

Abstract
The pentapeptide repeat is a recently discovered protein fold. Mycobacterium tuberculosis MfpA is a founding member of the pentapeptide repeat protein (PRP) family that confers resistance to the antibiotic fluoroquinolone by binding to DNA gyrase and inhibiting its activity. The size, shape, and surface potential of MfpA mimics duplex DNA. As an initial step in a comprehensive biophysical analysis of the role of PRPs in the regulation of cellular topoisomerase activity and conferring antibiotic resistance, we have explored the solution structure and refolding of MfpA by fluorescence spectroscopy, CD, and analytical centrifugation. A unique CD spectrum for the pentapeptide repeat fold is described. This spectrum reveals a native structure whose β-strands and turns within the right-handed quadrilateral β-helix that define the PRP fold differ from canonical secondary structure types. MfpA refolded from urea or guanidium by dialysis or dilution forms stable aggregates of monomers whose secondary and tertiary structure are not native. In contrast, MfpA refolded using a novel “time-dependent renaturation” protocol yields protein with native secondary, tertiary, and quaternary structure. The generality of “time-dependent renaturation” to other proteins and denaturation methods is discussed.
The solution of the crystal structure of the Mycobacterium tuberculosis protein MfpA revealed a novel right-handed quadrilateral β-helix named the pentapeptide repeat fold (1, 2). The 183-amino acid protein is almost entirely composed of a right-handed β-helix. MfpA is dimeric in solution (see below) and in the crystal (Fig. 1). Its C-terminal α-helices form the dimer interface. The rod-shaped dimer is highly asymmetric, ∼100 Å long with a diameter of 18 Å at the dimer interface. The diameter increases to 27 Å at the N termini. The size, shape, and electrostatic surface potential are sufficiently similar to B-form DNA to suggest that these properties of MfpA mimic those of DNA (1, 2).
FIGURE 1.

A, the amino acid sequence of the MfpA chain A with DSSP (11) assigned secondary structure (H, α-helix; B, isolated β-bridge; E, extended strand participating in β-ladder; T, hydrogen bonded turn; S, bend; R (dots), no assigned structure). B, a ribbon diagram of the Mycobacterium tuberculosis MfpA dimer (2). The two α-helices at the C termini of each monomer are shown. The β-strands are shown as long arrows. The bends, isolated β-bridges, and residues with no assigned structure are depicted as “unordered” according to the secondary structure types calculated from CD spectra. C, the ribbon diagram of MfpA depicting the isolated β-bridges (short arrows) along with other types of the secondary structures shown in B. The tryptophan residues whose fluorescence was monitored are highlighted in red.
The antibacterial activity of fluoroquinolones results from their modification of the intermediate complexes of covalently linked DNA with DNA gyrase and DNA topoisomerase IV during their catalytic cycles (3). Amino acid substitutions in the putative fluoroquinolone binding region of the gyrA-encoded A subunit of DNA gyrase confers resistance to fluoroquinolones in laboratory strains of M. tuberculosis (4). MfpA confers bacterial resistance to fluoroquinolone; DNA mimicry is proposed to explain inhibition of DNA gyrase activity by MfpA with the resultant antibiotic resistance (1). Escherichia coli-expressed MfpA inhibits both the ATP-dependent DNA supercoiling and ATP-independent relaxation reactions catalyzed by E. coli DNA gyrase in a concentration-dependent manner (1). Direct binding of MfpA to DNA gyrase has been demonstrated. A model has been proposed in which MfpA competes with DNA for the catalytic site of DNA gyrase (1). Although the mimicry of DNA by proteins is intrinsically related to structure and surface properties (5, 6), the pattern of acidic residues displayed across the surface of MfpA mimics distorted rather than canonical B-form DNA (2). Thus, conformational malleability may influence MfpA binding to DNA gyrase in solution.
The novelty of a new protein fold, the link to antibiotic resistance and the potential of PRPs2 as cellular regulators conjoin as the stimulus for our investigations into the structure and function of MfpA. In the present study, we have explored the solution structure and refolding of MfpA with analytical ultracentrifugation, CD spectroscopy, and fluorescence spectroscopy.
The native solution structure revealed by CD spectroscopy surprisingly differs from that of the canonical secondary structure types. This difference is presumably due to apparent alteration of the β-strands and turns in the novel right-handed quadrilateral β-helix that defines the PRP fold; single-residue β-bridges and short β-strands may be dynamic in solution and thus fluctuate from the crystalline structure. The consequences of structural pliability for the proposed function of MfpA are discussed. The refolding of chemically denatured MfpA by dialysis or dilution yields aggregates whose monomers do not possess native secondary and tertiary structure. A “time-dependent renaturation” protocol was developed that efficiently yields native MfpA. The generality of “kinetic renaturation” is discussed.
EXPERIMENTAL PROCEDURES
Protein and Buffer Preparation—M. tuberculosis MfpA was expressed in E. coli and purified as described (1). Mutation of cysteine residue 179 to serine yields a protein with a single solvent-accessible cysteine residue located at the N terminus that is used for labeling the protein with an exogenous fluorophore.3 The C179S mutant is used in the present study to confirm the independence of MfpA aggregation on disulfide bond formation (see “Results”). The mutation was introduced by site-directed PCR mutagenesis to make TGA (cysteine) to TGC (serine). Two primers were used in the PCR: forward, CGC ACG GGC TGA GCT TGG CAG GGG GCT; backward, AGC CCC CTG CCA AGC TCA GCC CGT GCG. After the standard PCR (7), the DNA was digested with DpnI at 37 °C for 1 h to digest the parent DNA. The mutated DNA was ethanol-precipitated, resuspended, and transformed into Top 10-competent cells. The single expected mutation was verified by DNA sequencing. The mutant protein was expressed and purified as the wild type MfpA. All of the experiments were conducted in 25 mm Tris-HCl, 25 mm KCl, 0.1 mm EDTA, 6% glycerol, 2 mm dithiothreitol (Buffer A) or 10 mm KH2PO4, 25 mm KCl, 2 mm dithiothreitol (Buffer B) at pH 7.6 and 25.0 °C unless noted. Refolding of MfpA was conducted using variations of Buffers A and B as described below.
