

Abstract
The core human mitochondrial transcription machinery comprises a single subunit bacteriophage-related RNA polymerase, POLRMT, the high mobility group box DNA-binding protein h-mtTFA/TFAM, and two transcriptional co-activator proteins, h-mtTFB1 and h-mtTFB2 that also have rRNA methyltransferase activity. Recapitulation of specific initiation of transcription in vitro can be achieved by a complex of POLRMT, h-mtTFA, and either h-mtTFB1 or h-mtTFB2. However, the nature of mitochondrial transcription complexes in vivo and the potential involvement of additional proteins in the transcription process in human mitochondria have not been extensively investigated. In Saccharomyces cerevisiae, transcription and translation are physically coupled via the formation of a multiprotein complex nucleated by the binding of Nam1p to the amino-terminal domain of mtRNA polymerase (Rpo41p). This model system paradigm led us to search for proteins that interact with POLRMT to regulate mitochondrial gene expression in humans. Using an affinity capture strategy to identify POLRMT-binding proteins, we identified mitochondrial ribosomal protein L7/L12 (MRPL12) as a protein in HeLa mitochondrial extracts that interacts specifically with POLRMT in vitro. Purified recombinant MRPL12 binds to POLRMT and stimulates mitochondrial transcription activity in vitro, demonstrating that this interaction is both direct and functional. Finally, from HeLa cells that overexpress FLAG epitope-tagged MRPL12, increased steady-state levels of mtDNA-encoded transcripts are observed and MRPL12-POLRMT complexes can be co-immunoprecipitated, providing strong evidence that this interaction enhances mitochondrial transcription or RNA stability in vivo. We speculate that the MRPL12 interaction with POLRMT is likely part of a novel regulatory mechanism that coordinates mitochondrial transcription with translation and/or ribosome biogenesis during human mitochondrial gene expression.
The core machinery responsible for specific initiation of transcription from human mtDNA promoters has been elucidated. The minimal requirement is a three-component complex comprising a single subunit bacteriophage-related RNA polymerase, POLRMT, the high mobility group box DNA-binding protein h-mtTFA/TFAM, and two transcriptional co-activator proteins, h-mtTFB1 and h-mtTFB2 (1). The mtTFB factors interact directly with a carboxyl-terminal tail of h-mt-TFA and bridge interactions between the promoter-bound h-mtTFA and POLRMT to facilitate specific initiation (2). Despite these advances, our understanding of the nature of mitochondrial transcription complexes in vivo remains limited, especially in mammalian systems. For example, whether other factors interact with POLRMT to regulate transcription initiation and/or elongation or to couple additional RNA-related activities to transcription have not been determined. However, interesting in this regard, h-mtTFB1 and h-mtTFB2 are members of a conserved family of rRNA methyltransferases (3, 4) and can methylate small subunit rRNAs at a conserved stem loop (5, 6), representing a potential link between the transcription machinery and ribosome biogenesis and/or translation activity. Consistent with such a role, mtTFB1 from Drosophila melanogaster is required for efficient mitochondrial translation in Schneider cells (7).
A direct link between mitochondrial transcription and translation has been demonstrated in studies of the mitochondrial (mt) RNA polymerase of Saccharomyces cerevisiae (Rpo41p). Like many mtRNA polymerases, Rpo41p has an amino-terminal extension not present in the related bacteriophage enzymes (8–10). An amino-terminal domain of Rpo41p is the binding site for Nam1p (9) that is proposed to deliver newly synthesized RNAs (or active transcription complexes) to the inner mitochondrial membrane (11, 12) and promote subsequent interactions with gene-specific translational activators and ribosomes (13, 14). Thus, like in bacteria, the processes of transcription and translation are physically and functionally coupled. The amino-terminal extension of human POLRMT is not homologous to that of yeast (9), and therefore whether proteins (other than the core transcription factors required for initiation) interact with POLRMT and couple additional activities to transcription has not been examined.
The circular 16.5-kb human mtDNA molecule encodes thirteen essential protein components of the mitochondrial oxidative phosphorylation system responsible for the production of cellular ATP (15). These mRNAs are translated into protein by a dedicated set of ribosomes in the mitochondrial matrix made up of the 12 S and 16 S rRNAs, which are also encoded by mtDNA, and ∼80 mitochondrial ribosomal proteins that are the products of nuclear genes and must be imported into the organelle (16). Therefore, in contrast to bacterial or eukaryotic cytoplasmic ribosome biogenesis, mitochondrial ribosomal biogenesis requires coordination of rRNA synthesis from within the organelle by the mitochondrial transcription machinery with nuclear expression and import of ribosomal proteins from the cytoplasm by a separate set of regulatory proteins. Furthermore, some of the mitochondrial ribosomal proteins do not have homologs in bacterial or cytoplasmic ribosomes and likely provide unique functions specific for mitochondrial protein synthesis or perhaps have additional functions in the organelle (17). In the present study, we set out to identify proteins that interact with POLRMT that we hypothesized would be involved in new aspects of mitochondrial gene expression in humans. Here we describe our finding that a conserved mitochondrial ribosomal protein is bifunctional, acting both as a component of ribosomes and of transcription-related complexes via an interaction with POLRMT.
Materials and Methods
Construction of Expression Plasmids for Human POLRMT and MRPL12
The vector used to express POLRMT in bacteria was pProEX-Htb (Invitrogen). A portion of the human cDNA encoding amino acids 41–1250 and the stop codon was cloned into the BamH1 and XhoI of this vector via a BamH1-SalI restriction fragment. Amino acids 1–40 were deleted because they compose the mitochondrial localization sequence (MLS)2 that is predicted to be cleaved off by proteases during import into mitochondria (18). However, in place of the MLS, there are 29 unnatural amino acids fused to POLRMT that include an initiator methionine, a His6 tag, a spacer of 7 amino acids, a TEV protease cleavage site, and another spacer of 6 amino acids. The vector has an intact Escherichia coli lacIq gene allowing POLRMT expression from the trc promoter to be regulated by addition of isopropylthiogalactoside (Sigma).
A strategy similar to that described above for POLRMT was used to express MRPL12 in bacteria, except it was expressed as a glutathione S-transferase (GST) fusion using the vector pGEX4T-3. A portion of the MRPL12 cDNA encoding amino acids 46–198 and the stop codon was cloned directly into this vector via a BamH1-XhoI restriction fragment. Amino acids 1–45 were deleted because they compose the MLS that is predicted to be cleaved off during import (19). However, in its place is the GST peptide followed by a thrombin cleavage site. The plasmid also has an intact E. coli lacIq gene allowing MRPL12 expression from the tac promoter to be regulated by addition of isopropyl-1-thio-β-d-galactopyranoside.
The plasmid used for doxycycline-regulated expression of FLAG-tagged MRPL12 in human cells (pTRE2-FLAG-MRPL12) was constructed as follows. PCR primers were designed to amplify the entire MRPL12 cDNA (including the MLS; Fig. 1C) but to replace the normal stop codon with a FLAG epitope followed by a stop codon. The 5′- and 3′-primers were also designed to contain a BamH1 and SalI restriction site, respectively, allowing the PCR product to be cloned into these same sites in the plasmid pTRE2hyg (Clontech).
