

Abstract
Xanthine oxidoreductase (XOR) generates proinflammatory oxidants and
secondary nitrating species, with inhibition of XOR proving beneficial in a
variety of disorders. Electrophilic nitrated fatty acid derivatives, such as
nitro-oleic acid (OA-NO2), display anti-inflammatory effects with
pleiotropic properties. Nitro-oleic acid inhibits XOR activity in a
concentration-dependent manner with an IC50 of 0.6
μm, limiting both purine oxidation and formation of superoxide
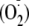 .
Enzyme inhibition by OA-NO2 is not reversed by thiol reagents,
including glutathione, β-mercaptoethanol, and dithiothreitol.
Structure-function studies indicate that the carboxylic acid moiety, nitration
at the 9 or 10 olefinic carbon, and unsaturation is required for XOR
inhibition. Enzyme turnover and competitive reactivation studies reveal
inhibition of electron transfer reactions at the molybdenum cofactor accounts
for OA-NO2-induced inhibition. Importantly, OA-NO2 more
potently inhibits cell-associated XOR-dependent
.
Enzyme inhibition by OA-NO2 is not reversed by thiol reagents,
including glutathione, β-mercaptoethanol, and dithiothreitol.
Structure-function studies indicate that the carboxylic acid moiety, nitration
at the 9 or 10 olefinic carbon, and unsaturation is required for XOR
inhibition. Enzyme turnover and competitive reactivation studies reveal
inhibition of electron transfer reactions at the molybdenum cofactor accounts
for OA-NO2-induced inhibition. Importantly, OA-NO2 more
potently inhibits cell-associated XOR-dependent
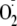 production than does allopurinol. Combined, these data establish a novel role
for OA-NO2 in the inhibition of XOR-derived oxidant formation.
production than does allopurinol. Combined, these data establish a novel role
for OA-NO2 in the inhibition of XOR-derived oxidant formation.
Xanthine oxidoreductase
(XOR)3 is a
molybdoflavin protein that serves as the rate-limiting enzyme in the terminal
steps of purine degradation in humans, catalyzing the oxidation of
hypoxanthine to xanthine and finally to uric acid. Intracellularly, XOR exists
primarily as a dehydrogenase (XDH) where the majority of substrate-derived
electrons reduce NAD+ to NADH. During inflammatory conditions,
reversible oxidation of critical cysteine residues or limited proteolysis
converts XDH to xanthine oxidase (XO), which reduces O2 to
superoxide
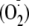 and hydrogen peroxide (H2O2)
(1,
2). Conversion to XO, however,
is not requisite for reactive oxygen species (ROS) production, as XDH displays
partial oxidase activity (3).
When such ROS production occurs in the vascular compartment, normal function
is altered by enhanced redox-dependent cell signaling reactions or by the
reduction of NO bioavailability due to reaction with
and hydrogen peroxide (H2O2)
(1,
2). Conversion to XO, however,
is not requisite for reactive oxygen species (ROS) production, as XDH displays
partial oxidase activity (3).
When such ROS production occurs in the vascular compartment, normal function
is altered by enhanced redox-dependent cell signaling reactions or by the
reduction of NO bioavailability due to reaction with
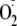 (4,
5). Both animal models and
clinical studies affirm a key role for XOR in pathophysiology, where
inhibition of XOR has proven beneficial in a variety of vascular inflammatory
processes (6).
(4,
5). Both animal models and
clinical studies affirm a key role for XOR in pathophysiology, where
inhibition of XOR has proven beneficial in a variety of vascular inflammatory
processes (6).
The splanchnic system, the principal site of XOR activity, readily releases
XOR into the circulation in response to ischemic or inflammatory insults
(7,
8). Once released into the
vascular compartment, XDH is rapidly converted to XO. Cationic amino acid
motifs present on the enzyme confer a high affinity (kd =
6 nm) for negatively charged glycosaminoglycans (GAGs) located on
the luminal face of the vascular endothelium
(9). This XO-GAG association
induces substantial sequestration and thus amplification of local endothelial
XO concentration, producing a microenvironment with enhanced
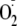 and H2O2 production. At the same time, GAG association
results in resistance to XOR inhibition by oxypurinol, the active metabolite
of allopurinol, increasing the Ki from 230 nm
for soluble XOR to 405 nm for GAG-bound XOR
(10,
11). Identification of more
efficacious inhibitors, especially with regard to endothelium-associated XOR,
will facilitate the treatment of vascular inflammatory conditions.
and H2O2 production. At the same time, GAG association
results in resistance to XOR inhibition by oxypurinol, the active metabolite
of allopurinol, increasing the Ki from 230 nm
for soluble XOR to 405 nm for GAG-bound XOR
(10,
11). Identification of more
efficacious inhibitors, especially with regard to endothelium-associated XOR,
will facilitate the treatment of vascular inflammatory conditions.
The nitration of unsaturated fatty acids represents a convergence of NO, ROS, and lipid-mediated signaling. Current data suggest that oxidative inflammatory conditions that include the generation of NO-derived species induce the nitration of unsaturated fatty acids (12–14). Two fatty acid nitroalkene derivatives, nitro-oleic acid (OA-NO2; 9- or 10-nitro-9-cis-octadecenoic acids) (26) and nitrolinoleic acid (LNO2; 9-, 10-, 12-, or 13-nitro-octadecadienoic acids) (27) display pleiotropic cell signaling capabilities. For example, these nitroalkenes preferentially activate peroxisome proliferator-activated receptor-γ, inhibit inflammatory cytokine secretion from macrophages, impede platelet activation, and, in aqueous milieu, decay via a Nef-like reaction to yield NO (16–19).
A central mechanism accounting for nitroalkene-mediated signaling is via post-translational modification of proteins (15). The alkenyl nitro configuration imparts electrophilic reactivity on the β-carbon adjacent to the nitro-bonded carbon, facilitating Michael addition reactions with nucleophiles such as cysteine and histidine residues of proteins (15). For example, adduction of OA-NO2 with the catalytic Cys-149 of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and promotes membrane localization due to enhanced hydrophobicity of the nitroalkene-modified enzyme (15). Compared with other biological electrophilic lipids, nitro-fatty acids react with thiols with a high rate constant (20).
Since electrophilic substrates and inhibitors of XOR both interact with the molybdenum cofactor, and long chain alkene modifications augment the effectiveness of XOR inhibitors, the effects of various nitro-fatty acid derivatives on XOR activity were evaluated (21, 22). We reveal that OA-NO2 is a potent and irreversible inhibitor of soluble and GAG-immobilized XOR. These data suggest that nitration of fatty acids to nitroalkene derivatives yields products that can down-regulate XOR-dependent generation of ROS and account for some of the anti-inflammatory actions observed for these lipid signaling mediators.
