

Abstract
Alignment of three fungal mtRNA polymerases revealed conserved amino acid sequences in an amino-terminal region of the Saccharomyces cerevisiae enzyme implicated previously as harboring an important functional domain. Phenotypic analysis of deletion and point mutations, in conjunction with a yeast two-hybrid assay, revealed that Nam1p, a protein involved in RNA processing and translation in mitochondria, binds specifically to this domain. The significance of this interaction in vivo was demonstrated by the fact that the temperature-sensitive phenotype of a deletion mutation (rpo41Δ2), which impinges on this amino-terminal domain, is suppressed by overproducing Nam1p. In addition, mutations in the amino-terminal domain result specifically in decreased steady-state levels of mature mitochondrial CYTB and COXI transcripts, which is a primary defect observed in NAM1 null mutant yeast strains. Finally, one point mutation (R129D) did not abolish Nam1p binding, yet displayed an obvious COX1/CYTB transcript defect. This mutation exhibited the most severe mitochondrial phenotype, suggesting that mutations in the amino-terminal domain can perturb other critical interactions, in addition to Nam1p binding, that contribute to the observed phenotypes. These results implicate the amino-terminal domain of mtRNA polymerases in coupling additional factors and activities involved in mitochondrial gene expression directly to the transcription machinery.
Expression of the mitochondrial genome occurs in the organelle matrix and involves both nuclear- and mtDNA-encoded factors. In addition to mRNAs (which usually encode protein subunits of the enzyme complexes involved in oxidative phosphorylation), mtDNA in many organisms also contains genes for tRNAs and ribosomal RNAs that are necessary for mitochondrial translation. The remaining factors required for expression and replication of the mitochondrial genome are encoded in the nucleus and imported into the organelle. Mitochondrial transcripts are often polycistronic and thus a large number of RNA processing reactions is required to liberate mature RNA species (1). In addition, RNA processing events have been implicated in the initiation of mtDNA replication (2). Thus, a complete understanding of mitochondrial gene expression and mtDNA replication requires broader understanding of how these RNA processing events are accomplished in vivo.
The mitochondrial transcription machinery in the budding yeast, Saccharomyces cerevisiae, is well characterized (3, 4) and involves a nucleus-encoded mtRNA polymerase (sc-mtRNA polymerase, encoded by the RPO41 gene) that is homologous to the single subunit Escherichia coli bacteriophage RNA polymerase (5, 6) and a transcription initiation factor sc-mtTFB (7–9). In addition, factors involved in mtRNA processing have also been identified in this organism, including those involved in liberating tRNAs, rRNAs, and mRNAs from polycistronic transcripts and excising introns from certain messages (see Ref. 10 for review). One such factor, Nam1p (also known as Mtf2p), was initially identified as a high copy suppressor of mtDNA point mutations that affect splicing of introns from the mitochondrial COX1 and CYTB messages (11) and independently as a temperature-sensitive mutation affecting mitochondrial transcript levels (12). Nam1p is localized to the mitochondrial matrix (13), and characterization of NAM1 null mutant strains has confirmed its involvement in COX1 and CYTB intron removal and elucidated potential roles for this protein in overall mitochondrial translation capacity and ATP6/8 mRNA processing and/or stability (14). In addition, crude mitochondrial transcription complexes isolated from one NAM1 mutant strain remain competent for transcription but exhibit an altered DNA binding activity profile (12), suggesting a potential link between Nam1p function and the mitochondrial transcription machinery.
We had shown previously that an amino-terminal domain of yeast mtRNA polymerase is dispensable for transcription initiation in vivo but nonetheless is required for stability and maintenance of the mitochondrial genome, suggesting that additional activities may be coupled to the transcription process in mitochondria (15). These studies implicated amino acids 29–208 of mtRNA polymerase as a minimal portion of the protein that harbors an independent functional domain of the enzyme. Here we have characterized this domain further and demonstrate that one of its functions is to provide an interaction point for Nam1p, that may provide the means to couple factors involved in additional aspects of RNA metabolism directly to the transcription machinery in yeast mitochondria. In principal, such a coupling phenomenon in mitochondria may be analogous to mechanisms of gene expression in the nucleus, where many aspects of mRNA processing are coupled functionally to transcription via interactions involving the carboxyl-terminal domain of the largest subunit of RNA polymerase II (16, 17).
Experimental Procedures
Plasmid Construction
Wild-type and mutated alleles of RPO41 used in the plasmid shuffle assay were expressed in yeast from the plasmid pGS348 (15), which contains a 7.2-kb,1 RPO41 gene-containing, SalI-SpeI restriction fragment of yeast genomic DNA cloned into the shuttle vector pRS314 (18). Construction of the rpo41Δ2- and rpo41Δ3-containing versions of pGS348 has been described elsewhere (15). All of the yeast two-hybrid bait plasmids utilized in this study are derivatives of pAS1 (19), which express the RPO41 reading frame as amino-terminal fusions to the Gal4p DNA-binding domain. Two parental RPO41-bait plasmids were used, pAS1-RPO and pAS1-RPO-FL, that contained amino acids 27–633 and amino acids 27–1351 of sc-mtRNA polymerase, respectively (numbered according to Ref. 6). To construct the corresponding mutated RPO41-bait plasmids, restriction fragments containing the mutated versions of RPO41 were isolated from PCR products (or from the corresponding pGS348 derivatives) and ligated into either pAS1-RPO or pAS1-RPO-FL (Table I). The plasmid used to overproduce Nam1p (pYES/GS-NAM1) was obtained from Invitrogen (Genestorm clone yDL044cy). This plasmid contains a V5 epitope-tagged version of the NAM1 gene under control of a galactose-inducible promoter.