FIGURE 1. Affinity capture of mitochondrial ribosomal protein L12 (MRPL12) in association with POLRMT, the human mitochondrial RNA polymerase.
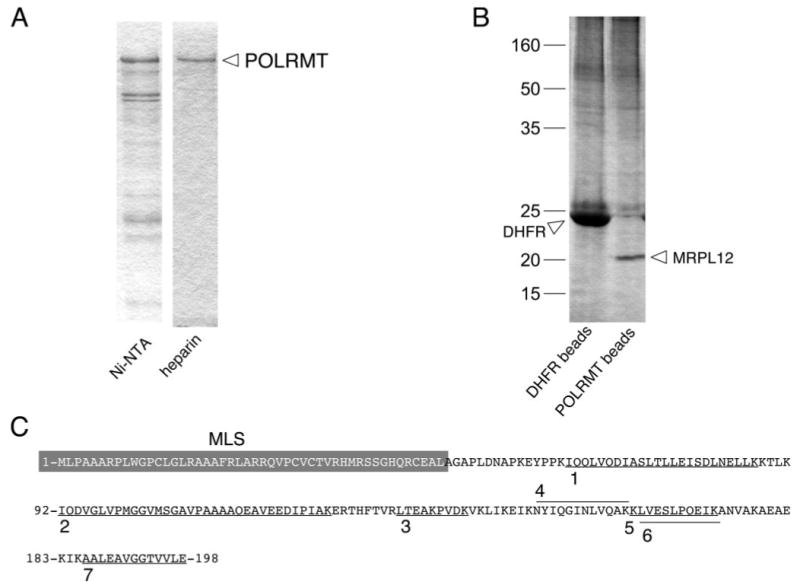
A, purification of His-tagged POLRMT by nickel-affinity and heparin-agarose chromatography. Shown are Coomassie-stained SDS-PAGE gels of representative fractions from a typical purification of recombinant POLRMT from E. coli lysate after nickel-affinity chromatography (Ni-NTA) and subsequent heparin-agarose (heparin) chromatography. The ∼130-kDa POLRMT band is indicated. B, results of affinity capture of POLRMT-binding proteins from HeLa cell mitochondrial lysates. Shown is a silver-stained SDS-PAGE gel of proteins eluted from control DHFR beads (left lane) or POLRMT beads (right lane). The major ∼20-kDa band that co-purified with POLRMT is labeled MRPL12, based on the mass spectroscopy-based identification performed on the excised band. C, mass spectroscopy-based peptide analysis identified the 20-kDa band in panel B as MRPL12. The seven peptides unambiguously identified as MRPL12 are underlined (or overlined) on the primary sequence of the human MRPL12 polypetide (amino acids 1–198) and labeled 1–7. The gray box indicates the mitochondrial localization sequence (MLS) of MRPL12. Note that no peptides were identified in this region as would be predicted if the MLS were cleaved off upon import.
Expression and Purification of Recombinant POLRMT
The expression and purification of POLRMT was a modification of that described (20). BL21-CodonPlus® E. coli (Stratagene) transformed with pProEX-Htb/POLRMT grown at 37 °C in 1 liter of Luria-Bertani medium containing 100 mg/ml ampicillin to an A600 of 0.4. Expression was induced at 25 °C for 4 h by addition of 0.4 mm isopropyl-1-thio-β-d-galactopyranoside. Cell pellets were collected and resuspended in a binding buffer consisting of 50 mm sodium phosphate, pH 8.0, 500 mm NaCl, 0.5 mm imidazole, 10 mm 2-mercaptoethanol, 10% glycerol, 0.5% Tween 20, and 1 mm phenylmethylsulfonyl fluoride (Sigma). Cells were sonicated using a microtip for six 20-s intervals on ice, and a cleared soluble lysate was obtained by centrifugation at 12,000 × g for 45 min. Lysates were loaded on a nickel-nitrilotriacetic acid-Sepharose column (Qiagen), pre-equilibrated with the binding buffer, and extensively washed with the binding buffer containing 20 mm imidazole. The bound His-tagged POLRMT was eluted using a step gradient of 50, 100, 150, and 200 mm imidazole in binding buffer. Eluted fractions containing POLRMT were combined, diluted by a factor of three with binding buffer without NaCl, and loaded on a heparin-agarose column (Sigma) pre-equilibrated with buffer A (50 mm sodium phosphate, pH 8.0, 150 mm NaCl, 10% glycerol, and 0.5% Tween 20). The column was washed with 50 ml of buffer A containing 150 mm NaCl. The bound proteins were eluted using a step gradient (0.3–1.0 m NaCl in buffer A) at 0.1-m intervals. The POLRMT peak eluted at ∼0.6 m NaCl as monitored by 12% SDS-PAGE and Coomassie blue staining (20). POLRMT-containing fractions were combined and dialyzed overnight at 4 °C against buffer B (50 mm sodium phosphate, pH 8.0, 50% glycerol). The resulting POLRMT preparation was used directly for in vitro assays or frozen at −80 °C in small aliquots. Protein concentrations were determined with a Bio-Rad protein assay kit using bovine serum albumin as a standard.
Expression and Purification of Recombinant MRPL12
BL21 CodonPlus® Ecoli transformed with pGEX4T-3-MRPL12 were grown and MRPL12 expression was induced exactly as described above for POLRMT. Cell pellets were collected by centrifugation and resuspended in 50 ml of ice-cold lysis buffer (20 mm Tris·Cl, pH 8.0,100 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride). Cells were then lysed by sonication, and the resulting cell lysate was cleared by centrifugation (10,000 × g, 10 min, 4 °C). The cleared supernatant was incubated for 1 h at room temperature with 1 ml of glutathione-Sepharose (Amersham Biosciences) that had been washed three times with lysis buffer and then resuspended in lysis buffer as a slurry (1:1 v/v). The MRPL12-bound beads were then loaded onto a column and washed five times with one bed volume of lysis buffer. GST-MRPL12 was eluted with 1.0 ml of elution buffer (50 mm Tris·Cl, pH 8.0, 10 mm reduced glutathione) per ml of bed volume. Thrombin-agarose (Sigma) was added to the eluted GST-MRPL12 and incubated for 4 h at room temperature with rotation. The suspension was then centrifuged at 500 × g for 5 min to remove the beads. The supernatant (containing a mixture of GST-MRPL12, GST peptide, and liberated MRPL12) was then loaded onto a glutathione column to which uncleaved GST-tagged MRPL12 and the GST peptide bind, but the liberated MRPL12 (without the GST tag) does not. Fractions from this column containing recombinant untagged MRPL12 were used for all in vitro assays. Protein concentrations were determined with a Bio-Rad protein assay kit using bovine serum albumin as a standard.
For some of the in vitro transcription experiments, we immunodepleted MRPL12 from the lysate by incubating it with 100 μl of protein A-Sepharose bound with either 20 μg of mouse IgG or anti-MRPL12 antibody (Abnova) at 4 °C for 2 h with rotation. Then mixtures were loaded to a Handee mini-spin column (Pierce) and centrifuged to remove the bead-bound immune complexes. The resulting supernatant was the immunodepleted lysate used in the indicated transcription reactions.