EXPERIMENTAL PROCEDURES
Materials—Xanthine, allopurinol, Chelex resin, diphenyleneiodonium chloride (DPI), 2,6-dichlorophenol indophenol (DCPIP), and uric acid were from Sigma. Medium 199 and fetal bovine serum were from Invitrogen. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was obtained from Alexis Biochemicals (San Diego, CA). Sodium dithionite was from J. T. Baker Inc.
XOR Purification and Activity—Enzyme was purified from fresh
bovine cream and stored in ammonium sulfate at 4 °C according to the
method of Rajagopalan (23).
Enzymatic activity was determined either spectrophotometrically by the rate of
uric acid formation monitored at 292 nm in 50 mm potassium
phosphate (KPi), pH 7.4 (ε = 11
mm–1 cm–1), or electrochemically
via reverse phase high pressure liquid chromatography analysis of uric acid
production (ESA Coul-Array System, Chelmsford, MA) (1 unit = 1 μmol of
urate/min), as previously
(11). XDH activity was
distinguished from XO activity by incubation with NAD+, as
previously described (24).
Formation of
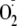 was assessed by the superoxide dismutase (SOD)-inhibitable reduction of
cytochrome c (550 nm)
(25).
was assessed by the superoxide dismutase (SOD)-inhibitable reduction of
cytochrome c (550 nm)
(25).
Nitroalkene Preparation—Nitrated lipids (Table 1) were synthesized by multiple methods. Nonspecific nitrolipids consist of equimolar proportions of single-nitration products at all possible nitration positions. Nonspecific nitro-oleic acid and methyl nitro-oleate (OA-NO2 and OA-NO2-methyl ester), nonspecific nitrolinoleic acid (LNO2), specific 9-nitro-oleic acid (9-OA-NO2), specific Z-isomer 9-nitro-oleic acid (9Z-OA-NO2), specific saturated 9-nitro-octadecanoic acid (9-SA-NO2, where “SA” represents stearic acid), specific alcohol 9-OA-NO2-OH, and specific positional isomer 12-isoOA-NO2 were prepared as previously reported (26–28). Nonspecific nitrooleamide (OA-NO2-amide) was synthesized from the corresponding native fatty acid by the nitroselenation method (26, 27). Biotinylated nitro-oleic acid (OA-NO2-biotin) was synthesized from nonspecific nitro-oleic acid and biotin hydrazide (29). Keto-containing diene 13-KODE was synthesized by oxidation of (9Z, 11E)-13-(S)-hydroxy-octadeca-9,11-dienoic acid (30). Oxime-containing derivative (13-OxODE) was synthesized from 13-KODE (31).
TABLE 1.
Effects of structural variation on enzyme inhibition
XOR was exposed to structural variants of OA-NO2, and enzymatic activity was assessed by monitoring the evolution of uric acid (292 nm) from xanthine. Values represent the mean and S.D. of at least three independent determinations. Compounds were used at a final concentration of 10 μm.

*, p < 0.5 compared with untreated control.
XOR Binding to GAGs—Xanthine oxidase was bound to heparin-Sepharose 6B (HS6B) as previously (11). Briefly, XOR (2 mg/ml) was added to a fixed amount of gel (0.05 g, dry weight), and the mixture was gently stirred in 5 mm KPi, pH 7.4 (2 ml final volume) at 25 °C for 30 min. The suspension was centrifuged at 10,000 × g for 5 min and washed, and the pellet was resuspended in 5 mm KPi, pH 7.4.
Anaerobic Experiments—Anaerobic analyses were performed in a table-top glove box (Coy Instruments, Grass Lake, MI) purged with N2. Oxygen concentrations were verified by monitoring the atmosphere in the chamber with an O2 monitor (Maxtec, Salt Lake City, UT). All buffers were equilibrated >12 h before use. Spectrophotometric determinations were carried out in gas-tight cuvettes, and FAD reduction/oxidation studies were performed by the method of Ichimori (32).
Oxygen Consumption Studies—Real time concentrations of molecular O2 were determined polarographically using an Apollo 4000 Free Radical Analyzer (World Precision Instruments, Sarasota, FL). Experiments were performed at standard temperature and pressure.
Cellular Studies—Bovine aortic endothelial cells were isolated as previously (11). Primary cell culture, routine passage, and experimental manipulations were all conducted in the absence of proteases. Cells were propagated by subculturing (1:4 ratios) in Medium 199 containing 5% fetal bovine serum and thymidine (10 μm). Cells were utilized between passages 4 and 8 and were monitored visually for typical cobblestone morphology indicative of endothelial cells and by staining for von Willebrand factor expression.
EPR Spectrometry—The spin trap DMPO was further purified with activated charcoal, and the concentration was verified spectrophotometrically at 228 nm and ε228 = 7,800 M–1 cm–1. EPR measurements were performed at 25 °C using a Bruker Elexsys E-500 spectrometer equipped with an ER049X microwave bridge and an AquaX cell. Studies were performed with 20 milliwatts microwave power, 100 kHz modulation frequency, 1 G modulation amplitude, 3510 G center field, 100 G sweep width, 164 ms time constant, 168 s sweep time, and 60 dB receiver gain with 1024 data points. Adventitious metals were removed from all buffers and water by treatment with Chelex resin. Buffers were then evaluated for the presence of metals by testing for the appearance of the ascorbyl radical in the EPR upon the addition of 100 μm ascorbic acid. The following constituents were added as indicated: DMPO (25 mm), allopurinol (50–100 μm), OA-NO2 (0–50 μm), SOD (10 units/ml), and xanthine (50 μm) to a bovine aortic endothelial cell suspension (3 × 106 cells/ml in Chelex-treated PBS, pH 7.4), and the spectrum was recorded after 10 min. Treatment of cells with XO and preparation is as follows. Confluent bovine aortic endothelial cells were exposed to XO (5 milliunits/ml) for 20 min at 25 °C, harvested by mechanical dissociation, washed thoroughly (three times with PBS, pH 7.4), resuspended as a single-cell suspension (3 × 106 cells/ml), and placed on ice (for less than 1 h) until warmed to 25 °C immediately before evaluation in the EPR.
Statistics—Data were analyzed using one-way analysis of variance followed by Tukey's range test for multiple pairwise comparisons. Significance was determined as p < 0.05.
