasma membrane in cells expressing CFP-TM-GαoA/i1. Average profiles of fluorescence intensity derived from lines drawn normal to the plasma membrane are plotted below the micrograph panels. Peaks of CFP intensity document comparable expression of CFP-TM-GαoA/i1 (n = 10 cells) and CFP-TM-GαoA/i1 N149I (n = 10 cells). a.u., arbitrary units.
FIGURE 10.

GoLoco-insensitive Gαil does not interact with GoLoco motif proteins in cells. COS-7 cells were transfected with cDNAs encoding indicated combinations of YFP-GPSM1, YFP-GPSM2, wild type Gαi1-HA, and N149I Gαi1-HA. Cells were lysed, and total YFP- and HA-tagged protein levels were analyzed by immunoblot (IB) with anti-HA and anti-green fluorescent protein antibodies (left panel). Anti-HA antibody was added to the cell lysates to immunoprecipitate (IP) C-terminal HA-tagged wild type or N149I Gαi1. Bound proteins were separated by SDS-PAGE and immunoblotted with anti-HA and anti-green fluorescent protein antibodies (right panel).
Structural Basis of the GoLoco-insensitivity Mutation—Our data illustrate that the GLi mutation abrogates the ability of Gαi subunits and GoLoco motif proteins to interact in vitro and in cells. Despite such an extreme loss-of-function in this one aspect of Gα biology, the GLi Gα subunits behave normally in all other biochemical and cellular assays we have conducted. There is ample precedent for finding such mutations within Gα subunits; the RGS-insensitivity mutation (G183S within Gαi1) was first isolated in Saccharomyces cerevisiae Gpa1 (60), shown to be transferable to mammalian Gαi, Gαo, and Gαq subunits (60, 61), and validated as affecting only the Gα/RGS domain interaction without affecting nucleotide, receptor, Gβγ, or effector interactions (62). To better understand the potent and highly selective nature of the GLi mutation, we re-analyzed the previously described Gαi1·GDP/RGS14 GoLoco motif structure (50). Structural analysis of this Gα·GoLoco peptide complex indicates that the predominant role of Asn149 within Gαi1 is to directly contact Gln515 of the RGS14 GoLoco motif. The side chain oxygen of Gln515 forms a hydrogen bonding network with both the side chain terminal amine (distance of 3.1 Å) and the backbone amine (distance of 2.9 Å) of Asn149 (Fig. 1B). Asn149 appears to be an important node in a network of Gαi1 amino acid residues, including Glu43, Asn76, Gln79, Ser80, Gln147, Leu148, and Arg178 that act to stabilize the position of Gln515 in the GoLoco motif (Fig. 1C and supplemental Fig. S5). Gln515 of the GoLoco motif is crucial in positioning Arg516 into direct contact with GDP, and to accomplish this positioning, Gln515 makes a number of stabilizing interactions with Gα residues in the P-loop, αA helix, switch I, and the αD/αE loop (supplemental Figs. S2 and S5). This network of residues, in which Asn149 is involved, is also important in stabilizing the “seatbelt” between Glu43 and Arg178 hypothesized to restrain the bound nucleotide within its binding pocket (supplemental Fig. S5) (63, 64). Although the only amino acid residue of the RGS14 GoLoco motif that directly contacts GDP is Arg516 (Fig. 1B), GoLoco motif binding to Gαi1 induces a tighter fit of GDP into the nucleotide binding pocket (39). The salt bridge interaction between the P-loop residue Glu43 and the switch I residue Arg178 likely stabilizes bound GDP (39, 63, 65) and, in cooperation with Arg516, accounts for the structural determinants of GDI activity. We also observed that the backbone amine of Asn149 contacts the side chain of Ala512 (distance of 3.8 Å); however, this interaction was not observable in all crystallographic models7 and so may be of uncertain significance.