Isolation of HeLa Cell Mitochondria
HeLa cells that were adapted for growth in suspension were used as the source of human mitochondria, which were isolated by a standard differential centrifugation procedure followed by Nycodenz gradient purification as follows. HeLa cells (6 liters) were grown in Joklik's minimal essential medium (Sigma) supplemented with 10% bovine growth serum to late exponential phase at 37 °C in spinner flasks. Cells were collected by centrifugation and washed with 100 ml of buffer A (10 mm HEPES-NaOH, pH 7.4, 1 mm EDTA, 0.25 m sucrose) and resuspended in 100 ml of buffer A. Cells in this suspension were lysed using a Dounce homogenizer (20 strokes) and then centrifuged at 800 × g for 10 min to remove nuclei, cell debris, and unbroken cells. The resulting supernatant was then centrifuged at 20,000 × g for 15 min to pellet mitochondria. The mitochondrial pellet was resuspended in 10 ml of 25% Nycodenz in buffer A. Mitochondria were then separated from other organelles using the following discontinuous Nycodenz gradient: from bottom to top, 4 ml of 34%, 7 ml of 30%, 7 ml of 23%, and 2 ml of 20%. Sealed tubes were centrifuged for 90 min at 52,000 × g at 4 °C, and mitochondria were removed from the 25/30% interface and stored frozen at −80 °C until needed.
POLRMT Affinity Capture Assays
Talon cobalt beads (BD Biosciences) were loaded with recombinant His-tagged POLRMT as described for the purification of POLRMT above. HeLa cell mitochondria (isolated as described in the previous section) were homogenized, sonicated, and lysed in 10 volume equivalents of lysis buffer (150 mm NaCl, 1.0% Nonidet P-40, 50 mm phosphate buffer, pH 8.0,1 mm phenylmethylsulfonyl fluoride). Lysates were treated with 200 units/ml RNase A (Sigma) and 40 units/ml DNase (Promega) for 2 h and then centrifuged for 15 min at 4 °C at 12,000 × g. The resulting soluble mitochondrial extract was used in the affinity capture assay as follows. The mitochondrial extract (800 μl) was first precleared with 100 μl of naked Talon affinity beads for 1 h at 4 °C. Then 400-μl samples of the precleared supernatant were incubated overnight at 4 °C with Talon beads bound with either 100 μg of purified His-tagged POLRMT or His-tagged dihydrofolate reductase (DHFR) (plasmid was a gift from Dr. E. Chu, Yale University, and the protein was purified exactly as described for POLRMT above) or with 20 μl of naked beads in binding buffer. After binding, the beads were washed five times in 400 μl of wash buffer (500 mm NaCl, 1.0% Nonidet P-40, 0.5% sodium deoxycholate, 1% SDS, 50 mm phosphate buffer, pH 8.0,1 mm phenylmethylsulfonyl fluoride, 50 mm imidazole) and bound proteins were removed with 30 μl of elution buffer (wash buffer + 300 mm imidazole). Samples (10 μl) were loaded on 12% SDS-PAGE gels and silver stained to identify bound proteins. The conditions used for the detection of direct binding of POLRMT and MRPL12 were the same as those used above, except purified recombinant MRPL12 (prepared as described above) was used instead of the mitochondrial lysate.
Mass Spectroscopy Identification of MRPL12
The ∼20-kDa protein identified in the POLRMT affinity capture experiment (Fig. 1A) was excised from a Coomassie-stained SDS-PAGE gel, and electrospray ionization mass spectrometry was carried out at the Biotechnology Resource Facility of the Howard Hughes Medical Institute Biopolymer Facility/W. M. Keck Foundation, Yale University, using an AB QSTAR instrument. Peptide identification was accomplished by the Mascot distiller and the Mascot data base search algorithm (Matrix Science).
Mitochondrial in Vitro Transcription Assays
Mitochondrial run-off transcription reactions were performed essentially as described previously (2, 4) but using different mtDNA promoter-containing templates. The template employed was a PCR product corresponding to nucleotides 242–825 of human mtDNA (21) encompassing the light-strand promoter (LSP) and heavy-strand promoter (HSP) that was cloned into the plasmid pGEMT-EZ (Promega). Digestion of this plasmid with EcoR1 results in linear transcription template from which specific initiation from the LSP and HSP1 promoters results in transcripts 168 and 288 nucleotides in length, respectively. Individual reaction mixtures (25 μl) contained 26 μg of EcoRI-digested template, 10 mm Tris·Cl, pH 8.0, 10 mm MgCl2, 1 mm dithiothreitol, 100 μg/ml bovine serum albumin, 400 μm ATP, 150 μm CTP and GTP, 10 μm UTP, 0.2 μm [α-32P]UTP (3,000 Ci/mmol), 40 units of RNase OUT™ (Invitrogen), 2.5 μl of a transcription-competent mitochondrial extract from HeLa cells that was prepared as described previously (2, 4), and the indicated concentrations of recombinant h-mtTFA or MRPL12. After 30 min at 32 °C, reactions were stopped by adding 200 μl of stop buffer (10 mm Tris·Cl, pH 8.0, 0.2 m NaCl, and 1 mm EDTA). Samples were treated with 0.5% SDS and 100 μg/ml proteinase K for 45 min at 42 °C and extracted twice with 225 μl of phenol:chloroform, and then RNA was precipitated by adding 0.6 ml of ice-cold ethanol and 1 μg of yeast tRNA (Sigma). The resulting RNA pellets were dissolved in 20 μl of gel loading buffer (98% formamide, 10 mm EDTA, pH 8.0, 0.025% xylene cyanol, 0.025% bromphenol blue), heated at 95 °C for 5 min, and then separated on 6% polyacrylamide/7 m urea gels in 1× TBE (Tris borate-EDTA) buffer. Radiolabeled 10-bp ladder DNA (Invitrogen) was run in parallel as markers to estimate RNA transcript sizes. Gels were dried and exposed to x-ray film using an intensifying screen at −80 °C.
Generation of Stable MRPL12-overexpressing HeLa Cell Lines
HeLa Tet-On cells (Clontech Laboratories, Inc.) were grown in Dulbecco's modified Eagle's medium supplemented with 10% bovine growth serum (10%) at 37 °C, 5% CO2. Cells were transfected with pTRE2-FLAG-MRPL12 using an Effectene kit (Qiagen, Inc.) as described by the manufacturer. After transfection, cells were incubated for 24 h and then plated in the presence of hygromycin B to select for growth of drug-resistant colonies. Single colonies were picked and expanded to generate pure stable cell lines. Cell lines were then grown in the presence of doxycycline (1 μg/ml) and screened for expression of FLAG-tagged MRPL12 by Western blot using an anti-FLAG epitope antibody (Sigma).