RESULTS
XOR Inhibition—When purified XOR is exposed to increasing
concentrations of either allopurinol or OA-NO2, there is a
concentration-dependent decrease in the initial rate of uric acid formation
(Fig. 1A).
OA-NO2 is significantly more potent than allopurinol in inhibiting
XOR activity with IC50 values of 0.6 versus 2.4
μm, respectively. Exposure to 10 μm native oleic
acid (18:1) or to 125 mm MeOH (vehicle control) did not alter XOR
activity (not shown). Similar to its effect on urate production,
OA-NO2 inhibits
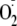 formation in a concentration-dependent manner, with an IC50 of 0.75
μm compared with 2.25 μm for allopurinol
(Fig. 1B). The
addition of up to 2 units/ml SOD does not alter inhibition of uric acid
formation, demonstrating that the mechanism of inactivation is not the result
of release of NO from nitroalkene
(17) and subsequent reaction
with
formation in a concentration-dependent manner, with an IC50 of 0.75
μm compared with 2.25 μm for allopurinol
(Fig. 1B). The
addition of up to 2 units/ml SOD does not alter inhibition of uric acid
formation, demonstrating that the mechanism of inactivation is not the result
of release of NO from nitroalkene
(17) and subsequent reaction
with
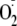 and ONOO– formation
(13). Furthermore, when the
consumption of oxygen was monitored, in the presence of SOD,
xanthine-dependent H2O2 production by XOR is inhibited
by OA-NO2 in the same manner as
and ONOO– formation
(13). Furthermore, when the
consumption of oxygen was monitored, in the presence of SOD,
xanthine-dependent H2O2 production by XOR is inhibited
by OA-NO2 in the same manner as
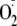 formation (not shown). pH dependence of OA-NO2 inhibition is shown
in Fig. 1C. A Dixon
plot (Fig. 1D)
indicates noncompetitive XOR inhibition by OA-NO2.
formation (not shown). pH dependence of OA-NO2 inhibition is shown
in Fig. 1C. A Dixon
plot (Fig. 1D)
indicates noncompetitive XOR inhibition by OA-NO2.
FIGURE 1.

Nitro-oleic acid inhibits XOR activity. A, following
exposure of XOR (10 milliunits/ml) to a 0–10 μm
concentration of either OA-NO2 (•) or allopurinol (▪) in
50 mm phosphate buffer (pH 7.4, 25 °C), enzymatic activity was
determined by the production of uric acid (λ = 292 nm). Reactions were
initiated by the addition of xanthine (100 μm). Control analyses
were conducted with 10 μm native oleic acid (18:1). The
concentration range from 0 to 1 μm for OA-NO2 is
expanded (inset). Dashed lines, IC50 values
(IC50 = 0.6 and 2.4 μm for OA-NO2 and
allopurinol, respectively). B, following the same protocol as in
A,
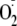 formation was determined by the reduction of cytochrome c (λ =
550 nm) upon OA-NO2 (•) and allopurinol (▪) addition.
Dashed lines demonstrate IC50 values (IC50 =
0.75 and 2.25 μm for OA-NO2 and allopurinol,
respectively). C, the effect of pH on XOR inhibition by
OA-NO2 (0.5 μm) was determined by normalizing the
activity (uric acid production upon the addition of 100 μm
xanthine) in the presence of OA-NO2 to XOR alone. A nonlinear pH
activity (y = (a0 +
a1·10(x –
a2))/(1 + 10(x
– a2)) curve fit produced
r2 = 0.92. D, shown is the Dixon plot
(1/V0 versus [inhibitor]) for OA-NO2
(0.25, 0.5, 1.25, and 2.5 μm) at varying concentrations (12.5
(▴), 25 (▪), and 100 μm (♦)) of xanthine. Data
represent the mean ± S.E. of at least three independent determinations;
*, p < 0.05.
formation was determined by the reduction of cytochrome c (λ =
550 nm) upon OA-NO2 (•) and allopurinol (▪) addition.
Dashed lines demonstrate IC50 values (IC50 =
0.75 and 2.25 μm for OA-NO2 and allopurinol,
respectively). C, the effect of pH on XOR inhibition by
OA-NO2 (0.5 μm) was determined by normalizing the
activity (uric acid production upon the addition of 100 μm
xanthine) in the presence of OA-NO2 to XOR alone. A nonlinear pH
activity (y = (a0 +
a1·10(x –
a2))/(1 + 10(x
– a2)) curve fit produced
r2 = 0.92. D, shown is the Dixon plot
(1/V0 versus [inhibitor]) for OA-NO2
(0.25, 0.5, 1.25, and 2.5 μm) at varying concentrations (12.5
(▴), 25 (▪), and 100 μm (♦)) of xanthine. Data
represent the mean ± S.E. of at least three independent determinations;
*, p < 0.05.
Influence of Reducing Agents and Other Electrophiles on XOR Inhibition—Inhibition of XOR by OA-NO2 was not reversible by the subsequent addition of reducing agents (Fig. 2A). Treatment of OA-NO2-inhibited XOR with GSH (20 mm), β-mercaptoethanol (20 mm), or dithiothreitol (20 mm) did not abolish inhibition. However, the addition of these reducing agents to OA-NO2 prior to exposure of the enzyme abrogated the inhibitory actions of OA-NO2. Control experiments in which NAD+ (50–500 μm) was added as an electron acceptor did not alter uric acid production, indicating that experimentally induced conversion of XO to XDH does not affect inhibition (not shown).
FIGURE 2.

Nitro-oleic acid inhibition of XOR is not reversible by reducing agents. A, XOR (10 milliunits/ml) was exposed to OA-NO2 (10μm) for 10 min at 25 °C in phosphate buffer (50 mm, pH 7.4). Next, GSH (20 mm), β-mercaptoethanol (BME; 20 mm), or dithiothreitol (DTT; 20 mm) was added for 15 min, and then enzymatic activity was determined by the production of uric acid (λ = 292 nm). B, XOR was incubated with 4-hydroxy-2-nonenol (4-HNE) (100 μm) and 15-deoxyprostaglandin J2 (15dPGJ2) (100 μm) or ethyl pyruvate (EP) (100 μm) for 10 min, and then enzymatic activity was determined as in A. Data points represent the mean ± S.E. of at least three independent determinations; *, p < 0.05.
The effect of other electrophilic species on XOR activity, 4-hydroxy-2-nonenol (100 μm), 15-deoxyprostaglandin J2 (100 μm), and ethyl pyruvate (100 μm) was also evaluated (Fig. 2B). Neither electrophilic lipids nor ethyl pyruvate induced XOR inhibition.