Through multiple experimental methods, we have demonstrated that the N149I mutation in Gαi does not perturb in vitro biochemical nor in cellulo signal transduction properties of Gαi subunits. Based on the structural analysis described above, substitution of the amide side chain of Asn with the aliphatic side chain of Ile would disrupt the hydrogen bonding network between the side chain nitrogen of Asn149 and the side chain carbonyl of Gln515 of the GoLoco motif. This is most likely responsible for the majority of the loss-of-function phenotype of the GLi mutation, as it appears that orientation of this highly conserved glutamine is critical to GoLoco motif function. The only two amino acid positions completely conserved in all functional GoLoco motifs (supplemental Fig. S1) (7) are the Gln and the Arg8 residues of the DQR triad. In light of this, we examined the role of Asn149 in Gαi class subunits. As described above, it has been noted that Asn149 is involved in stabilizing the seatbelt configuration between Gα residues Glu43 and Arg178 that is partially responsible for GoLoco motif- and Gβγ-mediated GDI activity (39, 63). However, in our studies, interaction between N149I mutant Gαi subunits and Gβγ subunits appeared to be normal, and this is consistent with Gβγ subunits having GDI activity toward Gαs despite the Ile substitution at this position in Gαs (66). Although Asn149 is conserved in all Gαi/o subunits, the closely related Gαz subunit contains a histidine at this position (Fig. 2A). Interestingly, Gαz is unique among Gα subunits in that it reportedly interacts with the truncated GoLoco motif of Rap1GAP1a in a GTP-selective manner (67) unlike the canonical Gα/GoLoco motif interaction, which is GDP-selective (7).
Gαi/GoLoco Motif Interaction Is Crucial for the Modulation of Microtubule Dynamics—In mammalian cells, overexpression of either GPSM2 or wild type Gαi1 has previously been shown to destabilize the processes of mitotic spindle orientation and metaphase chromosome segregation (32). In MDCK cells, this is characterized by an increase in the amplitude of spindle oscillations during metaphase (32). The presumed mechanism of action of Gαi or GPSM2 overexpression on spindle oscillations is an increased recruitment of force-generating Gαi·GoLoco motif complexes to the plasma membrane (32). Independently, it has also been observed that overexpression of Gαi3 or GPSM2 alters spindle pole positioning in mammalian cells (68). However, these observations are only suggestive of a critical function for a Gα·GoLoco protein complex, given that these results might alternatively reflect separate, distinct functions of Gα and GoLoco proteins in parallel pathways.
To delineate the precise role of Gαi/GoLoco motif interactions in ectopically induced spindle oscillations, we used MDCK cells transfected with either wild type or GLi Gαi1-YFP and measured simple spindle oscillations using time-lapse video microscopy. We observed that MDCK cells transfected with wild type Gαi1 underwent vigorous mitotic spindle oscillations during mitosis, as described previously (32) (Fig. 11 and supplemental movies 1 and 2), whereas at comparable expression levels, Gαi1(N149I)-transfected cells did not exhibit enhanced spindle rocking relative to untransfected cells (Fig. 11 and supplemental movies 3 and 4). To quantify these results, we measured the change of the long angle of metaphase chromosomal arrays during mitosis using image analysis. The amplitude of spindle oscillations induced by wild type Gαi1 expression was substantially higher than that found upon GLi Gαi1 expression (Table 1). To our knowledge, this result represents the first unambiguous demonstration that direct protein/protein interaction between Gαi subunits and GoLoco motifs is responsible for the modulation of cortical MT dynamics controlling mitotic spindle orientation.
FIGURE 11.

Wild type, but not GoLoco-insensitive, Gαil-YFP destabilizes metaphase chromosomes and spindle orientation. Time-lapse images of Hoechst 33342-stained chromosomes during metaphase alignment and segregation were recorded as described (32); supplemental movies are available. Representative consecutive fluorescence images taken from time-lapse sequences showing the motion of Hoechst-stained chromosomes in MDCK II cells expressing Gαi1(wild type)-YFP (upper panel) and Gαi1(N149I)-YFP (lower panel). The images were taken every 3 s as described (32). To show the movements of the metaphase chromosomal arrays, the position of the long axis of the chromosomal array along the metaphase plate in each image was marked by red (upper panel) or blue lines (lower panel), and positions of the axis in previous adjacent images are marked with white lines.