Co-immunoprecipitation Assays
A HeLa Tet-On cell line overexpressing FLAG-tagged MRPL12 in the presence of doxycycline (generated as described in the previous section) was used for co-immunoprecipitation (co-IP) experiments. Cells from one 100-mm plate were harvested on ice in the presence of 0.8 ml of lysis buffer (25 mm Tris·Cl, pH 7.4, 100 mm NaCl, 0.5% Nonidet P-40, and Sigma protease inhibitor mixture), lysed by sonication using a microtip for four, 15-s intervals on ice, and a cleared soluble lysate was obtained by centrifugation at 12,000 × g for 45 min. Precleared lysate (400 μl) was then incubated with 20 μl of anti-FLAG M2-agarose (Sigma) for 1 h. Antibody-linked complexes were collected via brief centrifugation in a microcentrifuge, washed three times in 400 μl of wash buffer (25 mm Tris·Cl, pH 7.4, 100 mm NaCl), and eluted from the matrix with SDS-PAGE sample buffer. Western blot analysis was used to probe for FLAG-tagged MRPL12 or POLRMT using the monoclonal M2 FLAG anti-mouse antibody (Sigma) or a POLRMT peptide antibody (22). Blots of the input and co-immunoprecipitate were also probed with control antibodies that recognize HSP60 (a mitochondrial matrix protein) and COX1 (a mitochondrial inner membrane protein) obtained from Santa Cruz Biotechnology, Inc. and Molecular Probes, Inc., respectively.
Northern Analysis of mtDNA-encoded Transcripts
RNA for Northern blot analysis was isolated from 1 × 106 cells from the indicated cell lines using the RNeasy kit (Qiagen) according to the manufacturer's instructions. RNA was eluted in the final step using RNase-free dH2O, quantified by absorbance at 260 nm, and stored frozen at −80 °C until used. RNA (2 μg) was separated by size on 1.2% agarose/formaldehyde gels and transferred to uncharged nylon membranes (Osmonics) via upward capillary flow. RNA was cross-linked to the blots by UV irradiation using a Stratalinker (Stratagene), stained with 0.1 μg/ml ethidium bromide/100 mm ammonium acetate, and destained with 100 mm ammonium acetate. The stained membrane was photographed using the Bio-Rad VersaDoc, and load was quantified using Quantity One software. The ethidium-stained 28 S rRNA band was used as a loading control for all experiments as described by others (23–25). Body-labeled DNA hybridization probes for mtDNA-encoded 12 S and 16 S rRNAs and the ND2 and ND6 mRNAs were generated by PCR with [32P]dCTP using the following gene-specific primers: h16 S-5′, 5′-CCCTCAACTGTCAACCCAACACAGG-3′; h16 S-3′, 5′-CCGGGCTCTGCCATCTTAACAAAC-3′; h12 S-5′, 5′-GACCCAAACTGGGATTAGATACCCCAC-3′; h12 S-3′, 5′-GACCCAAACTGGGATTAGATACCCCAC-3′; ND2-5′, 5′-GGCCCAACCCGTCATCTAC-3′; ND2-3′ 5′-GAGTGTGGGGAGGAATGGGG-3′; ND6-5′, 5′-GGGGTTTTCTTCTAAGCCTTCTCC-3′, and ND6–3′, 5′-CTAATCAACGCCCATAATCATAC-3′. PCR products were purified using the Qiagen PCR purification kit, denatured at 95 °C, and then placed on ice until hybridized to blots.
RNA blots were pre-hybridized with 20 ml of Rapid-Hyb buffer (GE Healthcare) for 1 h at 68 °C in a hybridizing oven with horizontal rotating cylindrical jars (Techne). After pre-hybridization, 20 ml of fresh Rapid-Hyb containing the desired radiolabeled probe was added, and incubation was carried out at 68 °C overnight. Probe solution was then removed, and hybridized blots were washed once with 2× SSC, 0.1% SDS at room temperature for 10 min, then three times with 1× SSC, 0.1% SDS at 68 °C for 10 min. The blots were wrapped in plastic wrap and exposed to x-ray film with intensifying screens at −80 °C. Films were photographed using a Bio-Rad VersaDoc and quantified using Quantity One software. When blots were analyzed serially for multiple transcripts, they were first stripped with 10 mm Tris·Cl, pH 7.4, 0.2% SDS at 72 °C for 2 h to remove previously hybridized probe.
Results
Identification of a Protein in HeLa Cell Mitochondrial Extracts That Binds to Human Mitochondrial RNA Polymerase (POLRMT) as Mitochondrial Ribosomal Protein L7/L12 (MRPL12)
We chose an affinity capture strategy to attempt to identify proteins that interact with POLRMT, the human mitochondrial RNA polymerase. To do this we first generated a recombinant amino-terminal His-tagged version of POLRMT that could be expressed in E. coli and bound stably to cobalt-containing beads. Using this metal affinity-based purification method, we were able to obtain highly purified recombinant POLRMT from soluble bacterial lysates (Fig. 1A). We next incubated soluble HeLa mitochondrial extracts with beads loaded with the recombinant POLRMT, and after several stringency washes we eluted proteins bound to the POLRMT affinity matrix using imidazole. The eluted proteins were separated by SDS-PAGE, and the gel was silver-stained, after which we detected one major band of ∼20 kDa that appeared to bind to POLRMT under these conditions (Fig. 1B). In parallel, we also analyzed beads containing a His-tagged control protein, human DHFR. The candidate ∼20-kDa protein was not co-purified with DHFR-loaded beads (Fig. 1B), indicating that the observed binding interaction was POLRMT-specific. We note that the elution conditions we used (300 mm imidazole) were not optimal for the elution of POLRMT from the matrix, which is why a ∼130-kDa POLRMT band was not clearly visible on the silver-stained gel (Fig. 1B, lane 2). However, these conditions were optimal for the elution of DHFR (Fig. 1B, lane 1) and the 20-kDa protein (presumably by disrupting its interaction with POLRMT). Virtually identical results, but with substantially higher background, were obtained when we boiled the beads in SDS-PAGE loading buffer to elute the bound proteins (instead of using imidazole), and in this case we were able to elute the bound POLRMT (data not shown).
To identify the ∼20-kDa putative POLRMT-binding protein, we carefully excised the band from the gel and had it subjected to tryptic digestion and subsequent liquid chromatography tandem mass spectroscopic analysis. Mass-based sequence analysis of the resulting peptide mixture revealed the presence of seven different peptides that matched exactly the amino acid sequence of human mitochondrial ribosomal protein L7/12 (Fig. 1C). Early work on bacterial ribosomes revealed that ribosomal proteins L7 and L12 are the same protein, with L7 representing an amino-terminal acetylated form of L12. Therefore, from here on, we will simply refer to this protein as mitochondrial ribosomal protein L12 (MRPL12), although at present we do not know the acetylation status of the protein. No peptides corresponding to the MLS of MRPL12 were identified (Fig. 1C), consistent with amino acids 1–45 of the protein being cleaved off upon import into mitochondria (19).
Recombinant MRPL12 Binds Directly to POLRMT
Based on the mass spectroscopy analysis, we hypothesized that MRPL12 was most likely the major protein we identified binding to POLRMT in the previous experiment. In line with this hypothesis is the predicted molecular mass of the mature MRPL12 of 16.4 kDa that is consistent with its migration at ∼20-kDa based on molecular mass standards (Fig. 1B). However, to confirm this, we generated recombinant MRPL12 (Fig. 2A), corresponding to amino acids 46–198, from E. coli and tested it for the ability to bind directly to immobilized recombinant POLRMT. Similar to the 20-kDa protein from the HeLa cell lysates (Fig. 1B), recombinant MRPL12 remained bound to immobilized POLRMT after several stringency washes, but not to unloaded beads or to control DHFR-laden beads (Fig. 2B), the two negative controls. Therefore, we conclude that MRPL12 is the protein from HeLa mitochondrial lysates we identified originally as a potential POLRMT binding partner and that it binds directly to POLRMT (i.e. without the need for additional proteins in a complex).