Inhibition Is Specific to Site of Fatty Acid Nitration and Conformation—Nitro-oleic acid preparations are an equimolar mixture of 9-cis-octadecaenoic acid nitrated at carbon 9 or 10. The extent of XOR inhibition with pure 9-nitro-9-cis-octadecaenoic was similar to that induced by a mixture of both 9- and 10-nitro-9-cis-octadecaenoic acids (Table 1). Nitro-linoleic acid (LNO2), a nitrated fatty acid preparation consisting of regioisomers of linoleic acid nitrated at carbon 9, 10, 12, or 13, also inhibited XOR but to a lesser extent (76%) than equimolar concentrations of OA-NO2 (98%). Several other OA-NO2 derivatives, differing in vinyl versus allylic NO2 adduction and carboxylic acid derivatization, were ineffective in inhibiting XOR (Table 1).
Nitro-oleic Acid Does Not Affect the FAD Cofactor—In the presence of purine substrate, the addition of DPI prevents FAD-dependent electron transfer, leading to reduction of the enzyme and an inability to reoxidize the molybdenum cofactor, ultimately resulting in inhibition of purine oxidation (Fig. 3A, line 4). DCPIP can accept electrons from the reduced molybdenum of DPI-inhibited XOR and so restores the capacity to oxidize xanthine to uric acid, (Fig. 3A, line 2). However, when DPI-inhibited XOR is first exposed to OA-NO2 before the addition of DCPIP and xanthine, uric acid production remained significantly depressed (Fig. 3A, line 3). Control studies revealed no reactions between OA-NO2 and DPI or DCPIP.
FIGURE 3.

The FAD cofactor of XOR is not affected by OA-NO2. A, XOR (10 milliunits/ml) enzymatic activity was assayed as in Fig. 1 with additions made in the order listed: XOR + xanthine (line 1), XOR + DPI + DCPIP + xanthine (line 2), XOR + DPI + OA-NO2 + DCPIP + xanthine (line 3), XOR + DPI + xanthine (line 4). Concentrations were as follows: xanthine (100 μm), DPI (50 μm), DCPIP (15 μm), and OA-NO2 (10 μm). B, anaerobic absorption spectra of FAD were assessed under the following conditions: 1) XOR (30 milliunits/ml) + dithionite (420 μm), 2) XOR + OA-NO2 (50 μm), 3) XOR + OA-NO2 + dithionite (300 μm), 4) XOR + OA-NO2 + dithionite (420 μm), 5) XOR + OA-NO2 + dithionite (420 μm) and then 30 min exposure to room air. C, XOR (30 milliunits/ml) was placed in a closed system O2 monitor, and O2 levels were monitored over time. At 4 min, NADH (500 μm) was added, and then OA-NO2 (50 μm) and finally DPI (100 μm) were added (solid line). The dashed line represents O2 consumption in the absence of enzyme.
Potential reactions of OA-NO2 with the FAD cofactor were also
addressed (Fig. 3B).
Spectrum 1 represents reduced XOR in the absence of
OA-NO2. When XOR was treated with concentrations of
OA-NO2, inducing full inhibition under anaerobic conditions, a
pronounced absorption (spectrum 2) of the oxidized FAD (450 nm),
similar to XOR in the absence of OA-NO2 (not shown), was
observed. A significant loss (spectrum 3) and near abolition
(spectrum 4) of absorption was observed upon reduction of the
XOR-OA-NO2 complex with increasing dithionite concentration.
Moreover, when a reduced sample (spectrum 4) was exposed to room air
for 30 min, a robust absorption appears (spectrum 5), indicating
oxidation of the FAD. Analysis of NADH-reduced XOR affirmed this conclusion,
since XOR-dependent NADH-mediated O2 consumption was not affected
by OA-NO2 (Fig.
3C, solid line). The addition of 500
μm NADH reduces the FAD in XOR, which then transfers electrons
to O2, inducing the generation of
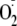 and consumption of O2. The addition of DPI immediately inhibits
this O2 consumption, confirming that the reactions are
FAD-dependent. Control studies in the absence of enzyme show no
OA-NO2 autoxidation and consequent O2 consumption in the
presence or absence of DPI (dashed line). There was also no effect of
preincubation, in the absence of substrate, of XOR with OA-NO2 on
O2 consumption (not shown).
and consumption of O2. The addition of DPI immediately inhibits
this O2 consumption, confirming that the reactions are
FAD-dependent. Control studies in the absence of enzyme show no
OA-NO2 autoxidation and consequent O2 consumption in the
presence or absence of DPI (dashed line). There was also no effect of
preincubation, in the absence of substrate, of XOR with OA-NO2 on
O2 consumption (not shown).
Nitro-oleic Acid Interaction with the Molybdenum Cofactor of XOR—Xanthine can anaerobically reduce the FAD of XOR following the initial reduction of the molybdenum cofactor (Fig. 4A, line 2). However, when XOR was inhibited by OA-NO2, xanthine reduction of the FAD was prevented (line 4). The effect of OA-NO2 on XOR during enzyme turnover is presented in Fig. 4B (1 and 2). When OA-NO2 (10 μm) was added 15 s after XOR (10 milliunits/ml), turnover was initiated by xanthine addition, and formation of uric acid was inhibited 97%. Under the same conditions in the presence of DCPIP (15 μm), the addition of OA-NO2 yielded 30% inhibition.
FIGURE 4.

Nitro-oleic acid affects the molybdenum cofactor of XOR. A, anaerobic absorption spectra of FAD: 1) XOR, 2) XOR + xanthine, 3) XOR + OA-NO2, 4) XOR + OA-NO2 + xanthine. The data represent three replicate determinations. B(1), enzyme activity was measured as in Fig. 1. At t = 15 s after the initiation of enzyme turnover with xanthine (100 μm), 10 μm OA-NO2 was added. The solid line represents conditions in the absence of DCPIP, and the dashed line indicates the presence of DCPIP (15 μm) added before the initiation of turnover with xanthine. B(2), rates of uric acid production before and after the addition of OA-NO2 in B were calculated for four independent experiments; *, p < 0.05. C(1), XOR was exposed to either OA-NO2 or allopurinol (AP) for 5 min, separated from free inhibitor by size exclusion column chromatography (G25 Sephadex), and reactivated by the addition of xanthine (100 μm), and uric acid production was determined. Allopurinol and then OA-NO2 indicates that enzyme was sequentially exposed to allopurinol and then OA-NO2. C(2), rates of uric acid production for the three conditions in C(1) were calculated for three independent experiments. Data points represent the mean ± S.E. of at least three independent determinations; *, p < 0.05.