Table I. Results of a yeast two-hybrid analysis of the interaction of Nam1p with the amino-terminal domain of sc-mtRNA polymerase.
| RPO41-bait plasmid | mtRNA polymerase amino acids present | LacZ assay |
|---|---|---|
| pAS1-RPO-FL | 27–1351 | Positive |
| pAS1-RPO | 27–633 | Positivea |
| pAS1-RPOΔ2 | 118–633 | Positive |
| pAS1-RPOΔ3 | 212–633 | Negative |
| pAS1-RPOΔ4 | 400–633 | Negativea |
| pAS1-RPO (E119A/C121A) | 27–633 | Positive |
| pAS1-RPO (R129D) | 27–633 | Positive |
| pAS1-RPO-FL (N152A/Y154A) | 27–1351 | Negative |
| pAS1-RPO-FL (E179A) | 27–1351 | Positive |
Also shown in Fig. 3A.
Site-directed Mutagenesis
Specific point mutations in the RPO41 gene were generated by a two-step, megaprimer PCR protocol as follows. The PCR template (pBS348) consisted of a 2.1-kb SalI-BamHI restriction fragment from pGS348, which contains the amino-terminal extension of RPO41 and upstream sequences, cloned into pBSII KS+ (Stratagene, Inc). Promoters for T7 and T3 RNA polymerase flank the RPO41 insert in this plasmid. For each mutation, a PCR was performed that utilized a specific mutagenic oligonucleotide (synthesized by Midland Certified Reagent Co., Midland, TX) as one primer and an oligonucleotide corresponding to the T7 promoter as the second primer to generate an ∼500-base pair product. This PCR product was gel-purified and used as the source of a megaprimer (after denaturation, the 500-nucleotide strand containing the T7 primer sequence serves as the megaprimer) in a second PCR in conjunction with a T3 primer. The 2.2-kb product of this second PCR, which now had the desired mutation fixed in the RPO41 gene, was digested with SalI and BamHI and ligated into pGS348 to form a full-length mutated RPO41 allele that can be expressed in yeast. All PCRs were performed using Pfu Turbo DNA Polymerase (Stratagene, Inc.) in the buffer supplied by the manufacturer and typically consisted of 25–30 amplification cycles (95 °C, 30 s; 55 °C, 1 min; 68 °C, 8 min) followed by a 12-min, 68 °C extension period at the end of the last cycle. In all cases, the nucleotide sequence of the portion of RPO41 open reading frame in pGS348 that was amplified during the mutagenesis protocol was determined to ensure that the desired site-directed mutation was the only mutation introduced during the PCR procedure.
Phenotypic Analysis of RPO41 Mutations by Plasmid Shuffle
Routine growth and transformation of yeast strains, as well as standard growth media preparation, were accomplished as described by Sherman (20). The yeast strain GS112 (α his3-Δ200 leu2-3,-112 ura3-52 trp1-Δ1 ade2, rpo41Δ1∷HIS3 +pGS347 (URA3, RPO41)) was used to analyze RPO41 gene mutations by plasmid shuffle (21). After transformation of GS112 with the desired mutant RPO41 allele on a pGS348 plasmid, two rounds of growth on synthetic dextrose (SD) medium containing 5-fluoroorotic acid were used to select for strains that express the mutated version of the RPO41 gene as their only source of mtRNA polymerase, as described previously (15). To characterize the growth phenotypes of the mutant strains in detail, serial 10-fold dilutions were plated onto solid YPD (glucose-containing medium) and YPG (glycerol-containing medium) using a 48-pin multiplex plating tool (“Frogger,” Dankar, Inc.) and grown at both 30 and 37 °C. Growth on YPG medium requires mitochondrial respiration, whereas growth on YPD does not; therefore, defective growth on YPG medium was scored as a mitochondrial petite phenotype.
Yeast Two-hybrid Analysis
The yeast strain Y190 (a gal4 gal80 his3 trp1-901 ade2-101 ura3-52 leu2-3, -112 + URA3∷GAL-lacZ + LYS2∷GAL-HIS3 cyhr) was used for all two-hybrid analyses. This strain contains two Gal4p-inducible reporter genes integrated in its genome that allow two-hybrid positives to be selected based on a His+ growth phenotype and screened using a blue color-generating β-galactosidase (LacZ) assay (19). Y190 was transformed with pAS1-RPO to generate an RPO41-bait strain for the initial two-hybrid screen for proteins that interact with an intact amino-terminal domain. The resulting strain, Y190 (pAS-RPO), was subsequently transformed with a library of yeast cDNA fragments fused to the Gal4p activation domain in the LEU2 plasmid pACT (19). Leu+, His+ colonies were selected on SD medium supplemented with adenine (20 mg/liter) and containing 3-amino-1,2,4-triazole (Sigma) at 30–50 mm. We pooled the Leu+, His+ colonies from a screen of ∼70,000 library plasmid transformants and isolated plasmids from these strains en masse using a smash and grab protocol (22). In this manner, an enriched library of potential positives (library plasmids that yielded a His+ phenotype) was generated. This library was used to transform the yeast strain Y190 pAS1-RPOΔ4 that contains an RPO41-bait plasmid specifically missing the amino-terminal domain. Leu+ transformants from this transformation were replica-plated onto both Leu−, His+ medium and Leu−, His− medium to select for strains that were Leu+ (i.e. contained a library plasmid), yet His− due to lack of interaction with the amino-terminal truncated bait. The 52 strains that met all the above criteria were kept for further analysis. Next, the library plasmid from each of these strains was isolated and used to transform a fresh Y190 (pAS1-RPO) strain. A LacZ color assay (19) was used as our final screen for library plasmids that encoded proteins that interacted specifically with the amino-terminal domain. Plasmids that conferred a blue color in Y190 (pAS1-RPO) but not in Y190 (pAS1-RPOΔ4) were subjected to nucleotide sequence analysis to determine the identity of the gene implicated in the interaction.