FIGURE 2. Recombinant MRPL12 binds directly to POLRMT in vitro.
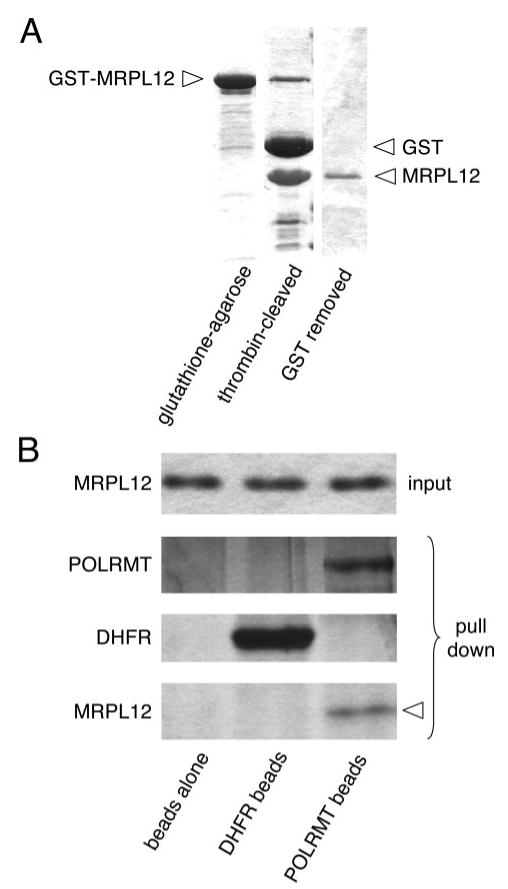
A, purification of recombinant MRPL12 from E. coli. Shown is a Coomassie-stained SDS-PAGE gel of three stages of the GST-based purification of MRPL12. The GST-MRPL12 fusion protein after glutathione-agarose chromatography is shown in the left lane, cleavage of the fusion protein with thrombin to remove the GST tag is shown in the middle lane, and the MRPL12 protein after separation from the cleaved GST peptide is shown in the right lane (see “Materials and Methods” for details). The latter fraction was used in all of the in vitro experiments in this report involving recombinant MRPL12. B, shown are silver-stained gels that represent the results of an in vitro binding experiment. The same amount of recombinant MRPL12 (input) was added to beads alone (left lane; negative control), DHFR beads (middle lane; negative protein control), or POLRMT beads (right lane). After several stringency washes, in the resulting “pull down” of bound proteins, MRPL12 only was found associated with POLRMT beads (indicated by the arrowhead).
Recombinant MRPL12 Stimulates Transcription from mtDNA Promoters in Vitro
The direct interaction between MRPL12 and POLRMT, the mitochondrial RNA polymerase, led us to hypothesize that MRPL12 has a role in regulating mitochondrial gene expression and, in particular, a function related to transcription. To test this hypothesis, we added recombinant MRPL12 to our standard in vitro mitochondrial transcription reaction. Adding MRPL12 to a partially purified POLRMT-containing lysate that alone is capable of low levels of transcription initiation from the LSP and HSP1 promoters in vitro greatly stimulated the abundance of both transcripts (Fig. 3A). This effect was not synergistic with the addition of h-mt-TFA, a well characterized mitochondrial transcription factor (1), despite the fact that, as predicted and previously reported (4), adding h-mtTFA did have some stimulatory activity when added alone to the reaction under these conditions (Fig. 3A). To confirm that the observed stimulatory activity of recombinant MRPL12 was not due to contaminating bacterial proteins that happen to co-purify with MRPL12, we performed transcription reactions with our MRPL12 recombinant fractions that were immunodepleted of MRPL12. As predicted, recombinant fractions immunodepleted of MRPL12 were no longer able to stimulate production of the LSP transcript in vitro, while mock depletion of the extract with preimmune serum had a minimal effect (Fig. 3B). The minimal reduction in activity observed in the mock-depleted reactions is likely because of alterations in buffer conditions due to the addition of serum. We conclude from these results that MRPL12 is most likely stimulating transcription from mitochondrial promoters via its interaction with POLRMT.
FIGURE 3. Recombinant MRPL12 stimulates mitochondrial transcription from the mitochondrial LSP and HSP1 promoters in vitro.
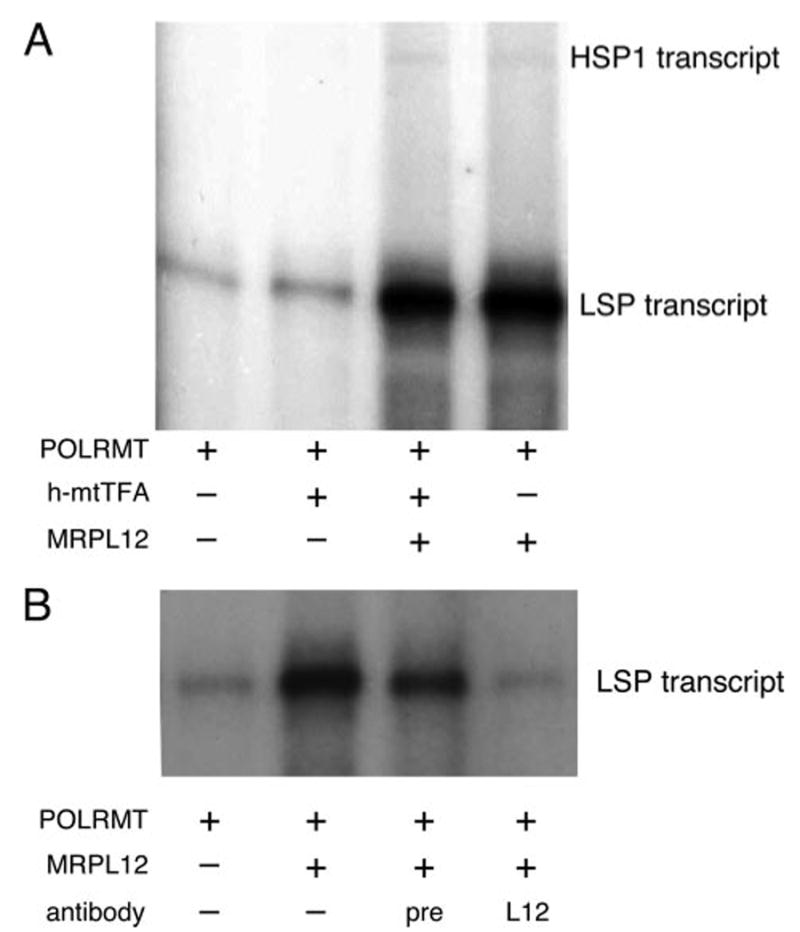
A, shown is an autoradiogram of labeled RNA transcripts from an in vitro mitochondrial transcription assay. The two run-off transcripts generated from initiation at the human mtDNA LSP and HSP promoters are labeled. The presence (+) or absence (−) of ∼200 ng of partially purified POLRMT fraction from HeLa cell mitochondria (POLRMT), recombinant h-mtTFA (40 ng), or recombinant MRPL12 (200 ng) in each reaction is indicated at the bottom. B, immunodepletion of MRPL12 from the recombinant MRPL12 preparation eliminates its transcription stimulatory activity. Shown is the LSP run-off transcript only. The presence (+) or absence (−) of a partially purified POLRMT fraction from HeLa cell mitochondria (POLRMT), recombinant MRPL12, or the antibody used (preimmune, “pre”; immune “L12”) to treat the recombinant MRPL12 preparation prior to addition to the transcription reaction is indicated at the bottom.