TABLE 1.
Ectopic expression of wild type, but not GoLoco-insensitive, Gαil-YFP destabilizes spindle orientation
Time-lapse images of Hoechst 33342-stained chromosomes during metaphase of MDCK II cells expressing Gαil(wild type)-YFP or Gαil(N149I)-YFP were recorded as described (32). Consecutive fluorescence images were taken every 3 s during mitosis (see supplemental movies 1-4 for example). The angles of the long axis of the chromosomal arrays in each frame (see Fig. 11 for examples) were measured using Metamorph software. The absolute angle changes of at least 100 sets of adjacent frames were binned into three categories: <5°, between 5 and 10°, and >10°. Data are the mean (S.E.M.) values obtained from three independent time-lapse analyses for either wild type or N149I Gαil-YFP-expressing cells.
|
Angle change during spindle rocking
|
Relative frequency of observation (%)
|
|
|---|---|---|
| Wild type Gαil-YFP | N149I Gαil-YFP | |
| 0-5° | 45 (3) | 94 (2) |
| 5-10° | 38 (4) | 6 (2) |
| >10° | 17 (2) | 0 |
The precise mechanism of Gαi·GoLoco motif complex-mediated regulation of cortical MT dynamics and spindle positioning during cell division is not clear. The use of our newly described GLi Gαi mutant in various model systems of symmetric and asymmetric cell division should help to clarify some of the molecular mechanisms of these processes. In particular, there are several important questions that remain unresolved. First, what is the nature of the Gαi nucleotide binding/hydrolysis cycle that occurs during cell division? What is the active Gαi species (Gα·GDP, Gα·GDP/GoLoco complex, or Gα·GTP), and what is the hierarchy of participating Gα regulatory proteins such as Gβγ, RIC-8, RGS proteins, and GoLoco motif proteins? A consensus within the field appears to be that Gαi·GDP/GoLoco motif complexes represent the “active” species during MT dynamics in cell division (69). However, the order in which the nucleotide binding and hydrolysis cycle of Gαi progresses has not been resolved. A recent paper has described RIC-8 as being able to act as a GEF on GoLoco motif-liganded Gαi subunits, thereby implying that GoLoco motif-bound Gαi may be the physiological substrate for RIC-8 (22). However, this same paper also demonstrated that the GoLoco motif is a noncompetitive inhibitor of RIC-8 GEF activity (22), in concordance with earlier results observed within the nematode system (9).
Second, what is the direct mechanism by which Gαi subunits modulate MT dynamics? This latter question is beginning to be understood. It appears that Gαi proteins act to relieve intramolecular auto-inhibition of “Pins-like” GoLoco motif proteins (e.g. GPSM1/AGS3 and GPSM2/LGN). Gαi·GDP binding to the GoLoco motifs of these multidomain proteins is believed to act as a conformational switch, allowing the subsequent binding of members of the nuclear mitotic apparatus/mushroom body defect (NuMA/MUD) family of proteins (21, 32, 70). The nuclear mitotic apparatus/mushroom body defect (NuMA/MUD) proteins are MT-binding and -regulating proteins; thus, their association with Gαi·GoLoco motif protein complexes at the cell cortex most likely modulates the dynamics of plus-end astral MTs (71-73).