Fluorescence and Light Scattering Spectroscopy—All fluorescence measurements were made using a Fluoromax-3 spectrofluorometer (Jobin Yvon Inc.). Spectra are routinely corrected for the spectral sensitivity of the instrument. Measurements were made at the magic angle of 55° between the excitation and emission polarization vectors. The intensity of the Raman scattering band of water is the internal standard of spectrofluorometer sensitivity. Fluorescence anisotropy was calculated by Equation 1,
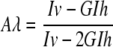 |
(Eq. 1) |
where Aλ is the fluorescence anisotropy measured at a combination of the excitation and emission wavelengths with the excitation polarizer in the vertical position, Iv and Ih are the fluorescence intensities measured at the vertical and horizontal position of the emission polarizer, and G is the instrumental factor accounting for the bias of the detection system for vertically versus horizontally polarized light. Values of λmax were calculated from the first derivative of the fluorescence spectrum by polynomial fitting. The accuracy of these values is not less than ±0.2 nm.
Raleigh (quasielastic) light scattering was also recorded using the Fluoromax-3 spectrofluorometer. Scattered light (350 nm) was collected at an angle of 90° to the incident light.
CD Spectroscopy—CD measurements were performed with a JASCO J720 spectropolarimeter. Spectra were acquired from five replicates between 240 to 190 nm using a 1 nm resolution in 1 mm path length cells. The 1–4 μm protein samples were prepared in buffer as described in the figure legends. Predicted secondary structure contributions were determined by SELCON3, CONTINLL, and CDSSTR programs (8) available as the CDPro package, available on the World Wide Web. This software uses reference data bases of 43 soluble native proteins and 48 proteins composed of 43 soluble native and five denatured proteins. The program CLUSTER in CDPro was used to estimate the tertiary class of MfpA from the CD spectra (9). The CDPro package results were compared with those obtained from the DICHROWEB Web server, using the protein reference data base SP175, consisting of more than 70 proteins representing a wide range of secondary structures, protein folds and architectures (10). The CD spectra are expressed as the mean residue ellipticity, [Θ]mrw in degrees·cm2·dmol-1 or as difference in molar absorbance, Δε in cm-1·m-1 with the numerical relationship at any wavelength as [Θ]mrw = 3298 ×Δε.
Ultracentrifuge Experiments—Sedimentation equilibrium experiments were performed using the absorption optics of a Beckman XL-I analytical ultracentrifuge with 6-channel centerpieces in the Ti-60 rotor. Three concentrations each of samples of MfpA in Buffer A and Buffer B and time-dependent renatured MfpA in Buffer A were equilibrated at 22 °C for 24 h at 15,000 and then again at 20,000 rpm. The absorbance scans obtained at the two speeds were globally analyzed using HeteroAnalysis version 1.0.114 (J. L. Cole and J. W. Lary, Analytical Ultracentrifugation Facility, Biotechnology Services Center, University of Connecticut) for the weight average molecular weight. The resolved molecular weight and the 95% joint confidence intervals are reported. The values of v̄ (from the amino acid composition), density, and viscosity were calculated using Sednterp version 1.06 (Hayes, B., T. Laue, and J. Philo, Sedimentation Interpretation Program, University of New Hampshire).
Protein Denaturation and Refolding—MfpA was denatured by the addition of either urea or guanidinium HCl to concentrations of 8.5 and 6 m, respectively.
Attempts were made to refold 8.5 m urea denatured MfpA by direct dilution and one-step and multistep dialysis using a variety of solutes and/or solute concentrations that are variants of Buffers A and B. Among the solute conditions explored were 4 versus 25 °C, 0–10 mm dithiothreitol, 25–500 mm KCl, and 6–20% glycerol for Buffer A or 0.5 m l-arginine and 0–1000 mm KCl for Buffer B. Since these buffer variants did not affect the structure of refolded MfpA, all subsequent experiments were conducted in Buffer A or B, as noted in the figure legends.
Time-dependent Denaturation—An 8.5 m urea stock solution was added to a solution of MfpA to final concentrations of urea and protein of ∼6.4 m and 1–2 μm, respectively. The mixed solution was stirred, and fluorescence intensity and anisotropy were measured as a function of time progressively at excitation and emission wavelengths of 280 and 324 nm, respectively. The dead time of the experiment was <30 s. The denaturation time courses were fit to Equation 2,
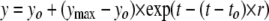 |
(Eq. 2) |
where yo and ymax represent the initial and infinite values of y (either fluorescence intensity or anisotropy at time t), r is the rate of the denaturation, and to is the reaction dead time. Values of yo, ymax, and r were determined by nonlinear least squares analysis (Origin version 6.1).
Time-dependent Refolding—The native tertiary structure of MfpA is stable in solution up to 0.8 m urea, as judged by the fluorescence intensity, position of λmax and anisotropy, CD spectra, and light scattering profiles in Buffers A and B without urea (data not shown). Thus, refolding solutions contained <0.8 m urea. MfpA at concentrations of >20 μm was denatured by the addition of urea to 6.4 m. Aliquots of MfpA denatured for 5–45 min were diluted 10–20-fold into buffer without urea and mixed, resulting in 1–2 μm MfpA and 0.3–0.6 m urea (MfpA is fully denatured in <5 min under these conditions; Fig. 6). Following incubation for 30 min in the refolding buffer, the samples were centrifuged for 15 min at 80,000 rpm and 4 °C, and the precipitate was discarded (TLA-120 rotor; Beckman Optima TLX).
FIGURE 6.

A, time progress curve of the denaturation of MfpA upon the addition of 8.5 m urea (to a final concentration of 6.4 m), as evidenced by the fluorescence anisotropy (▪) and intensity (○) measured at 324 nm upon excitation at 280 nm. The solid line depicts the best fit to a monoexponential equation with a rate constant, r = 0.97 ± 0.046 min-1. The arrow marks the minimum time at which “time-dependent renaturation” was initiated (see below and “Experimental Procedures”). B–D track the changes in the optical parameters of the solutions containing MfpA during “time-dependent renaturation” as a function of refolding time. B, λmax of fluorescence emission following excitation at 280 nm; C, Raleigh light scattering recorded at 350 nm; D, quantitative yield of native MfpA. The x axis plots the denaturation time prior to which refolding was initiated by the addition of a 10-fold excess of Buffer A. The samples were then incubated for 30 min in Buffer A prior to the measurement of the optical parameters (•). Samples in which high molecular weight aggregates were cleared by ultracentrifugation are shown in B and C by an open circle. The protein concentration is 1 μm.