RNA Isolation and Northern Analysis of Mitochondrial Transcripts
A 7-ml culture of each yeast strain analyzed was grown at 30 °C in YPG medium to stationary phase. This culture was used to inoculate 450 ml of SD medium supplemented with leucine (100 mg/liter), adenine (20 mg/liter), and uracil (20 mg/liter) to a starting A600 of 0.025. This culture was subsequently grown at 37 °C on an orbital shaker (200 rpm) to a final A600 of 0.8. Yeast cells were harvested by centrifugation and resuspended in spheroplasting buffer (1.35 m sorbitol, 0.1 m EDTA, 0.1% β-mercaptoethanol, pH 7.4). Zymolyase 20T (U. S. Biochemical Corp.) was added to a concentration of 3 mg/g wet weight of cells and incubated at 37 °C for 10 min. Six volumes of 1 m sorbitol were added to the sample, and the resulting spheroplasts were harvested by centrifugation (3000 × g for 5 min at 4 °C). The spheroplast pellet was then resuspended in 2 volumes of MT buffer (400 mm mannitol, 2 mm EDTA, 50 mm Tris-Cl, pH 7.4). An equal volume of glass beads was added, and the spheroplasts were lysed by vortexing (two 30-s pulses separated by a 1-min incubation on ice). The resulting cell lysate was removed from the glass beads and transferred to a fresh centrifuge tube, and 10 volumes of MT buffer were added. The lysate was then subjected to a low speed centrifugation (1900 × g for 5 min at 4 °C) to remove cellular debris and nuclei. The resulting supernatant was transferred to a fresh centrifuge tube and subjected to a final high speed centrifugation (10,500 × g for 10 min at 4 °C) to pellet mitochondria. RNA was extracted immediately from this mitochondrial pellet as described (23) and purified further using RNeasy columns (Qiagen). Five μg of column-purified mitochondrial RNA from each sample was separated on a 1.3% agarose-formaldehyde gel as described (24). Northern blotting was performed essentially as described (15), except the random prime radiolabeled probes used for these experiments were generated using PCR products corresponding to the mitochondrial COX1, COX3, and COB genes as templates. The PCR products used as probe templates were generated using purified yeast mtDNA as a template and the following gene-specific primers: COX1, 5′-CCATTAATAATTGGAGCTACAG-3′ and 5′-CCAAAGAATCAAAATAAATGCTCG-3′; COB, 5′-GGCATTTAGAAAATCAAATGTG-3′ and 5′-CTGTCCATAAACACAACAATAACC-3′; COX3, 5′-ATGACACATTTAGAAAGAAGTAG-3′ and 5′-TTAGACTCCTCATCAGTAGAAGA-3′.
Results
A Conserved Amino-terminal Domain in sc-mtRNA Polymerase
The mtRNA polymerase of S. cerevisiae is related to bacteriophage RNA polymerases (e.g. T7, T3, and SP6) but contains a unique amino-terminal extension (Fig. 1). In an earlier study (15), we reported the analysis of a series of RPO41 deletion mutations that had no effect on protein stability or localization, and ultimately revealed the existence of a functional amino-terminal domain in mtRNA polymerase between amino acids 28 and 208 that is required for mtDNA maintenance and can function in trans. Whereas mtRNA polymerases from most species contain significant amino acid identity in those regions involved in catalytic activity (i.e. the bacteriophage T7 family-like domains), the amino-terminal extensions are not well conserved between species. However, we were able to align the amino acid sequence of the amino-terminal extension of the S. cerevisiae enzyme with that from two other fungal species, Neurospora crassa and Schizosaccharomyces pombe, and found that the most conserved region corresponded to amino acids 110–205 of the S. cerevisiae mtRNA polymerase (Fig. 1). In particular, the region encompassing amino acids 117–155 exhibited 18% amino acid identity (identical residues in all three species) and 38% amino acid similarity (identical or similar amino acids in all three species). The degree of similarity in this region is emphasized further if pairwise comparisons are made. For example, the S. cerevisiae and N. crassa proteins are 35% identical and 56% similar in this region.
Fig. 1. A region of the sc-mtRNA polymerase amino-terminal extension is conserved in two other fungal species.
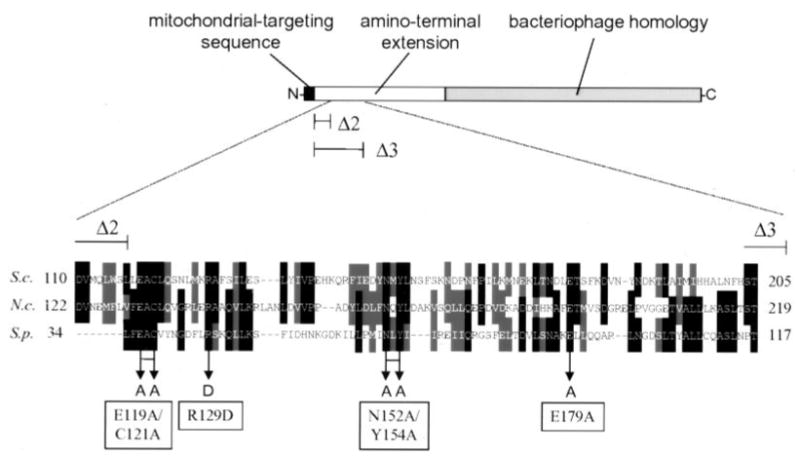
A linear representation of sc-mtRNA polymerase (encoded by the RPO41 gene) is presented at the top of the figure. The carboxyl-terminal portion of the enzyme that is homologous to bacteriophage RNA polymerases is depicted as a gray box, the mitochondrial targeting sequence by a black box, and the amino-terminal extension by a white box. Expanded at the bottom is an alignment of the most conserved portion of the amino-terminal extensions from S. cerevisiae (S.c.), amino acids 110–205; N. crassa (N.c.), amino acids 122–219; and S. pombe (S.p.), amino acids 34–117. Amino acid residues that are identical are indicated by darker shading, and those that are similar are indicated by lighter shading. The residues in sc-mtRNA polymerase that were changed by site-directed mutagenesis are indicated by arrows, at the ends of the arrows the amino acid substitution is shown (as well as the RPO41 allele designations, which are boxed). Also indicated is the end point of two deletion mutations that were characterized previously (15). The rpo41Δ2 allele deletes amino acids 27–117 (end point labeled Δ2), and thus partially impinges on the conserved region. The rpo41Δ3 allele deleted amino acids 27–212 (end point labeled Δ3) and therefore completely removes the conserved region.