Conclusion—Our data presented here describe a single point mutation in Gαi/o subunits that selectively abrogates the ability of Gα and GoLoco motifs to interact in vitro and in a cellular context. This Asn to Ile mutation in the αD/αE loop of the helical domain of Gα prevents the conserved GoLoco motif glutamine residue from properly orienting the GDP-binding arginine of the GoLoco motif. We have demonstrated the utility of this mutant in interrogating the role of Gα proteins in the modulation of mitotic spindle orientation. We anticipate the widespread use of this GoLoco-insensitivity mutation in both cell culture and in in vivo settings to address the physiological roles of Gα·GoLoco motif complex formation in diverse cell division processes, akin to how the RGS-insensitivity mutation of Gα subunits has been used to identify the physiological roles of endogenous RGS proteins in 7TMR signaling strength and duration (62, 74). A particularly important ancillary use of the GoLoco-insensitivity mutation will be to delineate a potential role for GoLoco motif proteins in 7TMR-mediated signal transduction. Gain-of-function studies suggest that GPSM1/AGS3 can modulate the cellular levels of Gα subunits and thus indirectly affect 7TMR signal transduction (75); other studies have suggested that 7TMR signaling in vivo may be modulated by GPSM1 and GPSM2 function (76-78). Application of the GLi mutant to such studies will surely provide biochemical and structural insights into the biological function of this important class of Gα regulatory proteins.
Supplementary Material
Acknowledgments
We thank Chris McCudden (University of North Carolina) for helpful discussions and Miller B. Jones (University of North Carolina) for attempting initial experiments using the GLi mutant. We also thank Pierre Gönczy (ISREC, Lausanne, Switzerland) for providing the RGS-7 cDNA.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM074268 (to D. P. S.), GM079506 (to Q. D.), GM078319 (to N. A. L.), MH060397 (to V. J. W.), F32 GM07694 (to C. A. J.), and F30 MH074266 (to A. J. K.). This work was also supported by the NIAAA intramural program (to S. R. I.), American Cancer Society Grant RSG0717601CSM (to Q. D.), National Science Foundation Grant MCB 0620024 (to N. A. L.), and fellowships from the American Heart Association (to G. J. D.) and the PhRMA Foundation (to M. D. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S5, movies 1-4, and additional references.
Footnotes
The abbreviations used are: 7TMR, seven transmembrane domain receptor; aa, amino acid; CFP, cyan fluorescent protein; GL, GoLoco; GLi, GoLoco-insensitive; GPSM, G-protein signaling modulator; GST, glutathione S-transferase; GTPγS, guanosine 5′-3-O-(thio)triphosphate; HA, hemagglutinin epitope tag; KT3, SV40 large T antigen-derived epitope tag; mRFP, monomeric red fluorescent protein; MECA, 5-N-methylcarboxamidoadenosine; mP, millipolarization unit of measurement; MT, microtubule; PCP-2, Purkinje cell protein 2; PTX, pertussis toxin; RGS, regulator of G-protein signaling; SPR, surface plasmon resonance; YFP, venus yellow fluorescent protein; NE, norepinephrine; PDB, Protein Data Bank; FITC, fluorescein isothiocyanate; GDI, guanine nucleotide dissociation inhibitor; SCG, superior cervical ganglion; TEA-OH, tetraethylammonium hydroxide; MDCK, Madin-Darby canine kidney.
The GoLoco motif is also referred to as the G-protein regulatory motif (79).
The GoLoco motif of Rap1GAP2 was not tested in these experiments as it is devoid of GDI activity and incapable of functional interactions with Gαi1, Gαi2, Gαi3, and GαoA (40).
There is considerable confusion with regard to cross-organism RGS protein nomenclature. Our experiments used C. elegans RGS-7 (GenBank™ accession number AY569308), which is likely the nematode ortholog of mammalian RGS3, a PDZ- and C2-domain-containing RGS protein (3, 80).
The chimeric GαoA/i1 subunit, comprising the N-terminal 33 amino acids of GαoA and the remainder of the polypeptide sequence from Gαi1, was originally created to facilitate kinetic imaging and functional assays not described in this manuscript (G. J. Digby and N. A. Lambert, unpublished data). Of the 33 GαoA-derived amino acids present within this Gα chimera, 22 are identical to those found in Gαi1, and 8 more are conservative substitutions (i.e., only three positions represent nonconservative differences in side chain character). This 33-amino acid N-terminal region composes the flexible first α-helix of Gα that does not participate in the GoLoco motif interaction (7).