The fluorescence intensity, anisotropy and λmax, and the light scattering intensity were measured for all samples before centrifugation and for the resultant supernatant. The fraction of refolded MfpA was calculated from the fluorescence intensity measurements. The quantitative yield of native MfpA in denaturant containing solution was obtained by fitting the fraction native protein versus denaturation time to Equation 2.
Secondary Structure Analysis—The programs DSSP (11) and STRIDE (12) assign the structure of soluble and membrane proteins with comparable accuracy (13, 14). The fraction of secondary structure types estimates from CD spectra by DSSP has overall performance indexes slightly superior to STRIDE (root meant square deviation (RMSD) and correlation coefficient (13)). Therefore, the secondary structure of crystalline MfpA (Protein Data Bank code 2bm4) was assigned using the digital shape sampling and processing algorithm DSSP (11). The α-helices and β-strands were divided into regular (αR and βR) and distorted (αD and βD) classes (13), yielding six classifications of secondary structure: regular α-helix (αR), distorted α-helix (αD), regular β-strand (βR), distorted β-strand (βD), turn, and disordered. The values of total α-helix and total β-strand were used for secondary structure comparison of the crystal structure and CD spectra.
Fig. 1A shows the amino acid sequence of MfpA with the DSSP assigned secondary structure types. Two approaches are typically used to group DSSP classes into the classes derived from solution CD spectra. The first approach treats all residues in isolated β-bridges and bends as disordered in the crystal structure assignment. Fitting may improve when the residues in isolated β-bridges are combined with the β-sheets (15). The CDPro package programs use the first approach by combining isolated β-bridges, bends, and residues with no assigned structure as “unordered” (B, S, and R in Fig. 1A). Fig. 1, B and C, show ribbon diagrams of the MfpA crystal structure that depict these two approaches. We used the assignment shown in Fig. 1B as it is more physically grounded.
Secondary structure calculations were characterized by the RMSD and the normalized RMSD (NRMSD) between the crystal and the average solution CD estimates of the secondary structure content. The definitions of RMSD and NRMSD used in this study are as follows,
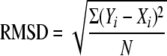 |
(Eq. 3) |
and
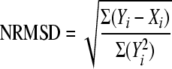 |
(Eq. 4) |
respectively, where Xi and Yi are crystallographic and CD estimates of a given type of secondary structure, i, in N reference samples (14, 16). RMSD and NRMSD of the average secondary structures values were calculated for the secondary structures (Yi), determined by the programs SELCON3, CONTINLL, and CDSSTR (see above), using the two protein reference sets. Six values were determined for each structure. Although RMSD measures the predictive power of the method, NRMSD may be a better indicator of the goodness of the calculations. It is generally accepted that calculated and experimental data are in good agreement when NRMSD is <0.1, are similar if 0.1 < NRMSD < 0.2, and in poor agreement if NRMSD is >0.2. Overall analysis performance was determined by collectively considering the calculations made by all of the CDPro package programs.
RESULTS
Each MfpA monomer contains three tryptophan residues that are the protein's only source of fluorescence (Fig. 1). The fluorescence emission maximum, λmax, is a sensitive measure of the relative polarity of the chromophore environment. Fluorescence anisotropy reports on the relative mobility of chromophores and is thus also sensitive to the size and shape of proteins. Rayleigh (quasielastic) light scattering is sensitive to the size and shape of the scattering particles and thus reports protein aggregation. CD is a particularly valuable measure of structure for proteins such as PRPs that are composed almost entirely of a repeating fold. We have used these measures to describe the native and denatured structure of MfpA in solution and develop a method by which it can be refolded following chemical denaturation. Figs. 2 and 3 summarize these measures for native, denatured, and renatured by dialysis MfpA.
FIGURE 2.

A, the fluorescence emission spectra upon excitation at 280 nm of native MfpA (solid line, dashed and dotted line, and dashed line) and MfpA-C179S (dotted line) in Buffer A (solid line and dotted line) and Buffer B (dashed and dotted line). Denatured MfpA and MfpA-C179S were incubated in Buffers A and B to which was added 8.5 m urea (thick line). Additional spectra show MfpA in Buffer A (solid red line) and Buffer B (dashed red line) refolded by dialysis. N, R, and D, native, refolded by dialysis, and denatured MfpA, respectively. Inset, the derivative of the R spectrum from which the exact position of λmax is determined; B, the CD spectra of native MfpA in Buffer A (○) and Buffer B (•), MfpA refolded by dialysis of the urea into Buffer B (red square]), and denatured MfpA (▴). The protein concentration is 1 and 4 μm for the fluorescence and CD measurements, respectively. Buffers A and B are described under “Experimental Procedures.”
FIGURE 3.

Values of λmax of fluorescence spectrum of protein excited at 280 nm (A), anisotropy of fluorescence excited at 280 nm and measured at 324 nm (B), and Raleigh light scattering at 350 nm measured at a right angle to the direction of the incident beam that characterize changes in tertiary and quaternary structures of MfpA and its mutant C179S at experimental conditions comparable with those utilized in Fig. 2 (C). Native protein is depicted by white bars, denatured protein by gray bars, and dialysis-renatured protein by right slanted bars. Samples denoted as “diluted” were diluted from a stock solution to the 1 μm concentration. Samples denoted as “dialyzed” were dialyzed from the stock solution into the indicated buffer. Protein denoted as “denatured” was diluted from the stock solution into buffer containing 8.5 m urea. Protein denoted “dialysis-renatured” was denatured in buffer containing 8.5 m urea and then dialyzed against the sample buffer without urea. The S.D. value of triplicate measurements for each measured parameter is comparable with the thickness of the lines. The only exception is the low values of light scattering for the denatured protein, where the error reached 20%. The protein concentration is 1 μm. Buffers A and B are described under “Experimental Procedures.”