Overexpression of MRPL12 in HeLa Cells Results in Increased Steady-state Levels of mtDNA-encoded mRNAs
Given that all of the data generated thus far describe the MRPL12-POLRMT interaction in vitro only, we sought to confirm that this interaction occurs in vivo and is biologically relevant. To address this we generated HeLa cell lines that overexpress a carboxyl-terminal FLAG-tagged version of MRPL12. Several cell lines were isolated that overexpress MRPL12 (Fig. 4). The cell lines we analyzed overexpressed MRPL12 by ∼2-fold based on Western quantitation with MRPL12 antibody and the tagged MRPL12 localized to mitochondria based on indirect immunofluorescence microscopy (data not shown). Using these cell lines, we were able to co-immunoprecipitate POLRMT with MRPL12 using an anti-FLAG antibody directed against the tagged MRPL12. Importantly, we did not co-immunoprecipitate a mitochondrial matrix protein, HSP60, with MRPL12 (Fig. 4). Furthermore, contamination from the inner mitochondrial membrane (as revealed by the presence of COX1) was not the reason for co-IP of POLRMT with MRPL12 as both the negative control and MRPL12 co-IP reactions were contaminated equally (Fig. 4), presumably due to some adherence of membranes to the beads during the co-IP procedure. These results confirm the presence of complexes of MRPL12 and POLRMT in HeLa cells. Based on the results that MRPL12 augments mitochondrial transcription reactions in vitro (Fig. 3), we next determined whether the steady-state levels of mtDNA-encoded mRNAs are affected by increased expression of MRPL12 in HeLa cells. Consistent with the in vitro results, we observed an ∼1.5–2.0-fold increase in abundance of two mtDNA-encoded transcripts, ND2 and ND6, that represent mRNAs generated from each mtDNA strand (Fig. 5). We also observed a small (∼25–50%) increase in the mtDNA-encoded 16 S rRNA (data not shown); however, this was not as reproducible as the effects observed with the mRNAs. These effects were consistent with the ∼2-fold overexpression of MRPL12 in these cell lines.
FIGURE 4. Co-immunoprecipitation of POLRMT-MRPL12 complexes from HeLa cells.
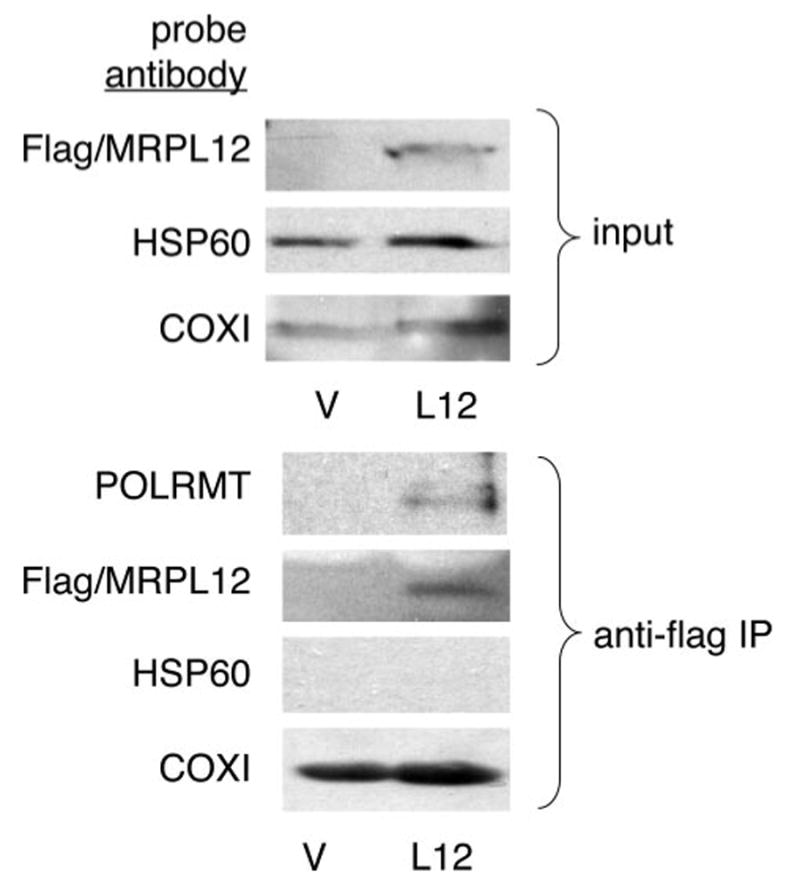
Shown are Western blots of the input (top panels) and anti-FLAG antibody-mediated immunoprecipitations (bottom panels) of MRPL12 from HeLa whole cell lysates from an empty vector transfected negative control cell line (V) or a FLAG-tagged MRPL12 overexpression cell line (L12). The input lanes were probed with the FLAG antibody to detect FLAG-tagged MRPL12 and with antibodies that recognize HSP60 (a mitochondrial matrix marker) or COX1 (a mitochondrial inner membrane protein) as controls. The immunoprecipitations were probed with these same antibodies, as well as with a POLRMT antibody to assay for co-immunoprecipitation of POLRMT by FLAGtagged MRPL12.
FIGURE 5. Overexpression of MRPL12 in HeLa cells enhances the steadystate level of mtDNA-encoded transcripts.