The suicide inhibitor allopurinol is oxidized to oxypurinol by XOR at the molybdenum cofactor, where oxypurinol then noncompetitively inhibits enzyme activity. Exposure of XOR (30 milliunits/ml) to allopurinol (100 μm) for 5 min (25 °C/room air) followed by removal of unbound inhibitor by size exclusion chromatography produced linear rates of uric acid production 12 min after reactivation with xanthine. In contrast, OA-NO2 (40 μm) produced no observable reactivation (Fig. 4C, 1 and 2). However, when XOR was exposed first to allopurinol (5 min) and then OA-NO2 (5 min), significant reactivation was observed.
Inhibition of Cell-associated XOR by OA-NO2—To
examine the impact of XOR association with GAGs on the inhibitory actions of
OA-NO2, XOR was immobilized on a heparin-Sepharose 6B complex
(XOR-HS6B) and then exposed to either allopurinol (10 μm) or
OA-NO2 (10 μm)
(Fig. 5A). Although 10
μm allopurinol completely inhibited XOR in solution, inhibition
of XOR-HS6B was only 50%. In contrast, OA-NO2 inhibited XOR to
similar extents in both free and heparin-bound states. In order to address
possible differences in allopurinol and OA-NO2 inhibition in a
model of cell-bound XOR, the impact of OA-NO2 on cell-associated
XOR-dependent
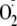 formation was assessed by EPR spectrometry
(Fig. 5B). Control
cells, without added XOR, displayed minimal DMPO radical signal in the
presence of xanthine (Xan). However, upon cellular GAG association
with exogenous XOR, a robust increase in
formation was assessed by EPR spectrometry
(Fig. 5B). Control
cells, without added XOR, displayed minimal DMPO radical signal in the
presence of xanthine (Xan). However, upon cellular GAG association
with exogenous XOR, a robust increase in
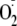 generation was observed (XO + Xan). This spectrum contains a
combination of both DMPO-OOH and DMPO-OH radical adducts. When XOR-treated
cells were exposed to OA-NO2, there was a concentration-dependent
decrease in radical adduct signal intensity. In contrast, allopurinol
treatment only partially inhibited cell-associated
generation was observed (XO + Xan). This spectrum contains a
combination of both DMPO-OOH and DMPO-OH radical adducts. When XOR-treated
cells were exposed to OA-NO2, there was a concentration-dependent
decrease in radical adduct signal intensity. In contrast, allopurinol
treatment only partially inhibited cell-associated
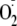 formation. Cell treatment with oleic acid (18:1) demonstrated minimal effects
on radical formation. The addition of SOD (10 units/ml) completely eliminated
the radical signal, confirming that
formation. Cell treatment with oleic acid (18:1) demonstrated minimal effects
on radical formation. The addition of SOD (10 units/ml) completely eliminated
the radical signal, confirming that
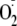 was the proximal radical detected under these conditions. Controls in which
XOR-treated cells (cells that were previously washed to remove free,
non-cell-associated-XOR) were spun down, and the supernatant evaluated for
radical formation produced no detectable DMPO signal (not shown). In addition,
when XOR-treated and washed cells were exposed to trypsin (0.25%) for 3 min at
37 °C to remove/inactivate cell-associated XOR, no DMPO spin adduct was
observed, demonstrating that the source of
was the proximal radical detected under these conditions. Controls in which
XOR-treated cells (cells that were previously washed to remove free,
non-cell-associated-XOR) were spun down, and the supernatant evaluated for
radical formation produced no detectable DMPO signal (not shown). In addition,
when XOR-treated and washed cells were exposed to trypsin (0.25%) for 3 min at
37 °C to remove/inactivate cell-associated XOR, no DMPO spin adduct was
observed, demonstrating that the source of
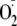 trapped in our experiments was extracellular XOR (not shown).
trapped in our experiments was extracellular XOR (not shown).
FIGURE 5.

Nitro-oleic acid inhibits GAG-associated XOR
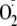 production. A, free and HS6B-bound XOR were exposed to 10
μm allopurinol (AP) or OA-NO2, and activity
was determined by monitoring the production of uric acid (292 nm) following
the addition of xanthine (100 μm). Data points represent the
mean ± S.E. of at least three independent determinations; *, p
< 0.05. B, bovine aortic endothelial cells were exposed to XOR (5
milliunits/ml) for 20 min and then harvested as described under
“Experimental Procedures”. Cell suspensions (3 ×
106 cells/ml) were exposed to OA-NO2 for 10 min and then
analyzed by EPR for
production. A, free and HS6B-bound XOR were exposed to 10
μm allopurinol (AP) or OA-NO2, and activity
was determined by monitoring the production of uric acid (292 nm) following
the addition of xanthine (100 μm). Data points represent the
mean ± S.E. of at least three independent determinations; *, p
< 0.05. B, bovine aortic endothelial cells were exposed to XOR (5
milliunits/ml) for 20 min and then harvested as described under
“Experimental Procedures”. Cell suspensions (3 ×
106 cells/ml) were exposed to OA-NO2 for 10 min and then
analyzed by EPR for
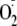 production with the spin trap DMPO (25 mm) at 25 °C. Reactions
were initiated by the addition of xanthine (100 μm). Samples for
all spectra contain cells plus DMPO and are identified beginning from the top
as follows: control without added XOR and xanthine (Xan) and added
XOR and xanthine (XOR + Xan). The remainder of spectra
consist of cells, added XOR, xanthine, and the constituents indicated.
production with the spin trap DMPO (25 mm) at 25 °C. Reactions
were initiated by the addition of xanthine (100 μm). Samples for
all spectra contain cells plus DMPO and are identified beginning from the top
as follows: control without added XOR and xanthine (Xan) and added
XOR and xanthine (XOR + Xan). The remainder of spectra
consist of cells, added XOR, xanthine, and the constituents indicated.
DISCUSSION
The pyrazolo-based suicide inhibitor allopurinol has been in clinical use for several decades to treat gout and more recently has been employed to reduce the effect of XOR-derived reactive species in ischemic tissue injury and cardiovascular disease (6). However, a limited ability of allo-/oxypurinol to inhibit cell-associated XOR has been reported (11). The nature of this limitation, combined with the emerging significance of vessel wall-associated XOR in cardiovascular disease, supports the need for development of new strategies to achieve XOR inhibition. The substrate promiscuity of XOR, combined with the post-translational modification properties of electrophilic nitroalkenes, prompted investigation of OA-NO2 as a potential inhibitor of XOR.