Native MfpA—The λmax of MfpA following excitation at 280 nm is ∼332 nm for the native protein (Figs. 2A (N) and 3A). λmax is invariant for both MfpA and C179S4 MfpA in all analyzed denaturant free buffers. The measured anisotropy of ∼0.12 is characteristic of a native protein with the size and shape of MfpA (Fig. 3B). The CD spectrum of native MfpA is characterized by two peaks at ∼193 and ∼218 nm (Fig. 2B). Although peaks of these wavelengths are characteristic of unordered and β-sheet structures, respectively (17, 18), their combination and relative magnitudes within a single spectrum are unusual (Fig. 2B). The fractions α-helix, β-strands, turns, and unordered structure were calculated from the CD spectra as described under “Experimental Procedures” (Table 1). Minimal α-helix is predicted for the native MfpA with the bulk of the structured protein as β-strands and turns. A surprisingly large fraction of the structure is predicted to be unordered.
TABLE 1.
The secondary structure contents (SSC) estimated from the CD spectra of native and dialysis-renatured MfpA
|
Fractionsa
|
NAAb(x-ray)
|
Native MfpA
|
Dialysis-renatured MfpA
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Fractionc | RMSDd | NRMSDd | NAAb(CD) | Fraction | RMSD | NRMSD | NAA (CD) | ||
| Htotal | 16 | 0.035 | 0.033 | 0.681 | 1–12 | 0.152 | 0.039 | 0.249 | 21–35 |
| Stotal | 40 | 0.188 | 0.096 | 0.454 | 17–52 | 0.317 | 0.008 | 0.027 | 57–59 |
| Turns | 40 | 0.221 | 0.026 | 0.118 | 36–45 | 0.216 | 0.011 | 0.049 | 38–42 |
| Unordered | 87 | 0.574 | 0.179 | 0.298 | 72–138 | 0.312 | 0.039 | 0.125 | 50–64 |
| Overalle | 0.104 | 0.307 | 0.029 | 0.11 | |||||
Secondary structure types: Htotal, total helixes, Stotal, total strands, Turns, turns; Unordered, the remainder of the protein
Number of amino acids included in the given type the secondary structure calculated from DSSP assignment of the crystal structure, NAA (x-ray) or from the CD spectra of MfpA, NAA (CD)
Calculated fraction of each secondary structure type
The values of r.m.s.d. and n.r.m.s.d. in this table reflect the analysis of the CD spectrum by the different programs and protein reference sets as described under “Experimental Procedures”
The overall performance of the CD analysis determined by considering all secondary structures collectively
Application of a reference set of 72 proteins available in DICHROWEB yields results qualitatively similar to Table 1 (data not shown). The average RMSD value as well as the values for the secondary structure content calculated for native MfpA is in reasonable agreement with expected accuracy of the method (13, 18, 19). However, the dispersion of the number of the residues within each secondary structure type predicted from the CD spectra is large (Table 1). Accordingly, the NRMSD values characterizing the goodness of the calculations are poor, resulting in the large errors associated with the calculated fractions (Fig. 4).
FIGURE 4.

The SSC of native and dialysis-renatured MfpA. The data reflects the performance of CD spectral analysis made by three programs with two reference protein sets included in the CDPro package (see “Experimental Procedures”). The secondary structure elements depicted are Htotal (white bars), Stotal (left slanted bars), turns (gray bars), and unordered (cross-hatched bars). The error bars are the NRSMD shown in Table 1 for the mean values of SSC obtained from the CDPro package programs.
The number of amino acid residues included in α-helix, β-strands, turns, and unordered structure were calculated from the crystal structure of MfpA (Table 1) and mapped on the structure of MfpA (Fig. 1, B and C). Table 2 quantitatively compares the secondary structure fractions calculated from the CD spectrum and the crystal structure. The RMSD values comparing the crystal and CD spectrum calculations reflect the expected quality of the predictions (17, 18). In contrast, the NRMSD values show a poor correspondence that results in the large errors associated with the calculated fractions (Table 2). To illustrate this point, we simulated a CD spectra from the calculated fractions; the simulation does not coincide with the experimental spectra of native MfpA (Fig. 5A).
TABLE 2.
Comparison of secondary structure contents (SSC) in native and renatured MfpA estimated from CD (Table 1) with x-ray data revealed by DSSP algorithm combined into four secondary structure types SSC (x-ray) is an assignment of different DSSP classes to different secondary structures revealed from x-ray data; SSC (rd) is a random assignment (0.25 each) of all secondary structure types. Definitions of r.m.s.d. and n.r.m.s.d. are described under “Experimental Procedures.” The secondary structure types are defined as in Table 1.
|
Secondary structure
|
SSC (x-ray)
|
SSC (x-ray) versus CD
|
SSC (rd) (simulated)
|
SSC (rd) versus CD
|
||
|---|---|---|---|---|---|---|
| RMSD | NRMSD | RMSD | NRMSD | |||
| Native MfpA | ||||||
| Htotal | 0.087 | 0.061 | 1.284 | 0.250 | 0.217 | 4.551 |
| Stotal | 0.219 | 0.101 | 0.478 | 0.250 | 0.114 | 0.541 |
| Turns | 0.219 | 0.026 | 0.119 | 0.250 | 0.039 | 0.175 |
| Unordered | 0.475 | 0.205 | 0.340 | 0.250 | 0.370 | 0.616 |
| Overall | 0.119 | 0.351 | 0.223 | 0.577 | ||
| Dialysis refolded MfpA | ||||||
| Htotal | 0.087 | 0.075 | 0.482 | 0.250 | 0.106 | 0.677 |
| Stotal | 0.219 | 0.099 | 0.311 | 0.250 | 0.068 | 0.214 |
| Turns | 0.219 | 0.011 | 0.052 | 0.250 | 0.036 | 0.167 |
| Unordered | 0.475 | 0.167 | 0.532 | 0.250 | 0.074 | 0.234 |
| Overall | 0.104 | 0.401 | 0.109 | 0.289 | ||
FIGURE 5.