To test the hypothesis that the conserved region of the mtRNA polymerase amino-terminal extension composes all or part of the important functional domain (aa 28–208) we identified previously (15), we made a series of point mutations in the S. cerevisiae RPO41 gene by site-directed mutagenesis that changed residues that are conserved in all three of the fungal mtRNA polymerases (Fig. 1). The mitochondrial phenotype (assessed by growth on YPG medium) of each of these mutations was determined using a standard plasmid-shuffle strategy, and the effects of these mutations were compared with the phenotypes of two previously characterized deletion mutations in this region (rpo41Δ2 and rpo41Δ3, Fig. 2A). Two individual point mutations were introduced to generate two mutant RPO41 alleles, arginine 129 changed to aspartic acid (denoted R129D) and E179A. In addition, two double point mutant alleles were created, E119A/C121A and N152A/Y154A. Both double-point mutant alleles, as well as the R129D allele, exhibited a mitochondrial petite phenotype (slow growth on YPG medium) at both 30 and 37 °C, whereas the E179A mutations resulted in no discernible phenotype. Of the three mutant strains that had a mitochondrial phenotype, the E119A/C121A mutant was the only one capable of sustained growth on YPG at 37 °C, although its growth rate was still markedly reduced compared with the wild-type strain at both 30 and 37 °C. This growth phenotype was almost identical to that observed for the rpo41Δ2 mutation. The N152A/Y154A mutant strain grew slowly on YPG at 30 °C and was dramatically impaired on YPG at 37 °C. This growth phenotype was similar to that observed for the rpo41Δ3 mutation. The R129D mutation was the most severe mutation, resulting in the slowest growth rate on YPG at 30 °C and virtually no growth on YPG at 37 °C. It is also noted that two of the point mutations (R129D and N152A/Y154A) exhibited a more severe phenotype than the rpo41Δ3 mutation, suggesting that this domain when present and mutated perturbs mtRNA polymerase function more severely than a mutation that deletes this region altogether. To confirm that the observed phenotypes of the point mutations were not due to altered protein stability, the expression level of each of the mutant proteins was analyzed by western analysis (Fig. 2B). All of these proteins were expressed at levels similar to that of the wild-type protein. Again, these results are consistent with our earlier analysis of the rpo41Δ3 mutation that removes this domain completely but does not affect protein localization or stability (15).
Fig. 2. Analysis of sc-mtRNA polymerase amino-terminal domain mutations.

A, growth phenotypes. Shown to the left is the RPO41 allele expressed in each strain after plasmid shuffle in GS112. At the top of each column of panels, the growth medium (YPG or YPD) and growth temperature (30 or 37 °C) are indicated. For example, the strain that is expressing wild-type allele (RPO41) after plasmid shuffle is indicated. Serial 10-fold dilutions are plated from left to right within each panel and, in each row of panels, identical cultures were plated. B, western analysis of rpo41 point-mutant strains after plasmid shuffle. The blot was first probed with an anti-RPO41p antibody and the location of the 150-kDa, full-length mtRNA polymerase (Rpo41p) is indicated. The RPO41 null and point-mutant strains analyzed are indicated above each lane. The identical blot was stripped and probed again with an antibody against Yrb1p (yeast Ran-binding protein, a nuclear transport protein) to serve as a control for the amount of protein loaded in each lane.
Nam1p Interacts with the Amino-terminal Domain of Yeast mtRNA Polymerase
The observed phenotypes of the amino-terminal domain mutations suggest that this region of the protein is involved in coupling some critical function to the transcription process in mitochondria (15). This coupling capacity could occur directly, by virtue of a structural or catalytic role for this domain, or indirectly through the binding of other mitochondrial regulatory factors, or perhaps both. To test the hypothesis that other mitochondrial factors are involved, we screened a library of yeast genomic DNA fragments for proteins that bind to the amino-terminal portion of yeast mtRNA polymerase using a yeast two-hybrid assay. Our strategy was to identify proteins that interact specifically with the amino-terminal domain of mtRNA polymerase that was implicated here and in our previous studies (15). To accomplish this, we created two yeast two-hybrid bait plasmids. The first, pAS-RPO, contained RPO41 sequences that encompassed an intact amino-terminal domain (encoding aa 27–633) and the second, pAS-RPOΔ4, that does not encodes the amino-terminal domain but does contain other RPO41 sequences (encoding aa 392–633). The screen involved several steps that ultimately selected for plasmids encoding proteins that exhibited an interaction specifically with the amino-terminal portion of sc-mtRNA polymerase (see “Experimental Procedures”). From an initial screen of ∼70,000 transformants, four library plasmids were identified that met all selection criteria. The yeast genomic DNA fragment in each of these was determined by sequencing both ends of the insert and using this information to search the Saccharomyces Genome Database. Two of these isolates contained the same insert containing the entire NAM1 open reading frame (Fig. 3), a gene encoding a mitochondrial matrix protein involved in mtRNA processing and/or translation (11, 12, 14). The specificity of the Nam1p interaction for the amino-terminal extension of sc-mtRNA polymerase is shown in Fig. 3A. The other two isolates corresponded to YTA5 (or RPT2), an ATPase component of 26 S of the proteasome complex, and YGL037c, a yeast ORF of unknown function. We believe that YTA5 is probably of no functional significance because proteasome subunits are commonly found as false positives in yeast two-hybrid screens, and our preliminary experiments with YGL037c suggest that this gene product does not localize to mitochondria (data not shown). For these reasons, and because Nam1p exhibits characteristics compatible with a protein that might interact with mtRNA polymerase, we focused our attention on NAM1.