The RGS14 GoLoco motif residue Ala512 was observed to interact with Asn149 of Gαi1 in both asymmetric units in the PDB 2OM2 structure and the chain A/chain B asymmetric unit of the PDB 1KJY structure.
References
- 1.Pierce, K. L., Premont, R. T., and Lefkowitz, R. J. (2002) Nat. Rev. Mol. Cell Biol. 3 639-650 [DOI] [PubMed] [Google Scholar]
- 2.Johnston, C. A., and Siderovski, D. P. (2007) Mol. Pharmacol. 72 219-230 [DOI] [PubMed] [Google Scholar]
- 3.Siderovski, D. P., and Willard, F. S. (2005) Int. J. Biol. Sci. 1 51-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabrera-Vera, T. M., Vanhauwe, J., Thomas, T. O., Medkova, M., Preininger, A., Mazzoni, M. R., and Hamm, H. E. (2003) Endocr. Rev. 24 765-781 [DOI] [PubMed] [Google Scholar]
- 5.Wettschureck, N., and Offermanns, S. (2005) Physiol. Rev. 85 1159-1204 [DOI] [PubMed] [Google Scholar]
- 6.Hampoelz, B., and Knoblich, J. A. (2004) Cell 119 453-456 [DOI] [PubMed] [Google Scholar]
- 7.Willard, F. S., Kimple, R. J., and Siderovski, D. P. (2004) Annu. Rev. Biochem. 73 925-951 [DOI] [PubMed] [Google Scholar]
- 8.Betschinger, J., and Knoblich, J. A. (2004) Curr. Biol. 14 R674-R685 [DOI] [PubMed] [Google Scholar]
- 9.Afshar, K., Willard, F. S., Colombo, K., Johnston, C. A., McCudden, C. R., Siderovski, D. P., and Gonczy, P. (2004) Cell 119 219-230 [DOI] [PubMed] [Google Scholar]
- 10.Colombo, K., Grill, S. W., Kimple, R. J., Willard, F. S., Siderovski, D. P., and Gonczy, P. (2003) Science 300 1957-1961 [DOI] [PubMed] [Google Scholar]
- 11.Gotta, M., and Ahringer, J. (2001) Nat. Cell Biol. 3 297-300 [DOI] [PubMed] [Google Scholar]
- 12.Hess, H. A., Roper, J. C., Grill, S. W., and Koelle, M. R. (2004) Cell 119 209-218 [DOI] [PubMed] [Google Scholar]
- 13.Srinivasan, D. G., Fisk, R. M., Xu, H., and van den Heuvel, S. (2003) Genes Dev. 17 1225-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanada, K., and Tsai, L. H. (2005) Cell 122 119-131 [DOI] [PubMed] [Google Scholar]
- 15.Konno, D., Shioi, G., Shitamukai, A., Mori, A., Kiyonari, H., Miyata, T., and Matsuzaki, F. (2008) Nat. Cell Biol. 10 93-101 [DOI] [PubMed] [Google Scholar]
- 16.Morin, X., Jaouen, F., and Durbec, P. (2007) Nat. Neurosci. 10 1440-1448 [DOI] [PubMed] [Google Scholar]
- 17.Lechler, T., and Fuchs, E. (2005) Nature 437 275-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Afshar, K., Willard, F. S., Colombo, K., Siderovski, D. P., and Gonczy, P. (2005) Development (Camb.) 132 4449-4459 [DOI] [PubMed] [Google Scholar]
- 19.Johnston, C. A., Afshar, K., Snyder, J. T., Tall, G. G., Gonczy, P., Siderovski, D. P., and Willard, F. S. (2008) J. Biol. Chem. 283 21550-21558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilkie, T. M., and Kinch, L. (2005) Curr. Biol. 15 R843-R854 [DOI] [PubMed] [Google Scholar]