The CD spectra of native (A) and dialysis-renatured (B) MfpA. The experimentally determined CD spectra (▪) in Buffer B at the conditions similar to those of Fig. 2B are shown along with spectra calculated from the secondary structure contents shown in Table 1 (ContinLL, ⋆; Selcon3, ▴). The calculated spectra are based on the 48-protein reference set available in CDPro. Reference sets of 43 proteins (CDPro) or 72 proteins (DICHROWEB) provide similar results.
Native MfpA does not detectably form high order aggregates; the Raleigh light scattering of a solution of native MfpA is low (Fig. 3C). Sedimentation equilibrium analysis confirms that native MfpA is a stable dimer in Buffers A and B (molecular mass = 39,185 (37,302 and 41,116) and 40,819 (38,509 and 43,166) Da, respectively.
Denatured MfpA—Upon denaturation in the solutions containing either 8.5 m urea (Fig. 2A, annotated D; Fig. 3, gray bars) or 6 m guanidinium HCl (data not shown), the fluorescence parameters of MfpA change to those expected of proteins with fully exposed and mobile tryptophan side chains (20). The CD spectrum of denatured MfpA could be determined down to 217 nm, revealing the expected disappearance of regular secondary structure (Fig. 2B). The Raleigh light scattering of the solution of denatured MfpA remains low; the unfolded protein does not aggregate in denaturant-containing solution (Fig. 3C). Thus, denatured MfpA shows the absence of regular secondary structure and full solvent accessibility of tryptophan residues in solutions expected for a fully denatured protein.
Dialysis-refolded MfpA—Neither single nor multistep dialysis utilizing a wide range of solution conditions (see “Experimental Procedures”) elicited recovery of the native fluorescence spectrum following removing of denaturant (Fig. 2A, annotated R). That denatured MfpA aggregates during refolding by these protocols is evident by the dramatic increase in Raleigh light scattering of these solutions (Fig. 3C, dashed bars). These aggregates do not precipitate during 72 h at room temperature; sedimentation velocity analysis revealed a heterogeneous distribution of S values ranging from 5 to 100 (data not shown). The anisotropy of the aggregate solution is higher than that of native MfpA reflecting the larger size and decreased mobility of the tryptophan residues relative to the native dimer (Fig. 3B).
Reducing agent has no effect on the formation or stability of the aggregates. The MfpA mutant C179S has one of two solvent-exposed cysteine residues removed. This protein's aggregation upon refolding is identical to that of the wild type protein.
The CD spectrum of dialysis refolded MfpA (MfpA) is characteristic of the α/β and α + β secondary structure classes (9, 21). The spectrum contains two peaks, positive at ∼192 nm and the negative one at ∼218 nm (Fig. 2B). All of the programs used to calculate the secondary structures of this refolded MfpA (see “Experimental Procedures”) perform well. The overall RMSD = 0.029 predicts the secondary structure content with reasonable accuracy; the overall NRMSD = 0.11 yields small dispersion in the calculated numbers of amino acid residues in the given type of secondary structures (Table 1). The high accuracy of these calculations yields low error of the calculated fractions (Fig. 4) and good agreement between the experimental and calculated CD spectra (Fig. 5B).
In contrast, these calculations are in poor agreement with the predictions derived from the crystal structure of native MfpA (overall NRMSD = 0.401; Table 2). This value is higher than that of the protein with a random distribution of structural types (0.401 versus 0.289; Table 2). Thus, the structure of directly refolded MfpA is clearly different from the native protein crystal structure.
“Time-dependent Refolding” Yields Native MfpA—The differences between the secondary structures of native and dialysis-refolded MfpA (Fig. 2B) are accompanied by changes in tertiary structure and aggregation. Since reversibility of denaturation and renaturation is a prerequisite for thermodynamic analysis, we considered how this hysteresis could be overcome. Since various solution conditions and dialysis protocols failed to solve the problem (see “Experimental Procedures”), we explored a novel approach. We measured the rate at which MfpA denatures upon the addition of urea, reasoning that a slow process within the denatured ensemble might facilitate aggregation.
The denaturation reaction is characterized by coincident fluorescence intensity and anisotropy monoexponential time progress curves with a decay rate of 0.97 ± 0.05 min-1. Thus, MfpA denaturation is relatively fast; less than ∼2% of the native protein remains 4 min after the addition of denaturant (Fig. 6A; the arrow marks the time at which the fluorescence intensity and anisotropy progress curves plateau at their minimum values).
Based on this result, we efficiently refold MfpA by exploiting the difference in rates between intra- and intermolecular processes. At intervals commencing 5 min after unfolding was initiated, the denaturant and protein are diluted to initiate renaturation. These aliquots are incubated for 30 min, after which the λmax of the fluorescence spectra and Raleigh light scattering are separately measured to separately track the changes in MfpA tertiary structure and aggregation, respectively (Fig. 6, B and C).
When MfpA denatured for 5 min is refolded, the position of λmax is close to that of the native protein (Figs. 2 and 6B). As the denaturation time increases, the position of λmax and the magnitude of the Raleigh light scattering of the renatured protein gradually approach the values characteristic of protein refolded by dialysis (Figs. 3 and 6, B and C). The intermediate scattering values reflect different proportions of native and aggregated forms of MfpA present in solution.
Removal of the aggregates from a renatured sample by high speed centrifugation restores the position of fluorescence λmax to that of native MfpA and eliminates the light scattering (compare solid and open circles in Fig. 6, B and C). Homogeneous native MfpA is thus obtained from the time-dependent refolding solutions following centrifugation of the aggregate (Fig. 6).
The fraction of refolded MfpA is proportional to the fluorescence intensity of the supernatant of the solution clarified by centrifugation. The quantitative yield of native MfpA shown in Fig. 6D was obtained by fitting the fraction native protein versus denaturation time to Equation 2, as described under “Experimental Procedures.”
The fluorescence spectra, anisotropy (data not shown), and CD spectra of the time-dependent renatured MfpA solutions clarified by centrifugation completely recapitulate native MfpA (compare Figs. 2 and 7). Sedimentation equilibrium analysis of clarified refolded MfpA yields a molecular mass of 40,317 (37,811 and 41,887) Da, consistent with a native dimer. Thus, denatured MfpA refolded by the time-dependent renaturation protein acquires secondary, tertiary, and quaternary structure that matches the native protein.