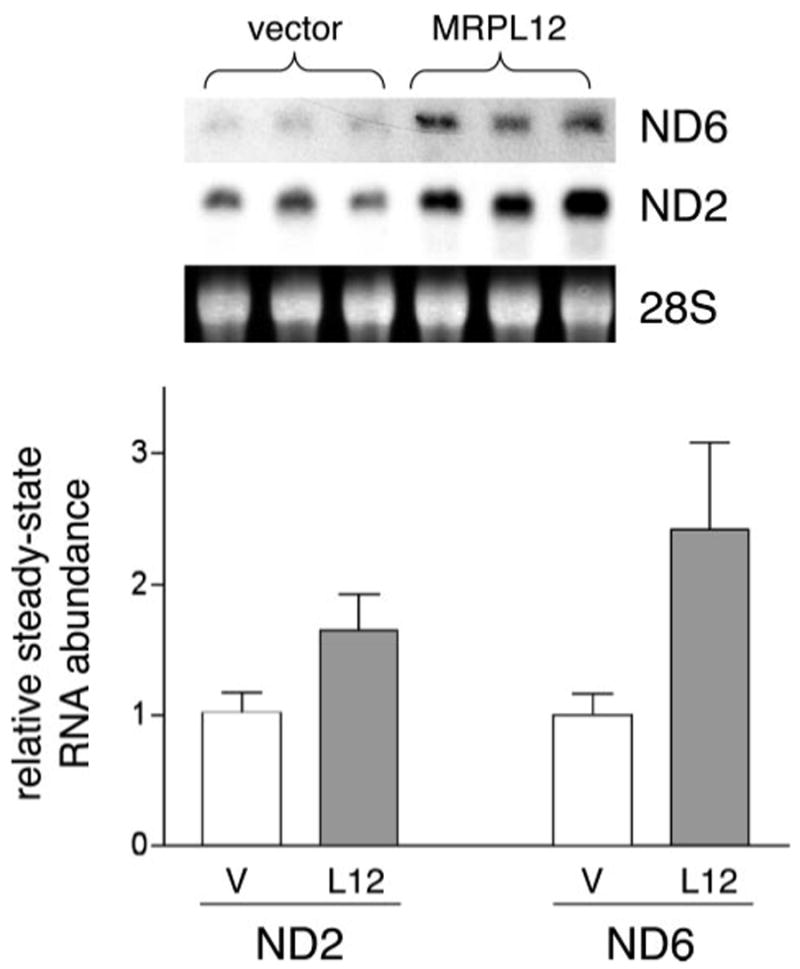
Shown is a Northern analysis of the mtDNA-encoded mRNAs from HeLa cells. The top is an autoradiogram of the blots probed for either the ND2 or the ND6 mRNA in three biological replicates of RNA isolated from empty vector control (vector) or MRPL12 overexpression HeLa cells (same lines as described in Fig. 4). The 28 S cytoplasmic rRNA from the ethidium-stained gel is also shown and was used as the loading control. The relative abundance of the ND2 and ND6 RNA was quantified based on the 28 S loading control and graphed (bottom). The ratio of the ND2/28 S or ND6/28 S in the vector control cell lines (V) was given a value of 1.0 (white bars) and used to normalize the signals from the MRPL12 overexpression lines (L12). Thus, the gray bars represent the fold up-regulation of ND2 and ND6 mRNA in the MRPL12 overexpression cell line. All data represent the average of six values ± S.D. (brackets), that is, two experiments done in triplicate (one representative experiment is shown).
Discussion
We previously demonstrated in budding yeast that mitochondrial transcription and translation are coupled at the inner mitochondrial membrane via interactions mediated by the translation-coupling factors Nam1p and Sls1p (14). Mechanistically, this involves a direct binding of Nam1p to the amino-terminal domain of yeast mtRNA polymerase (Rpo41p) and subsequent interactions of transcription complexes with Sls1p in the inner mitochondrial membrane (9–12). Thus, whether factors, in addition to those known to be required for transcription initiation, associate with the human mtRNA polymerase POLRMT to coordinate transcription and translation in human mitochondria remained an open question.
Here, we report that human MRPL12 binds to POLRMT and provide evidence that this interaction enhances transcription in vitro and the production of mtDNA-encoded RNAs in vivo. As we will discuss, our results show that, like in yeast, human mitochondrial transcription-related complexes contain proteins involved in translation and/or ribosome function that act to coordinate these processes during mitochondrial gene expression.
This study began with our attempts to identify potential POLRMT-binding proteins from HeLa cell mitochondrial extracts using an in vitro affinity capture strategy. Using this method, we isolated one major protein of ∼20-kDa that associates specifically with POLRMT in vitro (Fig. 1B). Mass spectrometry-based peptide analysis of this band excised from an SDS-PAGE gel revealed seven peptides that matched mitochondrial ribosomal protein L12 (Fig. 1C). We confirmed that this assignment was correct by producing recombinant MRPL12 in bacteria and demonstrating that the purified protein is able to bind directly to recombinant POLRMT in vitro (Fig. 2A) using the same assay we used to identify the 20-kDa protein. Thus, we were confident that we had identified MRPL12 as a bona fide POLRMT-interacting protein and proceeded with experiments geared toward determining whether the interaction of these proteins was of regulatory significance.
Given that the primary function of POLRMT is transcription of mtDNA (1), we next tested the ability of MRPL12 to modulate mitochondrial transcription. Addition of recombinant MRPL12 enhanced the production of RNA transcripts from both the LSP and HSP1 promoters of human mtDNA in vitro (Fig. 3A). In this experiment, transcription is directed by a partially purified POLRMT-containing fraction from HeLa cells that is responsive to addition of the high mobility group box transcription factor h-mtTFA. Under the conditions of this assay, MRPL12 had a stimulatory activity greater than that of h-mtTFA and there was not a major synergistic enhancement observed when both h-mtTFA and MRPL12 were added (Fig. 3A). These results are most consistent with a direct role for MRPL12 in enhancing transcription initiation. However, to conclude this unequivocally requires additional mechanistic studies in vitro, and we have not discounted a role for MRPL12 in enhancing transcription elongation or in stabilizing RNA transcripts. However, a function in RNA stability based on direct RNA binding seems less likely due to the predicted acidic pI of 5.37 of the mature form of MRPL12 in mitochondria (and used in this study) that is missing amino acids 1–45. Furthermore, MRPL12 does interact with rRNA in the context of the ribosome, likely for the same reason.
Entirely consistent with the in vitro transcription results and the conclusion that MRPL12 is involved in a mitochondrial RNA transcription-related function is the increase in the steady-state level of mtDNA-encoded transcripts we observe in HeLa cells overexpressing MRPL12 (Fig. 5). In these cells we also are able to detect POLRMT-MRPL12 protein complexes (Fig. 4). Taking all of these results together, we conclude that there is a functional interaction between MRPL12 and POLRMT in human mitochondria that is involved in regulating mitochondrial gene expression.
Our results point to a novel dual function for MRPL12 in mitochondrial gene expression. Obviously, MRPL12 is a critical component of the mitochondrial ribosome, having an evolutionarily conserved function in elongation factor binding and GTP hydrolysis (26–30). However, we show here that it is also a component of mitochondrial transcription-related complexes and has a role in regulating mitochondrial transcription and/or RNA stability. Our results demonstrate that the interaction between POLRMT and MRPL12 is direct and does not require the presence of the latter in intact mitochondrial ribosomes (Fig. 2). This, coupled with the fact that MRPL12 was not isolated in stoichiometric amounts with any other mitochondrial ribosomal proteins from HeLa cell extracts (Fig. 1B), strongly suggests that it is “free” MRPL12 that interacts with POLRMT. The fact that free L12 has been shown to accumulate in E. coli is consistent with this proposal (31). If this is in fact also the case inhuman mitochondria, it is tempting to speculate that one role for this interaction is to coordinate the rate of mitochondrial transcription with the rate of mitochondrial ribosomal biogenesis. That is, the presence of free MRPL12 could indicate that expression of mtDNA-encoded RNAs (which include the 12 S and 16 S rRNAs) is occurring slowly compared with the rate of import of mitochondrial ribosomal proteins. This would lead to a situation where expression of mtDNA-encoded rRNAs is limiting ribosome biogenesis. Under these conditions, MRPL12 is predicted to associate with POLRMT to increase the rate of mtDNA transcription and rebalance the system. Similarly, import of MRPL12 may serve as a signal to increase expression of mitochondrial rRNAs and mRNAs when cells need to increase mitochondrial oxidative phosphorylation activity or biogenesis in response to changing cellular energy demands. Such a function in balancing ribosome biogenesis with transcription may be conserved. In bacteria, several ribosome proteins, including L12 (32), when not assembled into functional ribosomes, feedback inhibit their own expression and that of RNA polymerase (33). Finally, it is noteworthy that mitochondrial ribosomal protein L12 was initially identified as a growth-regulated gene in cultured cells that accumulates in the G1 phase of the cell cycle (19, 34). Furthermore, Drosophila mitochondrial L12 was recently identified as a target of Cyclin D-Cdk4 complex in the regulation of cell growth (35). Thus, MRPL12 appears to be a novel-acting mitochondrial regulatory protein that coordinates mitochondrial function with cell cycle cues. Our identification of MRPL12 as regulator of mitochondrial transcription-related complexes is very likely a key underlying factor in these unique roles of this multifunctional mitochondrial ribosomal protein. Understanding the mechanism of this new form of regulation will be an exciting area of future investigation.