- 21.Tall, G. G., and Gilman, A. G. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 16584-16589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas, C. J., Tall, G. G., Adhikari, A., and Sprang, S. R. (2008) J. Biol. Chem. 283 23150-23160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu, F., Cai, Y., Kaushik, R., Yang, X., and Chia, W. (2003) J. Cell Biol. 162 623-633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuse, N., Hisata, K., Katzen, A. L., and Matsuzaki, F. (2003) Curr. Biol. 13 947-954 [DOI] [PubMed] [Google Scholar]
- 25.Kimple, R. J., De Vries, L., Tronchere, H., Behe, C. I., Morris, R. A., Gist Farquhar, M., and Siderovski, D. P. (2001) J. Biol. Chem. 276 29275-29281 [DOI] [PubMed] [Google Scholar]
- 26.Kimple, R. J., Willard, F. S., Hains, M. D., Jones, M. B., Nweke, G. K., and Siderovski, D. P. (2004) Biochem. J. 378 801-808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda, S. R., and Jeong, S. W. (2004) Methods Enzymol. 389 170-189 [DOI] [PubMed] [Google Scholar]
- 28.Ikeda, S. R. (1996) Nature 380 255-258 [DOI] [PubMed] [Google Scholar]
- 29.Kammermeier, P. J., Davis, M. I., and Ikeda, S. R. (2003) Mol. Pharmacol. 63 183-191 [DOI] [PubMed] [Google Scholar]
- 30.Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K., and Miyawaki, A. (2002) Nat. Biotechnol. 20 87-90 [DOI] [PubMed] [Google Scholar]
- 31.Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., and Tsien, R. Y. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 7877-7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du, Q., and Macara, I. G. (2004) Cell 119 503-516 [DOI] [PubMed] [Google Scholar]
- 33.Vidi, P. A., Chemel, B. R., Hu, C. D., and Watts, V. J. (2008) Mol. Pharmacol. 74 544-551 [DOI] [PubMed] [Google Scholar]
- 34.Willard, F. S., Kimple, A. J., Johnston, C. A., and Siderovski, D. P. (2005) Anal. Biochem. 340 341-351 [DOI] [PubMed] [Google Scholar]
- 35.Willard, F. S., and Siderovski, D. P. (2004) Methods Enzymol. 389 320-338 [DOI] [PubMed] [Google Scholar]
- 36.De Vries, L., Fischer, T., Tronchere, H., Brothers, G. M., Strockbine, B., Siderovski, D. P., and Farquhar, M. G. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 14364-14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCudden, C. R., Willard, F. S., Kimple, R. J., Johnston, C. A., Hains, M. D., Jones, M. B., and Siderovski, D. P. (2005) Biochim. Biophys. Acta 1745 254-264 [DOI] [PubMed] [Google Scholar]
- 38.Willard, F. S., McCudden, C. R., and Siderovski, D. P. (2006) Cell. Signal. 18 1226-1234 [DOI] [PubMed] [Google Scholar]
- 39.Kimple, R. J., Kimple, M. E., Betts, L., Sondek, J., and Siderovski, D. P. (2002) Nature 416 878-881 [DOI] [PubMed] [Google Scholar]
- 40.Willard, F. S., Low, A. B., McCudden, C. R., and Siderovski, D. P. (2007) Cell. Signal. 19 428-438 [DOI] [PubMed] [Google Scholar]