FIGURE 7.

The fluorescence (A) and CD (B) spectra of native (▪) and “time-dependent refolded” MfpA (○) in Buffer B. The solid line tracks the CD spectrum of native MfpA smoothed by polynomial fitting. The experimental conditions are similar to those in Fig. 2.
DISCUSSION
The fluorescence of native MfpA reflects the apolar environment of the MfpA tryptophan residues inside the protein (1, 20). Identical spectra are obtained in the buffers in which the MfpA dimer exhibits its biological functions (1) and that are required for the acquisition of its far UV CD spectra. The consistency of the fluorescence intensity and anisotropy, Raleigh light scattering, and analytical ultracentrifugation results obtained in these two buffers demonstrates the identity of the native MfpA structure in them.
MfpA denatured in solutions containing high concentration of urea or guanidinium HCl shows the fluorescence properties typical of denatured proteins; λmax increases from ∼332 to ∼355 nm, and the anisotropy decreases from ∼0.12 to ∼0.06 (Figs. 2 and 3). Neither direct dilution nor dialysis from highly concentrated denaturant recovers the native structure of MfpA. Directly refolded MfpA has all of the attributes of a higher order aggregate whose secondary, tertiary, and quaternary structure differ significantly from that of the native protein (Figs. 2 and 3).
Unlike many misfolded proteins, directly refolded MfpA does not precipitate. The aggregate is stable in solution for at least 72 h. Whether this conformer of PRP has functional and biological relevance is unknown. In contrast, denatured MfpA renatured by our time-dependent protocol has secondary, tertiary, and quaternary structures matching those of the native protein (Figs. 2, 3, and 7). Below we explore our results pertaining to the solution structure of native MfpA and the reversibility of its denaturation and renaturation.
Structural Interpretation of the CD spectrum of MfpA—The composition of secondary structure types calculated from the CD spectra of native and renatured MfpA yields RMSD values consistent with the typical accuracy described in literature (Table 1) (13, 18, 19). In contrast, the poor NRMSD values calculated for native MfpA are reflected in the large errors associated with the secondary structure fractions (Fig. 4) and the large content differences calculated by the different analysis programs. Although a low NRMSD value is a necessary but not sufficient condition for an accurate result, a high NRMSD value (>0.2) indicates poor agreement between the calculated and experimental secondary structures. This is the case for native MfpA (Table 1 and Figs. 4 and 5A).
The “Cluster” program determines the tertiary structure class of a protein (9) based on a classification scheme that includes the secondary structure classes, all α, all β, α-β (including α + β and α/β), and “denatured” (21). Cluster analysis predicts that native MfpA is in the “denatured” class. Clearly, this label cannot be applied literally to native MfpA in solution, since its physical properties are characteristic of a folded protein, and it is biologically active at buffer conditions used in this study (1).
Thus, what does the Cluster-defined “denatured” mean in relation to the PRP fold of native MfpA? It means only that the shape of the MfpA CD spectrum resembles the spectrum of denatured proteins. The negative band of 193 nm present in the CD spectrum of the native MfpA (Fig. 2B) is similar to the negative band of 195–198 nm for denatured proteins (9, 17). This peak is probably why the program Selcon also predicts MfpA to be a denatured protein (Fig. 5A). A reason for the poor performance of these programs is that they appear to ignore the second peak at 218 nm (Fig. 2B). The combination CD spectral peaks at 218 and 193 nm are unprecedented. We interpret this result to reflect that the β-strands and turns of the native MfpA within the right-handed quadrilateral β-helix that define the PRP fold differ from the canonical secondary structure types represented in the CDPro reference protein set. An alternative explanation of the poor performance of the secondary structure calculations for native MfpA is that the PRP fluctuates in solution (Table 1 and Figs. 4 and 5A) (17).
Our preference for these explanations is that the performance of the secondary structure calculations is more accurate for dialysis-refolded MfpA than for the native protein (Table 1 and Fig. 4). Thus, we used dialysis-refolded MfpA to test the accuracy of the secondary structure calculation in aggregates where the protein's structure more likely resembles the reference proteins. In this case, the secondary structure predictions are robust, and the calculated and measured CD spectra are in good agreement (Table 1 and Fig. 5B). Cluster analysis also reports that dialysis-refolded MfpA is related to an “α + β” tertiary class of proteins (21). We hypothesize that the secondary structure of dialysis refolded MfpA is canonical without the alterations that can dramatically change the CD spectra of proteins, especially β-strands (9, 17).
Inspection of the structure of native MfpA reveals that amino acid residues 90–150 consist only of single-residue β-bridges and two residues turns (Fig. 1). Thus, it is possible that the β-bridges and turns observed in the crystal structure are not preserved in solution. In support of this hypothesis is the observation that integration of molecular dynamics with CD spectrum calculations yields better agreement between theory and experiment than that obtained with the static crystal structure (17). The malleability of the solution structure may allow MfpA to better accommodate its likely cell target, DNA gyrase, upon binding (1). Such conformational malleability may facilitate the ability of MfpA to confer bacterial resistance to antibiotics.
Chromophores other than amides are unlikely to influence the CD spectrum of MfpA. Although aromatic side chains, particularly tryptophan, can contribute to CD spectra and influence secondary structure calculations, tryptophan residues generally do not exhibit intrinsic optical activity; their optical activity is introduced by the protein chiral field (17). Of the three tryptophan residues present in each MfpA monomer, only one (Trp-154) is buried in the interior of the protein and thus potentially chiral (Fig. 1). This single tryptophan is insufficient to perturb the CD spectrum of MfpA produced by 182 amino acids.
Differences in the Solution and Crystalline Structure of Native MfpA—Based upon the NRSMD values (Table 2), we conclude that neither native MfpA nor dialysis-refolded MfpA in solution precisely recapitulates the crystal structure. The differences in structure are seen in the dispersion of the NAA (CD) values and their lack of correspondence to the NAA (x-ray) values (Table 1). Indeed, the discrepancy between the CD estimates and the x-ray structure is comparable with a random secondary structure distribution (Table 2). We believe that the poor correspondence between the solution and crystalline structure of native MfpA is partly due to the novelty of the PRP fold (1) and its absence from CD reference data bases.