On an equimolar basis, OA-NO2 inhibits both uric acid and
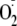 formation more potently than allopurinol, with IC50 values for
OA-NO2 ∼4 and ∼3 times lower, respectively. Inhibition is
maximal between pH 7 and 10, suggesting that OA-NO2 is less
effective when the carboxylic acid moiety is protonated
(Fig. 1C). Kinetic
analysis via Dixon plot reveals nearly parallel slopes for different substrate
concentrations, indicating noncompetitive inhibition
(Fig. 1D).
formation more potently than allopurinol, with IC50 values for
OA-NO2 ∼4 and ∼3 times lower, respectively. Inhibition is
maximal between pH 7 and 10, suggesting that OA-NO2 is less
effective when the carboxylic acid moiety is protonated
(Fig. 1C). Kinetic
analysis via Dixon plot reveals nearly parallel slopes for different substrate
concentrations, indicating noncompetitive inhibition
(Fig. 1D).
The alkenyl nitro configuration of OA-NO2 confers electrophilic reactivity on the β-carbon adjacent to the nitro-bonded carbon, allowing for post-translational modification of proteins via reaction with nucleophilic residues, such as cysteine and histidine (15). In the case of glyceraldehyde-3-phosphate dehydrogenase, this process is reversible by exposure of the OA-NO2-inhibited enzyme to reducing agents, such as GSH, that exchange OA-NO2 from the protein and restore enzymatic activity (15). In contrast to glyceraldehyde-3-phosphate dehydrogenase, high levels (20 mm) of GSH, β-mercaptoethanol, and dithiothreitol do not restore activity to OA-NO2-inhibited XOR (Fig. 2A), indicating an irreversible covalent reaction between OA-NO2 and XOR. However, when these nucleophiles first react with OA-NO2 before XOR, inhibition capacity is lost.
Inhibition of XOR by OA-NO2 is specific with respect to other electrophilic lipids, and ethyl pyruvate, a low molecular weight anionic electrophile, does not inhibit XOR activity (Fig. 2B). Furthermore, XOR is not inhibited by several structural variants of OA-NO2 that differ in the nitroalkenyl and carboxylic acid moieties (Table 1). The pure preparation of 9-OA-NO2 regioisomer produced similar inhibition to that observed using the mixture of 9- and 10-OA-NO2, indicating nitration of either carbon 9 or 10 is sufficient to inhibit XOR. LNO2, a mixture of linoleic acid nitrated at carbon 9, 10, 12, or 13, also inhibited XOR but to a lesser (76%) extent than equimolar concentrations of OA-NO2 (98%). This difference in inhibition may be proportional to the content of the LNO2 preparation nitrated at either carbon 9 or 10. Moving the nitro group away from carbon 9 or 10, removal of the double bond between carbons 9 and 10, and/or placing the nitro group in the Z configuration results in loss of inhibitory properties, since exposure of XOR to iso-OA-NO2 (0–30 μm), nitrostearic acid (0–30 μm), and elaidic acid (30 μm), respectively, did not block enzymatic activity. In addition, the presence of a ketone (KODE) or an oxine (OXDE) at carbon 13 resulted in the loss of ability to inhibit XOR. Another requirement for OA-NO2 inhibition of XOR is an intact carboxylic acid moiety. For example, modification of the carboxylic acid of OA-NO2 by replacement with an alcohol (OH), a methyl ester, an amide, or a biotin moiety eliminates the inhibitory properties. Together, these data indicate a high level of specificity regarding the structure of OA-NO2 with respect to its capacity to inhibit XOR, denoting that electrostatic interactions between OA-NO2 and particular domains on the XOR protein are crucial.
Inhibition of XOR by OA-NO2 does not occur by abrogation of
electron transfer at the FAD cofactor, since OA-NO2 inhibits uric
acid formation from xanthine, even when electrons are transferred to DCPIP,
bypassing the FAD (Fig.
3A). Additionally, OA-NO2 does not materially
alter reduction or oxidation spectra of the FAD, as shown in
Fig. 3B. These data
demonstrate that XOR maintains an electronically responsive FAD when purine
oxidation is completely inhibited by OA-NO2. Furthermore,
OA-NO2 did not alter the rate of NADH-driven O2
consumption or
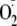 formation (Fig. 3C).
Combined, these data confirm that OA-NO2 inhibition of XOR is not
mediated via reaction with the FAD cofactor.
formation (Fig. 3C).
Combined, these data confirm that OA-NO2 inhibition of XOR is not
mediated via reaction with the FAD cofactor.
These results prompted investigation of possible effects of OA-NO2 on the molybdenum cofactor. Anaerobic reduction experiments were performed (as in Fig. 3) using xanthine instead of sodium dithionite to reduce the FAD. This method employs xanthine-derived electrons to sequentially reduce the molybdenum cofactor, the FeS centers, and finally the FAD. When XOR is exposed to OA-NO2, the ability of xanthine to reduce the FAD is abolished, suggesting that reaction of OA-NO2 with XOR affects the molybdenum cofactor (Fig. 4A). Enzyme turnover experiments in which OA-NO2 (10 μm) is added after XOR is primed with xanthine reduce the rate of uric acid formation by 80%, whereas only a 25% decrease in urate formation is observed when DCPIP is present (Fig. 4, B and B1). Since DCPIP associates with and accepts electrons directly from the molybdenum cofactor (33, 34), these data indicate that OA-NO2 competes with DCPIP in close proximity to the molybdenum cofactor. Reactivation studies were designed to further test this theory. Allopurinol is oxidized to oxypurinol at the molybdenum cofactor, where oxypurinol then inhibits enzyme activity. Incubation of XOR with allopurinol for 5 min, followed by size exclusion chromatography to remove free allopurinol, produced reversal of XOR inhibition and full enzyme reactivation within 12 min after the addition of xanthine (Fig. 4, C and C1). Reactivation of XOR could not be observed following treatment with OA-NO2. However, when XOR was sequentially treated with allopurinol followed by exposure to OA-NO2, a significant (75%) reactivation was observed, indicating that 1) allo-/oxypurinol may have easier access to the molybdenum cofactor and sequential exposure of allo-/oxypurinol followed by exposure to OA-NO2 results in significant reactivation of XOR, and 2) the molybdenum-allo-/oxypurinol association protects the molybdenum cofactor from inactivation by OA-NO2. Together, these data demonstrate that OA-NO2 inhibition of XOR is mediated via reaction with the molybdenum cofactor. Due to the electrophilic nature of OA-NO2, a possible target for adduction is the dithiolene of the pterin moiety, which serves in the coordination of the molybdenum molecule. Covalent modification of this site could lead to loss of the molybdenum and may explain our inability to reactivate the enzyme. Studies are ongoing to confirm the hypothesis.