Acknowledgments
We thank Jana Eaton for critical reagents and helpful advice on the project, Todd Gangelhoff and Dr. Mair Churchill for providing recombinant h-mtTFA, Dr. Edward Chu for providing the DHFR expression plasmid, and Dr. Thomas O'Brien for providing human MRPL12 antibodies.
Footnotes
This work was supported by NHLBI, National Institutes of Health Grant HL-059655 (to G. S. S.).
The abbreviations used are: MLS, mitochondrial localization sequence; GST, glutathione S-transferase; co-IP, co-immunoprecipitation; POLMRT, mitochondrial RNA polymerase; MRP, mitochondrial ribosomal protein; DHFR, dihydrofolate reductase; LSP, light-strand promoter; HSP, heavy-strand promoter.
References
- 1.Bonawitz ND, Clayton DA, Shadel GS. Mol Cell. 2006;24:813–825. doi: 10.1016/j.molcel.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 2.McCulloch V, Shadel GS. Mol Cell Biol. 2003;23:5816–5824. doi: 10.1128/MCB.23.16.5816-5824.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM. Nat Genet. 2002;31:289–294. doi: 10.1038/ng909. [DOI] [PubMed] [Google Scholar]
- 4.McCulloch V, Seidel-Rogol BL, Shadel GS. Mol Cell Biol. 2002;22:1116–1125. doi: 10.1128/MCB.22.4.1116-1125.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotney J, Shadel GS. J Mol Evol. 2006;63:707–717. doi: 10.1007/s00239-006-0075-1. [DOI] [PubMed] [Google Scholar]
- 6.Seidel-Rogol BL, McCulloch V, Shadel GS. Nat Genet. 2003;33:23–24. doi: 10.1038/ng1064. [DOI] [PubMed] [Google Scholar]
- 7.Matsushima Y, Adan C, Garesse R, Kaguni LS. J Biol Chem. 2005;280:16815–16820. doi: 10.1074/jbc.M500569200. [DOI] [PubMed] [Google Scholar]
- 8.Masters BS, Stohl LL, Clayton DA. Cell. 1987;51:89–99. doi: 10.1016/0092-8674(87)90013-4. [DOI] [PubMed] [Google Scholar]
- 9.Rodeheffer MS, Boone BE, Bryan AC, Shadel GS. J Biol Chem. 2001;276:8616–8622. doi: 10.1074/jbc.M009901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Shadel GS. Proc Natl Acad Sci U S A. 1999;96:8046–8051. doi: 10.1073/pnas.96.14.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryan AC, Rodeheffer MS, Wearn CM, Shadel GS. Genetics. 2002;160:75–82. doi: 10.1093/genetics/160.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodeheffer MS, Shadel GS. J Biol Chem. 2003;278:18695–18701. doi: 10.1074/jbc.M301399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naithani S, Saracco SA, Butler CA, Fox TD. Mol Biol Cell. 2003;14:324–333. doi: 10.1091/mbc.E02-08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shadel GS. Trends Genet. 2004;20:513–519. doi: 10.1016/j.tig.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Shadel GS, Clayton DA. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 16.Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Cell. 2003;115:97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien TW. Gene. 2002;286:73–79. doi: 10.1016/s0378-1119(01)00808-3. [DOI] [PubMed] [Google Scholar]
- 18.Tiranti V, Savoia A, Forti F, D'Apolito MF, Centra M, Rocchi M, Zeviani M. Hum Mol Genet. 1997;6:615–625. doi: 10.1093/hmg/6.4.615. [DOI] [PubMed] [Google Scholar]
- 19.Marty L, Fort P. J Biol Chem. 1996;271:11468–11476. doi: 10.1074/jbc.271.19.11468. [DOI] [PubMed] [Google Scholar]
- 20.Nam SC, Kang C. Protein Expression Purif. 2001;21:485–491. doi: 10.1006/prep.2000.1383. [DOI] [PubMed] [Google Scholar]
- 21.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 22.Seidel-Rogol BL, Shadel GS. Nucleic Acids Res. 2002;30:1929–1934. doi: 10.1093/nar/30.9.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Correa-Rotter R, Mariash CN, Rosenberg ME. BioTechniques. 1992;12:154–158. [PubMed] [Google Scholar]
- 24.Duhl DM, Gillespie DD, Sulser F. J Neurosci Methods. 1992;42:211–218. doi: 10.1016/0165-0270(92)90100-r. [DOI] [PubMed] [Google Scholar]
- 25.Eykholt RL, Mitchell MD, Marvin KW. BioTechniques. 2000;28:864–866. 868, 870. doi: 10.2144/00285bm07. [DOI] [PubMed] [Google Scholar]
- 26.O'Brien TW. IUBMB Life. 2003;55:505–513. doi: 10.1080/15216540310001626610. [DOI] [PubMed] [Google Scholar]
- 27.Liu M, Spremulli L. J Biol Chem. 2000;275:29400–29406. doi: 10.1074/jbc.M002173200. [DOI] [PubMed] [Google Scholar]
- 28.Dey D, Oleinikov AV, Traut RR. Biochimie (Paris) 1995;77:925–930. doi: 10.1016/0300-9084(95)80003-4. [DOI] [PubMed] [Google Scholar]
- 29.Savelsbergh A, Mohr D, Wilden B, Wintermeyer W, Rodnina MV. J Biol Chem. 2000;275:890–894. doi: 10.1074/jbc.275.2.890. [DOI] [PubMed] [Google Scholar]
- 30.Traut RR, Dey D, Bochkariov DE, Oleinikov AV, Jokhadze GG, Hamman B, Jameson D. Biochem Cell Biol. 1995;73:949–958. doi: 10.1139/o95-102. [DOI] [PubMed] [Google Scholar]
- 31.Ramagopal S. Eur J Biochem. 1976;69:289–297. doi: 10.1111/j.1432-1033.1976.tb10885.x. [DOI] [PubMed] [Google Scholar]
- 32.Johnsen M, Christensen T, Dennis PP, Fiil NP. EMBO J. 1982;1:999–1004. doi: 10.1002/j.1460-2075.1982.tb01284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nomura M, Gourse R, Baughman G. Annu Rev Biochem. 1984;53:75–117. doi: 10.1146/annurev.bi.53.070184.000451. [DOI] [PubMed] [Google Scholar]
- 34.Marty L, Taviaux S, Fort P. Genomics. 1997;41:453–457. doi: 10.1006/geno.1997.4691. [DOI] [PubMed] [Google Scholar]
- 35.Frei C, Galloni M, Hafen E, Edgar BA. EMBO J. 2005;24:623–634. doi: 10.1038/sj.emboj.7600523. [DOI] [PMC free article] [PubMed] [Google Scholar]