- 41.Soundararajan, M., Willard, F. S., Kimple, A. J., Turnbull, A. P., Ball, L. J., Schoch, G. A., Gileadi, C., Fedorov, O. Y., Dowler, E. F., Higman, V. A., Hutsell, S. Q., Sundstrom, M., Doyle, D. A., and Siderovski, D. P. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 6457-6462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimple, A. J., Yasgar, A., Hughes, M., Jadhav, A., Willard, F. S., Muller, R. E., Austin, C. P., Inglese, J., Ibeanu, G. C., Siderovski, D. P., and Simeonov, A. (2008) Comb. Chem. High Throughput Screen. 11 396-409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnston, C. A., Lobanova, E. S., Shavkunov, A. S., Low, J., Ramer, J. K., Blaesius, R., Fredericks, Z., Willard, F. S., Kuhlman, B., Arshavsky, V. Y., and Siderovski, D. P. (2006) Biochemistry 45 11390-11400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willard, F. S., and Siderovski, D. P. (2006) Biochem. Biophys. Res. Commun. 339 1107-1112 [DOI] [PubMed] [Google Scholar]
- 45.Guo, J., and Ikeda, S. R. (2004) Mol. Pharmacol. 65 665-674 [DOI] [PubMed] [Google Scholar]
- 46.Watts, V. J., Vu, M. N., Wiens, B. L., Jovanovic, V., Van Tol, H. H., and Neve, K. A. (1999) Psychopharmacology 141 83-92 [DOI] [PubMed] [Google Scholar]
- 47.Nguyen, C. H., and Watts, V. J. (2005) Biochem. Biophys. Res. Commun. 332 913-920 [DOI] [PubMed] [Google Scholar]
- 48.Digby, G. J., Lober, R. M., Sethi, P. R., and Lambert, N. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 17789-17794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heim, R., and Tsien, R. Y. (1996) Curr. Biol. 6 178-182 [DOI] [PubMed] [Google Scholar]
- 50.Sammond, D. W., Eletr, Z. M., Purbeck, C., Kimple, R. J., Siderovski, D. P., and Kuhlman, B. (2007) J. Mol. Biol. 371 1392-1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sobolev, V., Eyal, E., Gerzon, S., Potapov, V., Babor, M., Prilusky, J., and Edelman, M. (2005) Nucleic Acids Res. 33 W39-W43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Natochin, M., Gasimov, K. G., and Artemyev, N. O. (2002) Biochemistry 41 258-265 [DOI] [PubMed] [Google Scholar]
- 53.Slepak, V. Z., Quick, M. W., Aragay, A. M., Davidson, N., Lester, H. A., and Simon, M. I. (1993) J. Biol. Chem. 268 21889-21894 [PubMed] [Google Scholar]
- 54.Ferguson, K. M., Higashijima, T., Smigel, M. D., and Gilman, A. G. (1986) J. Biol. Chem. 261 7393-7399 [PubMed] [Google Scholar]
- 55.Ikeda, S. R., and Dunlap, K. (1999) Adv. Second Messenger Phosphoprotein Res. 33 131-151 [DOI] [PubMed] [Google Scholar]
- 56.Elmslie, K. S., Zhou, W., and Jones, S. W. (1990) Neuron 5 75-80 [DOI] [PubMed] [Google Scholar]
- 57.Ikeda, S. R. (1991) J. Physiol. (Lond.) 439 181-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeong, S. W., and Ikeda, S. R. (1999) J. Neurosci. 19 4755-4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeong, S. W., and Ikeda, S. R. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 907-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DiBello, P. R., Garrison, T. R., Apanovitch, D. M., Hoffman, G., Shuey, D. J., Mason, K., Cockett, M. I., and Dohlman, H. G. (1998) J. Biol. Chem. 273 5780-5784 [DOI] [PubMed] [Google Scholar]
- 61.Lan, K. L., Sarvazyan, N. A., Taussig, R., Mackenzie, R. G., DiBello, P. R., Dohlman, H. G., and Neubig, R. R. (1998) J. Biol. Chem. 273 12794-12797 [DOI] [PubMed] [Google Scholar]