Cell association of XOR significantly limits inhibition of XOR by
allopurinol or its active metabolite, oxypurinol, even at concentrations far
above those observed clinically
(11). However, when HS6B-bound
XOR was exposed to OA-NO2, enzyme activity was inhibited 95%
compared with 50% for an equimolar concentration of allopurinol
(Fig. 5A). At this
concentration (10 μm) both inhibitors were equally effective in
inhibiting XOR free in solution. To extend this observation to add cell
biological context, we exposed confluent bovine aortic endothelial cells to
exogenous XOR, using the cells as a platform to immobilize the enzyme on
extracellular GAGs, and then evaluated enzyme inhibition by OA-NO2
and allopurinol. Fig.
5B demonstrates that cell-associated
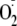 generation is more effectively inhibited by OA-NO2 than by
allopurinol, mirroring the results seen when XOR is immobilized by association
with HS6B. The spectra in Fig.
5B is composed of a combination of DMPO-OOH and DMPO-OH
radical adducts, since the –OOH can rapidly decompose to –OH,
especially in a cellular environment where DMPO-OOH can serve as a substrate
for glutathione peroxidase or react with adventitious metals
(35–37).
Under our experimental conditions, the
generation is more effectively inhibited by OA-NO2 than by
allopurinol, mirroring the results seen when XOR is immobilized by association
with HS6B. The spectra in Fig.
5B is composed of a combination of DMPO-OOH and DMPO-OH
radical adducts, since the –OOH can rapidly decompose to –OH,
especially in a cellular environment where DMPO-OOH can serve as a substrate
for glutathione peroxidase or react with adventitious metals
(35–37).
Under our experimental conditions, the
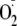 is generated extracellularly, since controls in which extracellular XOR was
removed/inactivated by treatment with trypsin produced no detectable DMPO spin
adduct. In addition, the absence of contaminating radical species such as
·OH that could serve to augment the spectra in
Fig. 5B is confirmed
by complete abrogation of the EPR signal in the presence of SOD. The SOD
control further demonstrates that the generation of the observed DMPO radical
adduct is a result of initial generation of
is generated extracellularly, since controls in which extracellular XOR was
removed/inactivated by treatment with trypsin produced no detectable DMPO spin
adduct. In addition, the absence of contaminating radical species such as
·OH that could serve to augment the spectra in
Fig. 5B is confirmed
by complete abrogation of the EPR signal in the presence of SOD. The SOD
control further demonstrates that the generation of the observed DMPO radical
adduct is a result of initial generation of
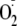 .
.
Nitroalkene derivatives of fatty acids have been detected (low nanomolar range) in human blood plasma, plasma-derived lipoproteins, and activated macrophages (13, 38, 39). Since fatty acid nitration is induced by oxidative and nitrative inflammatory conditions, these data suggest that fatty acid nitroalkene derivatives represent byproducts that may mediate adaptive anti-inflammatory reactions.
In summary, we have identified electrophilic OA-NO2 as an XOR inhibitor that is more effective at inhibiting endothelial cell-associated XOR, thus potentially enhancing treatment of vascular inflammation, where XOR-derived ROS alters redox-dependent cell signaling reactions and reduces NO bioavailability. The irreversible inhibition of XOR by OA-NO2 occurs with a lower IC50 than allopurinol and encourages further investigation.
Acknowledgments
We thank Dr. Garry R. Buettner and Brett A. Wagner at the University of Iowa Electron Spin Resonance Facility for use of the EPR and assistance with our studies. The technical assistance of Jose Toledo (University of Alabama at Birmingham) was greatly appreciated.
This work was supported, in whole or in part, by National Institutes of Health Grants HL58115 and HL64937 (to B. A. F.) and HL71189 and HL074391 (to J. L.). This work was also supported by the American Heart Association Beginning Grants-in-Aid 0665378U (to E. E. K.), 0665418U (to F. J. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: XOR, xanthine oxidoreductase; DCPIP, 2,6-dichlorophenolindophenol; DPI, diphenyleneiodonium; GAG, glycosoaminoglycan; ROS, reactive oxygen species; XDH, xanthine dehydrogenase; XO, xanthine oxidase; DMPO, 5,5-dimethyl-1-pyrroline-N-oxide; HS6B, heparin-Sepharose 6B; SOD, superoxide dismutase; LNO2, nitrolinoleic acid.