Accurate prediction of protein secondary structure from CD spectra depends on both the quality of the secondary structure fractions derived from x-ray structures and the quality of the experimental and reference spectroscopy data. The quality of crystal structure of the MfpA and reference proteins available in CDPro and DICHROWEB packages reports well resolved, accurate secondary structure and thus obviates their integrity as a source of the poor correspondence between x-ray and CD data.
Contemporary methods that estimate protein secondary structures from CD spectra assume the identity of the protein structure in crystal and solution (13, 17, 18). The validity of this assumption is particularly questionable for proteins with low helical content. The CD spectrum of MfpA is reminiscent of the small all β-sheet protein rubredoxin (15, 16), whose agreement between the secondary and tertiary structure calculated from its CD spectra and crystal structure is poor (9, 16). This discrepancy most likely results from unique features of individual β-strands and turns.
The reference CD spectrum of the β-sheet is difficult to characterize due to the variations in the geometry of β-strands in the proteins (17). Distorted portions of β-strands yield CD signatures unlike those of canonical strands (13). There are seven or eight types of turns ranging from α-like to β-like that cannot be distinguished by CD (13, 16, 17). The consideration of distortions that usually improves the performance of the CD secondary structure prediction (13) does not help the MfpA calculations.
All of the β-strands of MfpA are within a new structural motif, the PRP fold. Some strand segments in native MfpA are short and limited to two hydrogen-bonded residues. Many turns (T) often include only two amino acids rather than the three or four residues that is ideal (17). A great deal of the structure is represented by isolated β-bridges (B) and bends (S; Fig. 1). As noted above, such structures are presumably dynamic in solution and unlikely to preserve the precise crystalline structure.
These characteristics make agreement between the reference and MfpA CD spectra problematic. Thus, the poor performance of the CD calculations (Table 1 and Fig. 4) and the structural differences between crystalline native MfpA and the protein solution (Table 2) probably reflects alteration in solution of the secondary structure types in the right-handed quadrilateral β-helix along with the dynamics of some of the β-strands, β-bridges, and turns.
Correct Refolding of MfpA—The partitioning of denatured proteins between native and aggregated forms during folding is not well understood despite the practical and biological importance of the process. How tertiary and secondary structure effect the aggregation and conformational stability of a protein and vice versa is unclear (22). Denatured proteins are prone to form aggregates during refolding (23). These aggregates can result from rapid refolding and have been mistaken for folding intermediates (24), since formation of monomeric intermediates often precedes intermolecular interaction (24, 25). The aggregation of MfpA when refolded after extended incubation in denaturant is a major impediment to study of the thermodynamic stability of the PRP fold.
We have shown that MfpA undergoes a slow conformational change following rapid denaturation in chemical denaturant that increases its propensity to form an aggregate. Neither dilution nor long term dialysis prevents aggregation after 30 min of incubation in denaturant (Figs. 2, 3, and 6). The intermediates that lead to aggregation form at a rate of 0.14 ± 0.01 min-1. This rate is much slower than the 0.97 ± 0.05 min-1 rate by which the optical parameters change to values characteristic of the denatured state (Figs. 6 and 7). We hypothesize that MfpA exists as a heterogeneous ensemble of conformers following the addition of denaturant. Some conformers form nonnative aggregates during refolding. Some conformers form native protein. The conformational equilibrium shifts to the aggregation precursors during incubation in denaturant (Fig. 6).
Although the denatured state of proteins was historically viewed as a random coil (26), recent studies show that this is not the case. Denatured proteins can have native-like topology (27). NMR spectra of denatured proteins are typically far removed from that of a statistical random coil (28). The presence of six clusters of hydrophobic structure within the protein lysozyme was detected under strong denaturing conditions (29). The stability of the ribosomal protein L9 is increased by modulation of specific nonnative electrostatic interactions in the denatured state (30). Four hydrophobic segments cluster in the denatured state of staphylococcal nuclease (31). These hydrophobic clusters do not guide folding to the native structure (32). Conformational changes within the denatured state influence the folding of all β-sheet proteins (33).
Based upon these observations, a rationale for the partitioning of denatured MfpA between native and misfolded conformations upon refolding is the existence of a slowly equilibrating ensemble of different structures in denaturant-containing solution. Kinetic selection at early stages of denaturation preferentially selects the structure leading to formation of the native MfpA (Figs. 6 and 7). The yield of native structure depends on the starting time of refolding. Renaturation commencing after 30 min of denaturation results in the almost complete formation of the nonnative aggregates (Figs. 2 and 3).
Few studies have been conducted of the dependence of protein refolding on the structural nature of the denatured protein ensemble (33). Unsurprisingly, the mechanism(s) of this dependence are not understood. A well documented case is cis-trans isomerization of prolyl bonds (34). The prolyl bond in trans form is predominant in unfolded proteins. If a native protein contains a prolyl bond in cis-form, trans → cis isomerization during folding greatly slows the overall reaction due to its intrinsically slow nature (34). Pro-81 is cis in the native MfpA (2).
Thus, the time-dependent recovery of native protein by refolding from denaturant may be explained by incomplete cis-trans isomerization during the early stages of the MfpA denaturation (Fig. 6D). In addition, proline residues in the cis conformation introduce tight turns into the protein structure. The presence of the trans isomer of Pro-81 at late stages of denaturation (Fig. 6) may favor partitioning of MfpA into the nonnative aggregates (Fig. 2).
The selection of kinetically trapped intermediates that lead to formation of protein native structures demonstrated herein for MfpA may be important for crystallography and the studies of thermodynamics of the folding process for proteins when thermodynamic equilibrium cannot be reached using standard procedures, such as dialysis and dilution of denaturant.
Evidence suggests that MfpA binds to DNA gyrase, prevents binding of the enzyme to its natural substrate DNA, and thereby confers resistance to an antibiotic that targets the enzyme's catalytic cycle. The similarity to DNA of the size, shape, and distribution of negative charge on the surface of MfpA makes this hypothesis very appealing. However, “similar” is not “the same,” and nuances of the interacting surfaces of MfpA and DNA gyrase may be important in mediating the MfpA-gyrase interaction. The physical basis of this interaction and the function of MfpA as an inhibitor of antibiotic function are still to be conclusively determined. Alteration of canonical secondary structure within the right-handed quadrilateral β-helix formed and “random meander” of the PRP fold could provide MfpA in solution the necessary flexibility to adjust to the complementary surface of the DNA gyrase and thus be an important contributor to the biological function of MfpA and other PRPs.