Fig. 3. Nam1p interacts specifically with the amino-terminal extension of sc-mtRNA polymerase and suppresses a mutation in this region.

A, β-galactosidase activity (dark color in lower panel) indicates a two-hybrid interaction between Nam1p and sc-mtRNA polymerase in the lacZ-reporter strain Y190, which that contains a plasmid (pAS1-RPO) encoding an intact amino-terminal domain. The same strain containing a plasmid (pAS-RPOΔ4) that is missing the amino-terminal domain, but contains other sc-mtRNA polymerase sequences, produces no β-galactosidase activity (upper panel, no dark color). B, overproduction of Nam1p suppresses the YPG growth defects of a rpo41Δ2 strain but not a rpo41Δ3 strain. Five strains that were streaked onto a YPG plate and grown at 36 °C are shown. The yeast strain GS124 contains a plasmid encoding the rpo41Δ2 allele as its only source of sc-mtRNA polymerase; the yeast strain GS125 contains a plasmid encoding the rpo41Δ3 allele as its only source of sc-mtRNA polymerase; and GS122 is the isogenic wild-type strain that has the wild-type RPO41 gene provided on a plasmid as its only source of sc-mtRNA polymerase (15). GS124 and GS125 transformed with an empty URA3 plasmid (YEp352) or with a URA3 plasmid that overexpresses Nam1p (pYES/GS-NAM1) are indicated.
In our initial two-hybrid screen, the bait plasmid used (pAS-RPO) encoded only amino acids 27–633 of mtRNA polymerase, which primarily contains just the amino-terminal extension of mtRNA polymerase. To ensure that Nam1p interacts with full-length mtRNA polymerase, we constructed a two-hybrid bait plasmid (pAS-RPO-FL) that encodes the entire mature protein (amino acids 27–1351), and we tested it for the interaction with Nam1p in the two-hybrid assay. We found that Nam1p was able to interact with its amino-terminal target in the context of an intact mtRNA polymerase as well (Table I), thus this interaction could be analyzed using either the amino-terminal extension alone or in the context of the full-length mtRNA polymerase. We next determined the effect of the amino-terminal domain mutations on the ability of mtRNA polymerase to bind Nam1p in the two-hybrid assay (Table I). The rpo41Δ2 mutant, which retains a largely intact amino-terminal domain (Fig. 1), exhibited a reduced, yet significant, ability to interact with Nam1p, whereas the rpo41Δ3 mutant, which is deleted for the amino-terminal domain altogether, did not interact in this assay (Table I). Of the point mutated proteins, the N152A/Y154A mutant was the only one that did not interact with Nam1p to some degree in this assay. All of the mutated RPO41 fusion proteins shown in Table I were expressed at least as well as the wild-type fusion protein (data not shown); thus those mutations reported to disrupt the Nam1p interaction did not dramatically affect expression or stability of the two-hybrid bait protein.
Nam1p Overexpression Rescues the Temperature-sensitive Phenotype of the rpo41Δ2 Mutation
To determine whether the Nam1p interaction with the amino-terminal domain of mtRNA polymerase that we identified by two-hybrid analysis is of physiological significance, we next tested whether overexpression of Nam1p could rescue the phenotype of mutations in the amino-terminal domain. To accomplish this we utilized a high copy plasmid (pYES/GS-NAM1) that expresses an epitope-tagged version of the NAM1 gene from a galactose-inducible promoter (see “Experimental Procedures”). Expression levels of the tagged version of Nam1p from this promoter, even without galactose induction, are capable of complementing a chromosomal NAM1 disruption, confirming that the tagged Nam1p is functional in vivo (data not shown). Introduction of this Nam1p-overproducing plasmid resulted in significant rescue of the temperature-sensitive phenotype of the rpo41Δ2 mutation but not that of the rpo41Δ3 mutation (Fig. 3B). These results are consistent with the fact that the rpo41Δ2-encoded mtRNA polymerase is still capable of interacting with Nam1p to some extent in the two-hybrid assay, whereas that encoded by rpo41Δ3 is not (Table I). None of the amino-terminal domain point mutations was suppressed by overproducing Nam1p under these conditions (data not shown). In the case of the N152A/Y154A protein, this result is consistent with an inability to interact with Nam1p in the two-hybrid assay. However, the E119A/C121A and R129D proteins still interact with Nam1p to some degree in the two-hybrid assay, yet their mitochondrial defects cannot be rescued by overexpression of Nam1p, suggesting that there are other defects that are contributing to the observed phenotypes of these mutations (see “Discussion”).
Mutations in the Amino-terminal Domain of mtRNA Polymerase Result in Mitochondrial RNA Transcript Defects Consistent with Perturbation of Nam1p Function
A documented phenotype of NAM1 null mutations (14) is the specific reduction of the intron-containing mitochondrial transcripts encoding cytochrome b (COB) and cytochrome c oxidase subunit 1 (COX1). We had reported previously that a deletion of the amino-terminal extension of mtRNA polymerase (rpo41Δ3) did not affect transcription initiation from the mitochondrial ori5 promoter in vivo (15); however, we did not attempt to analyze other mitochondrial transcripts at that time. To address whether mutations in mtRNA polymerase amino-terminal domain exhibit a nam1-like phenotype, we analyzed mitochondrial transcripts in these strains by northern analysis (Fig. 4). Total mtRNA was analyzed from each strain after growth at 37 °C for five generations and compared with a NAM1 null strain grown at 30 °C, which exhibits the diagnostic COX1 and CYTB transcript defects. As observed in the NAM1 null strain, all of the mtRNA polymerase mutant strains exhibited a marked reduction in the steady-state levels of both CYTB and COX1 messages. In contrast, the steady-state level of COX3, a mitochondrial transcript that is largely unaffected by Nam1p function (14), was not dramatically altered under these conditions and served as an indicator that mitochondrial transcription per se was not globally affected by these mutations. This conclusion is supported by the additional observation that the overall mitochondrial transcript profile in all of the strains, as judged by ethidium bromide staining (data not shown), was virtually identical.