References
- 1.Fridovich, I. (1970) J. Biol. Chem. 245 4053–4057 [PubMed] [Google Scholar]
- 2.Harrison, R. (2002) Free Radic. Biol. Med. 33 774–797 [DOI] [PubMed] [Google Scholar]
- 3.Harris, C. M., and Massey, V. (1997) J. Biol. Chem. 272 8370–8379 [DOI] [PubMed] [Google Scholar]
- 4.White, C. R., Brock, T. A., Chang, L. Y., Crapo, J. D., Briscoe, P., Ku, D. D., Bradley, W. A., Gianturco, S. H., Gore, J., Freeman, B. A., and Tarpey, M. M. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 1620–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Granger, D. N., Rutili, G., and McCord, J. M. (1981) Gastroenterology 81 22–29 [PubMed] [Google Scholar]
- 7.Giler, S., Sperling, O., Brosh, S., Urca, I., and De Vries, A. (1975) Clin. Chim. Acta 63 37–40 [DOI] [PubMed] [Google Scholar]
- 8.Ramboer, C., Piessens, F., and De Groote, J. (1972) Digestion 7 183–195 [DOI] [PubMed] [Google Scholar]
- 9.Houston, M., Estevez, A., Chumley, P., Aslan, M., Marklund, S., Parks, D. A., and Freeman, B. A. (1999) J. Biol. Chem. 274 4985–4994 [DOI] [PubMed] [Google Scholar]
- 10.Radi, R., Rubbo, H., Bush, K., and Freeman, B. A. (1997) Arch. Biochem. Biophys. 339 125–135 [DOI] [PubMed] [Google Scholar]
- 11.Kelley, E. E., Trostchansky, A., Rubbo, H., Freeman, B. A., Radi, R., and Tarpey, M. M. (2004) J. Biol. Chem. 279 37231–37234 [DOI] [PubMed] [Google Scholar]
- 12.O'Donnell, V. B., Eiserich, J. P., Chumley, P. H., Jablonsky, M. J., Krishna, N. R., Kirk, M., Barnes, S., Darley-Usmar, V. M., and Freeman, B. A. (1999) Chem. Res. Toxicol. 12 83–92 [DOI] [PubMed] [Google Scholar]
- 13.Lima, E. S., Di, M. P., Rubbo, H., and Abdalla, D. S. (2002) Biochemistry 41 10717–10722 [DOI] [PubMed] [Google Scholar]
- 14.Napolitano, A., Camera, E., Picardo, M., and d'Ischia, M. (2000) J. Org. Chem. 65 4853–4860 [DOI] [PubMed] [Google Scholar]
- 15.Batthyany, C., Schopfer, F. J., Baker, P. R., Duran, R., Baker, L. M., Huang, Y., Cervenansky, C., Branchaud, B. P., and Freeman, B. A. (2006) J. Biol. Chem. 281 20450–20463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schopfer, F. J., Lin, Y., Baker, P. R., Cui, T., Garcia-Barrio, M., Zhang, J., Chen, K., Chen, Y. E., and Freeman, B. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 2340–2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schopfer, F. J., Baker, P. R., Giles, G., Chumley, P., Batthyany, C., Crawford, J., Patel, R. P., Hogg, N., Branchaud, B. P., Lancaster, J. R., Jr., and Freeman, B. A. (2005) J. Biol. Chem. 280 19289–19297 [DOI] [PubMed] [Google Scholar]
- 18.Coles, B., Bloodsworth, A., Eiserich, J. P., Coffey, M. J., McLoughlin, R. M., Giddings, J. C., Lewis, M. J., Haslam, R. J., Freeman, B. A., and O'Donnell, V. B. (2002) J. Biol. Chem. 277 5832–5840 [DOI] [PubMed] [Google Scholar]
- 19.Coles, B., Bloodsworth, A., Clark, S. R., Lewis, M. J., Cross, A. R., Freeman, B. A., and O'Donnell, V. B. (2002) Circ. Res. 91 375–381 [DOI] [PubMed] [Google Scholar]
- 20.Baker, L. M., Baker, P. R., Golin-Bisello, F., Schopfer, F. J., Fink, M., Woodcock, S. R., Branchaud, B. P., Radi, R., and Freeman, B. A. (2007) J. Biol. Chem. 282 31085–31093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masuoka, N., and Kubo, I. (2004) Biochim. Biophys. Acta 1688 245–249 [DOI] [PubMed] [Google Scholar]
- 22.Masuoka, N., Nihei, K., and Kubo, I. (2006) Mol. Nutr. Food Res. 50 725–731 [DOI] [PubMed] [Google Scholar]
- 23.Waud, W. R., Brady, F. O., Wiley, R. D., and Rajagopalan, K. V. (1975) Arch. Biochem. Biophys. 169 695–701 [DOI] [PubMed] [Google Scholar]
- 24.Tan, S., Zhou, F., Nielsen, V. G., Wang, Z., Gladson, C. L., and Parks, D. A. (1998) J. Neuropathol. Exp. Neurol. 57 544–553 [DOI] [PubMed] [Google Scholar]
- 25.McCord, J. M., and Fridovich, I. (1970) J. Biol. Chem. 245 1374–1377 [PubMed] [Google Scholar]
- 26.Baker, P. R., Lin, Y., Schopfer, F. J., Woodcock, S. R., Groeger, A. L., Batthyany, C., Sweeney, S., Long, M. H., Iles, K. E., Baker, L. M., Branchaud, B. P., Chen, Y. E., and Freeman, B. A. (2005) J. Biol. Chem. 280 42464–42475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker, P. R., Schopfer, F. J., Sweeney, S., and Freeman, B. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 11577–11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woodcock, S. R., Marwitz, A. J., Bruno, P., and Branchaud, B. P. (2006) Org. Lett. 8 3931–3934 [DOI] [PubMed] [Google Scholar]
- 29.Cui, T., Schopfer, F. J., Zhang, J., Chen, K., Ichikawa, T., Baker, P. R., Batthyany, C., Chacko, B. K., Feng, X., Patel, R. P., Agarwal, A., Freeman, B. A., and Chen, Y. E. (2006) J. Biol. Chem. 281 35686–35698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manini, P., Camera, E., Picardo, M., Napolitano, A., and d'Ischia, M. (2005) Chem. Phys. Lipids 134 161–171 [DOI] [PubMed] [Google Scholar]
- 31.Kerdesky, F. A., Schmidt, S. P., Holms, J. H., Dyer, R. D., Carter, G. W., and Brooks, D. W. (1987) J. Med. Chem. 30 1177–1186 [DOI] [PubMed] [Google Scholar]
- 32.Ichimori, K., Fukahori, M., Nakazawa, H., Okamoto, K., and Nishino, T. (1999) J. Biol. Chem. 274 7763–7768 [DOI] [PubMed] [Google Scholar]
- 33.Tamta, H., Kalra, S., and Mukhopadhyay, A. K. (2006) Biochemistry (Mosc.) 71 Suppl. 1, 49–54 [DOI] [PubMed] [Google Scholar]
- 34.Gurtoo, H. L., and Johns, D. G. (1971) J. Biol. Chem. 246 286–293 [PubMed] [Google Scholar]
- 35.Rosen, G. M., and Freeman, B. A. (1984) Proc. Natl. Acad. Sci. U. S. A. 81 7269–7273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finkelstein, E., Rosen, G. M., and Rauckman, E. J. (1982) Mol. Pharmacol. 21 262–265 [PubMed] [Google Scholar]
- 37.Britigan, B. E., Pou, S., Rosen, G. M., Lilleg, D. M., and Buettner, G. R. (1990) J. Biol. Chem. 265 17533–17538 [PubMed] [Google Scholar]
- 38.Lima, E. S., Di, M. P., and Abdalla, D. S. (2003) J. Lipid Res. 44 1660–1666 [DOI] [PubMed] [Google Scholar]
- 39.Ferreira, A. M., Ferrari, M., Troschansky, A., Batthyany, C. I., Souza, J. M., Alvarez, M. N., Lopez, G. V., Baker, P. R., Schopfer, F. J., O'Donnell, V. B., Freeman, B. A., and Rubbo, H. (2008) Biochem. J. 10.1042/BJ20080701 [DOI] [PMC free article] [PubMed]