- 62.Fu, Y., Zhong, H., Nanamori, M., Mortensen, R. M., Huang, X., Lan, K., and Neubig, R. R. (2004) Methods Enzymol. 389 229-243 [DOI] [PubMed] [Google Scholar]
- 63.Wall, M. A., Coleman, D. E., Lee, E., Iniguez-Lluhi, J. A., Posner, B. A., Gilman, A. G., and Sprang, S. R. (1995) Cell 83 1047-1058 [DOI] [PubMed] [Google Scholar]
- 64.Johnston, C. A., Willard, F. S., Jezyk, M. R., Fredericks, Z., Bodor, E. T., Jones, M. B., Blaesius, R., Watts, V. J., Harden, T. K., Sondek, J., Ramer, J. K., and Siderovski, D. P. (2005) Structure (Lond.) 13 1069-1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lambright, D. G., Sondek, J., Bohm, A., Skiba, N. P., Hamm, H. E., and Sigler, P. B. (1996) Nature 379 311-319 [DOI] [PubMed] [Google Scholar]
- 66.Brandt, D. R., and Ross, E. M. (1985) J. Biol. Chem. 260 266-272 [PubMed] [Google Scholar]
- 67.Meng, J., Glick, J. L., Polakis, P., and Casey, P. J. (1999) J. Biol. Chem. 274 36663-36669 [DOI] [PubMed] [Google Scholar]
- 68.Blumer, J. B., Kuriyama, R., Gettys, T. W., and Lanier, S. M. (2006) Eur. J. Cell Biol. 85 1233-1240 [DOI] [PubMed] [Google Scholar]
- 69.Gonczy, P. (2008) Nat. Rev. Mol. Cell Biol. 9 355-366 [DOI] [PubMed] [Google Scholar]
- 70.Nipper, R. W., Siller, K. H., Smith, N. R., Doe, C. Q., and Prehoda, K. E. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14306-14311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bowman, S. K., Neumuller, R. A., Novatchkova, M., Du, Q., and Knoblich, J. A. (2006) Dev. Cell 10 731-742 [DOI] [PubMed] [Google Scholar]
- 72.Izumi, Y., Ohta, N., Hisata, K., Raabe, T., and Matsuzaki, F. (2006) Nat. Cell Biol. 8 586-593 [DOI] [PubMed] [Google Scholar]
- 73.Siller, K. H., Cabernard, C., and Doe, C. Q. (2006) Nat. Cell Biol. 8 594-600 [DOI] [PubMed] [Google Scholar]
- 74.Fu, Y., Huang, X., Zhong, H., Mortensen, R. M., D'Alecy, L. G., and Neubig, R. R. (2006) Circ. Res. 98 659-666 [DOI] [PubMed] [Google Scholar]
- 75.Sato, M., Gettys, T. W., and Lanier, S. M. (2004) J. Biol. Chem. 279 13375-13382 [DOI] [PubMed] [Google Scholar]
- 76.Wiser, O., Qian, X., Ehlers, M., Ja, W. W., Roberts, R. W., Reuveny, E., Jan, Y. N., and Jan, L. Y. (2006) Neuron 50 561-573 [DOI] [PubMed] [Google Scholar]
- 77.Yao, L., McFarland, K., Fan, P., Jiang, Z., Inoue, Y., and Diamond, I. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 8746-8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bowers, M. S., McFarland, K., Lake, R. W., Peterson, Y. K., Lapish, C. C., Gregory, M. L., Lanier, S. M., and Kalivas, P. W. (2004) Neuron 42 269-281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takesono, A., Cismowski, M. J., Ribas, C., Bernard, M., Chung, P., Hazard, S., III, Duzic, E., and Lanier, S. M. (1999) J. Biol. Chem. 274 33202-33205 [DOI] [PubMed] [Google Scholar]
- 80.Willard, M. D., Willard, F. S., and Siderovski, D. P. (2009) in Handbook of Cell Signaling (Bradshaw, R., and Dennis, E., eds) 2nd Ed., Elsevier, San Diego, in press
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.