Fig. 4. Northern analysis of mitochondrial transcripts from amino-terminal domain mutant yeast strains.
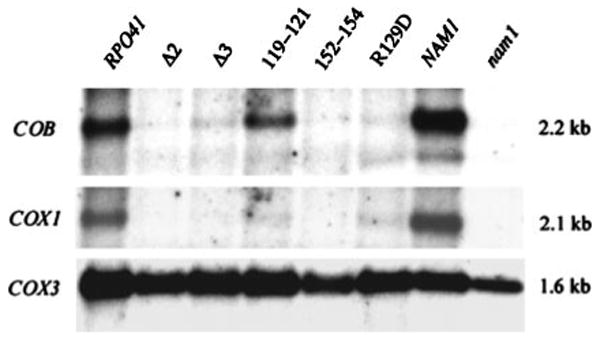
Five micrograms of mtRNA was analyzed from the yeast strains indicated at the top of each lane. RNA was isolated from the RPO41 mutant (Δ2, Δ3, E119A/C121A, N152A/Y154A, and R129D) and the corresponding isogenic wild-type strain (RPO41) after growing in SD medium for five generations at 37 °C. RNA was isolated from the NAM1 null mutant strain (nam1) and its corresponding isogenic wild-type strain (NAM1) after growing in SD medium at 30 °C. The resulting RNA blot was successively hybridized with COB, COX1, and COX3 exon probes (as indicated to the left of the figure). The size of each transcript detected is indicated in kb to the right of the figure.
Discussion
Amino-terminal extensions present in many mtRNA polymerases distinguish them from the related bacteriophage enzymes and provide the mitochondrial enzymes with localization information as well as additional function (6, 15). Previously, we demonstrated that the amino-terminal extension of sc-mtRNA polymerase harbors a functional domain involved in mtDNA stability, and we proposed that this domain could function by coupling additional activities to the transcription process (15). Because numerous RNA processing events are required for normal mitochondrial gene expression and mtDNA replication (1, 2), we hypothesized (15) that one process that may be coupled to transcription may be RNA processing. Here we report the characterization of a collection of RPO41 deletion and point mutations, the results of which demonstrate that Nam1p, a mitochondrial matrix protein implicated in translation and RNA processing events (11, 13, 14), interacts physically and functionally with an amino-terminal domain of sc-mtRNA polymerase. By using a two-hybrid protein interaction assay, we have demonstrated that Nam1p binds specifically to the amino-terminal extension of sc-mtRNA polymerase. The fact that the rpo41Δ2 mutation did not abolish Nam1p binding in this assay, whereas the rpo41Δ3 mutation did, suggests that the primary binding site being assayed here lies between the end points of these two deletions (amino acids 118–208). Consistent with this proposal, alignment of the amino-terminal extensions of S. cerevisiae, N. crassa, and S. pombe revealed that, although not highly conserved overall, the region exhibiting the highest degree of identity/similarity corresponds to amino acids 117–155 of the S. cerevisiae protein (Fig. 1).
The physiological significance the Nam1p/mtRNA polymerase interaction was demonstrated as follows. First, there is general correspondence between Nam1p binding and growth phenotypes of mtRNA polymerase mutants on YPG medium (growth on which requires mitochondrial respiration). Two mutations that eliminate Nam1p binding altogether, rpo41Δ3 and N152A/Y154A, exhibit a similar reduction of growth rate on YPG medium at 30 °C and a severe growth defect on YPG at 37 °C (Fig. 2A). Although less severe, mutations that retain some level of Nam1p binding, rpo41Δ2 and E119A/C121A, also exhibit significant YPG growth defects. In the rpo41Δ2 case, defective Nam1p binding appears to make a significant contribution to the phenotype because the YPG growth defect of this strain can be rescued by overproducing Nam1p (Fig. 3B). This is consistent with the fact that the end point of the rpo41Δ2 deletion impinges on, but does not eliminate, the most conserved residues identified in the fungal mtRNA polymerase alignment (Fig. 1). Thus, it is possible that this mutation reduces the affinity of the interaction but retains the major binding determinants for Nam1p. Also consistent with a defect in Nam1p binding is the fact that the rpo41Δ3 deletion completely removes the conserved domain and cannot be rescued by overexpression of Nam1p. Thus, suppression of YPG growth defects by Nam1p overexpression requires the presence of an intact amino-terminal domain, an observation most easily explained by a direct interaction between these two factors.
In addition to the growth data discussed above, our analysis of mtRNA species in RPO41 mutant strains also links amino-terminal domain function to Nam1p. All of our amino-terminal deletions and point mutations exhibit marked reductions in the steady-state levels of mature COX1 and CYTB mRNAs (Fig. 4). Although the precise function of Nam1p is unknown, it is clear that NAM1 null mutations affect a subset of mtRNA transcripts in yeast mitochondria. In particular, Nam1p appears to be involved in the processing and/or stability of the two intron-containing primary transcripts that contain COX1 and CYTB and, perhaps to a lesser degree, the 21 S rRNA (11, 14). However, Nam1p function is not limited to effects on intron removal because strains that contain an intronless mitochondrial genome still display a temperature-sensitive petite phenotype and instability of the mature ATP6 mRNA (14), which is co-transcribed with COX1. Finally, overall translation capacity is reduced in NAM1 null mutant strains (11), suggesting a dual role for Nam1p in RNA processing/stability and translation, or that these two processes are coupled in yeast mitochondria. Regardless of the precise activity of Nam1p in mtRNA metabolism, our data demonstrate that mutations that affect the amino-terminal domain of mtRNA polymerase lead to mtRNA transcript defects that are entirely consistent with disruption of Nam1p function. The simplest explanation for these data is that at least one function of Nam1p is carried out in association with mtRNA polymerase (i.e. Nam1p is coupled to transcription). The fact that COX3 transcripts are not grossly affected in our experiments is consistent with previously published data showing that mitochondrial transcription per se is not perturbed to large degree by loss of Nam1p function (11, 14) or by the rpo41Δ3 mutation (15).
This study also provides additional information regarding the function of the amino-terminal domain of mtRNA polymerase. First, none of the point mutations was rescued by overexpression of Nam1p, despite the ability of some of these to interact with Nam1p to some degree in the two-hybrid assay (Table I). Second, the R129D and N152A/Y154A mutations exhibited more severe YPG growth phenotypes than the rpo41Δ3 mutation (Fig. 2A), which is completely devoid of this region (Fig. 1). In fact, the R129D mutation was still capable of interacting with Nam1p, but exhibited the most severe mitochondrial phenotype (capable only of slow growth on YPG at 30 °C, Fig. 1). All of these data suggest that point mutations in the amino-terminal domain can be more deleterious than mutations that remove this domain altogether. Several explanations can potentially account for these observations. One possibility is that the amino-terminal domain is involved not only in Nam1p binding, but also in some other function. Although speculative at this point, it is tempting to consider that the amino-terminal domain of sc-mtRNA polymerase may be involved in binding the primary RNA transcript during transcription, perhaps to facilitate threading of the RNA into a coupled RNA-processing machinery during transcription. In this regard, it is noteworthy that the amino-terminal domain of T7 RNA polymerase, the closest relative to mtRNA polymerases, has been implicated in nascent RNA binding (25). If this is the case, then mutations in the amino-terminal domain could affect Nam1p binding, RNA binding, or both, resulting in the more severe phenotypes observed with particular point mutations. An alternative explanation is that a higher order complex is involved that contains not only mtRNA polymerase and Nam1p but also other factors involved in RNA processing/stability or translation. If this were the case, one could envision how point mutations result in the observed phenotypes by disrupting the binding of Nam1p, the binding of other factors, or both. Perhaps consistent with this idea is the observation that splicing of the group I intron (bI5) in the mitochondrial CYTB gene is facilitated by the Cbp2 protein in a transcription-dependent manner in vitro (26), suggesting that other factors involved in mtRNA processing events may indeed function in a transcription-coupled manner in vivo. Determination of the precise composition of the Nam1p-mtRNA polymerase complex and assignment of additional functions to the amino-terminal domain remain important goals.
Based on the apparent homology between the amino-terminal domain of sc-mtRNA polymerase and those of N. crassa and S. pombe (Fig. 1), we would predict that these organisms also contain a Nam1p homolog that is localized to the mitochondrial matrix. However, our searches of currently available data bases have yet to identify any obvious homologs of Nam1p in any organism. This brings into question whether the amino-terminal extensions found in mtRNA polymerases from other eukaryotes have similar functions to that proposed here for the S. cerevisiae protein. To begin to address this question, we have analyzed the sequences of the amino-terminal extensions of human (27) and Xenopus (GenBank™ accession number AF200705) mtRNA polymerases. As already mentioned, no obvious similarity exists between the vertebrate and the fungal enzymes in their amino-terminal extensions. However, like the fungal enzymes, the vertebrate proteins are similar to each other in this region (Fig. 5), exhibiting 34% amino acid identity and 53% similarity. Comparing these conserved amino-terminal regions of the vertebrate homologs to other known sequences in available data bases, we found a match of potential significance to a protein, CRP1, that is located in maize chloroplasts (28). Remarkably, the function of CRP1 in chloroplasts is similar to that documented for Nam1p in yeast mitochondria, that is, the processing and translation of specific mRNAs. The conserved amino-terminal region in both the human and Xenopus mtRNA polymerase is similar (20–25% identity, 40–45% similarity) to a block of amino acids that is repeated twice in CRP1 (Fig. 5). Similar data base searches reported by Fisk et al. (28), revealed similarity between CRP1 and a family of plant proteins of unknown function that are related to a salt-inducible protein in tobacco. Additionally, they found that the region held in common between CRP1 and this family of proteins was also related, although more distantly, to several other factors involved in post-transcriptional gene regulation in mitochondria including Pet309p, a translational activator; Rpm2p, a protein subunit of mitochondrial RNaseP (a tRNA processing factor); and threonyl-tRNA synthetase. Recently, the homologous regions in these proteins have been postulated to be composed of a tandemly repeated, 35-amino acid domain called a pentatricopeptide repeat (PPR) motif that is structurally similar to the well characterized tetratricopeptide repeat (TPR) motif (29). Based on comparisons with a proposed consensus sequence for a PPR motif (29), it appears that vertebrate mtRNA polymerases contain at least two PPR repeats in the amino-terminal extension (Fig. 5). Thus, the human and Xenopus mtRNA polymerases join this list of organelle regulatory proteins involved in RNA processing and translation that likely define a new PPR family of proteins. Although the function of these CRP1-like sequences in vertebrate mtRNA polymerases is unknown, it is interesting to speculate based on our results regarding the function of the S. cerevisiae amino-terminal domain. Perhaps, like yeast mtRNA polymerase, the CRP1-like domain in vertebrate mtRNA polymerases may be involved in coupling RNA processing or translation activities to transcription. Experiments are currently in progress to determine whether, in fact, the amino-terminal extension of human mtRNA polymerase has a role similar to that provided by the yeast enzyme and to determine whether the amino acid similarity to CRP1 (i.e. the PPR motifs) is of functional and evolutionary significance. Such experiments should lend new insights into the regulation of human mitochondrial genome expression and replication and its impact on human disease.
Fig. 5. The amino-terminal extensions of vertebrate mtRNA polymerases have a repetitive amino acid motif similar to that found in CRP1, a protein involved in RNA-processing and translation in chloroplasts.

The human mtRNA polymerase is diagrammed at the top in the same manner as the yeast enzyme in Fig. 1. A linear representation of the Zea mays CRP1 protein is also presented. The region of amino acid sequence similarity that is common to vertebrate (human and Xenopus) mtRNA polymerases and CRP1 is depicted as a hatched box. Shown at the bottom is a ClustalW (30) alignment of two regions in CRP1 (CRP-box1 and CRP-box2) with the analogous region of the human and Xenopus mtRNA polymerases. Black-boxed letters denote amino acid identity, and gray-boxed letters indicate amino acid similarity. Recent evidence suggests that these regions of CRP1 are composed of a tandemly repeated, 35-amino acid domain called a PPR motif that appears to define a new family of proteins involved in RNA processing and translation in organelles (29). Based on a proposed consensus sequence for a PPR motif (29), it appears that the vertebrate mtRNA polymerases contain at least two PPR repeats in the amino-terminal extension (indicated at the bottom of the figure).
Acknowledgments
We thank Yuan Wang for early contributions to this work, Melissa McKay for suggesting the PCR mutagenesis protocol, and Dr. Bonnie Seidel-Rogol for critical reading of the manuscript.
Footnotes
This work was supported by NHLBI Grant HL-59655 from the National Institutes of Health (to G. S. S.).
The abbreviations used are: kb, kilobase pair; PCR, polymerase chain reaction; aa, amino acids.
References
- 1.Shadel GS. Am J Hum Genet. 1999;65:1230–1237. doi: 10.1086/302630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shadel GS, Clayton DA. Annu Rev Biochem. 1997;66:409–435. doi: 10.1146/annurev.biochem.66.1.409. [DOI] [PubMed] [Google Scholar]
- 3.Jaehning JA. Mol Microbiol. 1993;8:1–4. doi: 10.1111/j.1365-2958.1993.tb01197.x. [DOI] [PubMed] [Google Scholar]
- 4.Shadel GS, Clayton DA. J Biol Chem. 1993;268:16083–16086. [PubMed] [Google Scholar]
- 5.Greenleaf AL, Kelly JL, Lehman IR. Proc Natl Acad Sci U S A. 1986;83:3391–3394. doi: 10.1073/pnas.83.10.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masters BS, Stohl LL, Clayton DA. Cell. 1987;51:89–99. doi: 10.1016/0092-8674(87)90013-4. [DOI] [PubMed] [Google Scholar]
- 7.Jang SH, Jaehning JA. J Biol Chem. 1991;266:22671–22677. [PubMed] [Google Scholar]
- 8.Shadel GS, Clayton DA. Mol Cell Biol. 1995;15:2101–2108. doi: 10.1128/mcb.15.4.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu B, Clayton DA. Nucleic Acids Res. 1992;20:1053–1059. doi: 10.1093/nar/20.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dieckman CL, Staples RR. Int Rev Cytol. 1994;152:145–181. doi: 10.1016/s0074-7696(08)62556-5. [DOI] [PubMed] [Google Scholar]
- 11.Asher EB, Groudinski O, Dujardin G, Altamura N, Kermorgant G, Slonimski PP. Mol Gen Genet. 1989;215:517–528. doi: 10.1007/BF00427051. [DOI] [PubMed] [Google Scholar]
- 12.Lisowsky T, Michaelis G. Mol Gen Genet. 1989;219:125–128. doi: 10.1007/BF00261167. [DOI] [PubMed] [Google Scholar]
- 13.Wallis MG, Groudinski O, Slonimski PP, Dujardin G. Eur J Biochem. 1994;222:27–32. doi: 10.1111/j.1432-1033.1994.tb18837.x. [DOI] [PubMed] [Google Scholar]
- 14.Groudinsky O, Bousquet I, Wallis MG, Slonimski PP, Dujardin G. Mol Gen Genet. 1993;240:419–427. doi: 10.1007/BF00280396. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Shadel GS. Proc Natl Acad Sci U S A. 1999;96:8046–8051. doi: 10.1073/pnas.96.14.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neugebauer KM, Roth MB. Genes Dev. 1997;11:3279–3285. doi: 10.1101/gad.11.24.3279. [DOI] [PubMed] [Google Scholar]
- 17.Steinmetz EJ. Cell. 1998;89:491–494. doi: 10.1016/s0092-8674(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 18.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai C, Elledge SJ. Methods Enzymol. 1997;283:141–156. doi: 10.1016/s0076-6879(97)83013-3. [DOI] [PubMed] [Google Scholar]
- 20.Sherman F. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 21.Sikorski RS, Boeke JD. Methods Enzymol. 1991;194:302–318. doi: 10.1016/0076-6879(91)94023-6. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman CS, Winston F. Gene (Amst) 1989;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- 23.Schmitt ME, Brown TA, Trumpower BL. Nucleic Acids Res. 1990;18:3091–3092. doi: 10.1093/nar/18.10.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd. Cold Springs Harbor Laboratory; Cold Springs Harbor, NY: 1989. pp. 7.37–7.51. [Google Scholar]
- 25.Sastry S, Ross BM. Proc Natl Acad Sci U S A. 1998;95:9111–9116. doi: 10.1073/pnas.95.16.9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewin AS, Thomas J, Jr, Tirupati HK. Mol Cell Biol. 1995;15:6971–6978. doi: 10.1128/mcb.15.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tiranti V, Savoia A, Forti F, D'Apolito MF, Centra M, Rocchi M, Zeviani M. Hum Mol Genet. 1997;6:615–625. doi: 10.1093/hmg/6.4.615. [DOI] [PubMed] [Google Scholar]
- 28.Fisk GF, Walker MB, Barkan A. EMBO J. 1999;18:2621–2630. doi: 10.1093/emboj/18.9.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Small ID, Peeters N. Trends Biochem Sci. 2000;25:46–47. doi: 10.1016/s0968-0004(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DJ, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]