Acknowledgments
We thank Dr. Sergei Venyaminov for insight into CD spectroscopy and helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grant R01-GM079618. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PRP, pentapeptide repeat protein; RMSD, root mean square deviation; NRMSD, normalized RMSD; SSC, secondary structure content.
S. Khrapunov, H. Cheng, S. Hegde, J. Blanchard, and M. Brenowitz, manuscript in preparation.
The C179S mutation leaves a single solvent-accessible cysteine residue at the N terminus of the protein. This mutant is being used in studies where the protein is extrinsically labeled, which will be presented elsewhere. Its intrinsic fluorescence characteristics, denaturation, and stability are included herein to document its properties and test whether disulfide linkages contribute to MfpA aggregation during refolding (Fig. 2).
References
- 1.Hegde, S., Vetting, M., Roderick, S., Mitchenall, L., Maxwell, A., Takiff, H., and Blanchard, S. (2005) Science 308 1480-1483 [DOI] [PubMed] [Google Scholar]
- 2.Vetting, M., Hegde, S., Fajaro, J., Fiser, A., Roderick, S., Takiff, H., and Blanchard, S. (2006) Biochemistry 45 1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drlica, K., and Malik, M. (2003) Curr. Topics Med. Chem. 3 249-282 [DOI] [PubMed] [Google Scholar]
- 4.Takiff, H., Salazar, L., Guerrero, C., Philipp, W., Huang, W., Kreiswirth, B., Cole, S., Jacobs, W. J., and Telenti, A. (1994) Antimicrob. Agents Chemother. 38 773-780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Putnam, D., and Tainer, J. (2005) DNA Repair 4 1410-1420 [DOI] [PubMed] [Google Scholar]
- 6.Dryden, D. (2006) Trends Biotechnol. 24 378-382 [DOI] [PubMed] [Google Scholar]
- 7.Li, F., and Mullins, J. (2002) Methods Mol. Biol. 182 19-27 [DOI] [PubMed] [Google Scholar]
- 8.Sreerama, N., and Woody, R. (2000) Anal. Biochem. 287 252-260 [DOI] [PubMed] [Google Scholar]
- 9.Venyaminov, S., and Vassilenko, K. (1994) Anal. Biochem. 222 176-184 [DOI] [PubMed] [Google Scholar]
- 10.Lees, J., Miles, A., Janes, R., and Wallace, B. (2006) Bioinformatics 2 1955-1962 [DOI] [PubMed] [Google Scholar]
- 11.Kabsch, W., and Sander, C. (1983) Biopolymers 22 2577-2637 [DOI] [PubMed] [Google Scholar]
- 12.Frishman, D., and Argos, P. (1995) Proteins 23 566-579 [DOI] [PubMed] [Google Scholar]
- 13.Sreerama, N., Venyaminov, S., and Woody, R. (1999) Protein Sci. 8 370-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace, B., Lees, J., Orry, A., Lobley, A., and Janes, R. (2003) Protein Sci. 12 875-884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sreerama, N., and Woody, R. (2003) Protein Sci. 12 384-388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brahms, S., and Brahms, J. (1980) J. Mol. Biol. 138 149-178 [DOI] [PubMed] [Google Scholar]
- 17.Sreerama, N., and Woody, R. (2004) Methods Enzymol. 383 318-351 [DOI] [PubMed] [Google Scholar]
- 18.Greenfield, N. (2006) Nat. Protocols 1 2876-2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuo, K., Yonehara, R., and Gekko, K. (2005) J. Biochem. (Tokyo) 138 79-88 [DOI] [PubMed] [Google Scholar]
- 20.Lakowicz, J. (1999) Principles of Fluorescence Spectroscopy, 2nd Ed., p. 452, Kluwer Academic/Plenum, New York
- 21.Manalavan, P., and Johnson, W. J. (1983) Nature 305 831-832 [Google Scholar]
- 22.Tartaglia, G., Pechmann, S., Dobson, C., and Vendruscolo, M. (2007) Trends Biochem. Sci. 32 204-206 [DOI] [PubMed] [Google Scholar]
- 23.Fink, A. (1998) Folding Des. 3 R9-R15 [Google Scholar]
- 24.Silow, M., and Oliberg, M. (1999) Proc. Natl. Acad. Sci. U. S. A. 94 6084-6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uversky, V., Karnoup, A., Khurana, R., Segel, D., Doniach, S., and Fink, A. (1999) Protein Sci. 8 161-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanford, C. (1968) Adv. Protein Chem. 23 12-282 [DOI] [PubMed] [Google Scholar]
- 27.Shortle, D., and Ackerman, M. (2001) Science 293 487-489 [DOI] [PubMed] [Google Scholar]
- 28.Dyson, H., and Wright, P. (2002) Adv. Protein Chem. 62 311-340 [DOI] [PubMed] [Google Scholar]
- 29.Klein-Seetharaman, J., Oikawa, M., Grimshaw, S., Wirmer, J., Elke Duchardt, E., Ueda, T., Imoto, T., Smith, L., Dobson, C., and Schwalbe, H. (2002) Science 295 1719-1722 [DOI] [PubMed] [Google Scholar]
- 30.Cho, J., Sato, S., and Raleigh, D. (2004) J. Mol. Biol. 338 827-837 [DOI] [PubMed] [Google Scholar]
- 31.Wang, Y., and Shortle, D. (1996) Protein Sci. 5 1898-1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baldwin, R. (2002) Adv. Protein Chem. 62 361-367 [DOI] [PubMed] [Google Scholar]
- 33.Kathir, K., Kumar, T., Rajalingam, D., and Yu, C. (2005) J. Biol. Chem. 280 29682-29688 [DOI] [PubMed] [Google Scholar]
- 34.Schmid, F. (2005) in Protein Folding Handbook (Buchner, J., and Kiefhaber, T., eds) pp. 916-945, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany