

Many recognition events in biological systems involve the interactions between extracellular carbohydrate ligands and protein receptors, with multivalency playing an important role in controlling both specificity and avidity.1-3 A wide variety of synthetically derived glycopolymers have been used to mimic natural glycoprotein and glycolipid assemblies and to mediate these multivalent binding interactions in an effort to better understand the recognition process at a molecular level.4-14 Methods such as traditional free-radical polymerization of glycosylated monomers and/or chemical attachment of simple mono- and oligosaccharides to previously synthesized polymers and polypeptides have been widely used for the production of various glycopolymers. In addition, ring-opening metathesis polymerization and dendrimer synthesis have been used to produce multivalent linear and branched glycopolymers with variation of molecular weights, valencies, and epitope density.
While these previous investigations have provided important information about the general architectural rules that govern the specificity and avidity of the multivalent interaction, the ability to more quantitatively understand the specific details of these interactions has been impeded by the inherent heterogeneity of the linear glycopolymeric materials produced via chemical methods. Therefore, the production of homogeneous and chemically defined glycopolymers would provide additional tools for probing the mechanism of glycopolymer-receptor interactions in more architectural detail. Additionally, the development of routes for the production of such well-defined glycopolymers would also provide important opportunities for purposefully tailoring polymeric materials for mediation of biological processes, such as pathogenesis, inflammation, and the immune response.
In this communication, we integrate chemical tools with biological technologies to synthesize a new type of monodisperse glycopolymer with controlled saccharide placement. Protein engineering methods15 have been employed to produce a series of alanine-rich, helical protein-based polymers with controlled conformational properties and varied placement of glutamic acid residues.16,17 Via the use of biosynthetic strategies, placement of glutamic acids residues in the polymer chain can be specified for desired applications, and the carboxylic acid side chain can be modified with a variety of ligands and organic groups.
In the specific example reported here, protein polymers with the sequence (AAAQAAQAQAAAEAAAQAAQAQ)6 (B6) were expressed from the bacterial expression strain BL21(DE3)pLysS/ pET19b-RF1-B6 via standard methods employing chemical induction with IPTG (isopropyl β-D-thiogalactopyranoside). These sequences have been previously shown to be highly helical,17 with controlled conformational behavior and, therefore, offer polymeric backbones that may be used not only for controlled presentation of saccharides but also to probe the role of backbone conformation in binding events. The target protein polymers carry an N-terminal fusion tag, MGH10SSGHINM-, to permit facile purification from cell lysate via Ni-NTA (nickel nitrilotriacetic acid) affinity chromatography under denaturing conditions. After purification, a monodisperse protein polymer with the desired molecular weight and amino acid composition was obtained (Supporting Information).
Because galactose binds to a variety of lectins and toxins, β-D-galactosylamine was initially employed to modify B6 to form the protein-based glycopolymer, G6. The reaction proceeded via activation of the pendant and C-terminal carboxylic acid groups of B6 with HBTU [N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl) uranium hexafluorophosphate], with subsequent coupling with the amine-functionalized saccharide (Scheme 1).
Scheme 1.

Chemical Modification of Helical Protein Polymers (a single site modification of a helical domain is shown for simplicity)
The coupling reaction was carried out under basic conditions in DMSO with excess β-D-galactosylamine (Supporting Information), and the glycosylated protein polymers were purified via a combination of precipitation and HPLC. The complete conversion of B6 to G6 could be easily monitored via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
The results for the SDS-PAGE analysis are shown in Figure 1. As illustrated in the figure, both unglycosylated B6 and fully glycosylated G6 migrate at a single molecular weight. G6 migrates near the expected molecular weight (15.2 kDa), while B6 electrophoreses at an anomalously high molecular weight, as previously observed for these polyanionic protein polymers.16,17 The partially glycosylated protein polymer electrophoreses over a range of molecular weights that vary between those of the unglycosylated protein to fully glycosylated protein, which was also confirmed via MALDI-TOF MS analysis (Supporting Information).
Figure 1.
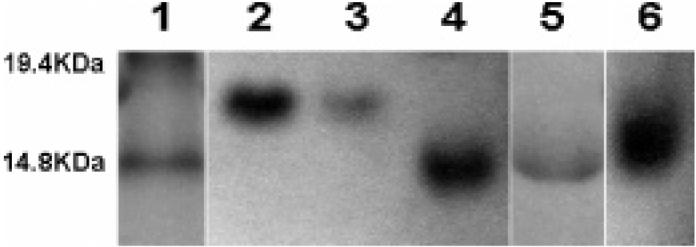
SDS-PAGE analysis of protein polymers. Lane 1: molecular weight marker. Lane 2: original protein. Lane 3: isolated protein from a control reaction without saccharide. Lanes 4 and 5: fully glycosylated protein (high and low concentrations, respectively). Lane 6: partially glycosylated protein.
The chemical composition of G6 was analyzed via amino acid analysis, NMR, and periodate assay (Supporting Information). Amino acid analysis shows the expected composition of the protein polymers. In corroboration of SDS-PAGE analysis, the complete glycosylation of G6 was indicated via 1H NMR spectroscopy, via comparison of the integrated area of the protons from galactose between δ 4.0-2.8 ppm with that of the α protons from the amino acids, near δ 4.3 ppm.18 The complete glycosylation of G6 was also indicated via periodate assay.19
Circular dichroic (CD) characterization of G6 and B6 confirm that glycosylation does not alter the conformational behavior of the protein polymer; the CD spectra of the two protein polymers are essentially identical and suggest a nonaggregating, α-helical structure of high fractional helicity (approximately 71%) under the specified experimental conditions (Figure 2A).16,17,20 The mean residue ellipticity values reported here for B6 are slightly lower than those previously reported,17 likely owing to the Tween 20, which was included in these PBS buffers for subsequent ELISA assays. As shown in Figure 2B, the conformational behavior of G6 is also thermally reversible, converting from α-helix to random coil at elevated temperatures; the helical structure is stable at 37 °C for at least 12 h (Supporting Information), suggesting the stability and utility of these macromolecules under physiologically relevant conditions.
Figure 2.

CD spectra of protein polymers (0.08 mg/mL) in pH 7.2 PBS buffer with 0.05% Tween 20. (A) Comparison of CD spectra of B6 and G6 at 7 °C. (B) CD spectra of G6 at different temperatures.
The nominal distance between glutamic acids on this protein polymer16,17 was chosen to be commensurate with that of the galactose-binding sites of the cholera toxin (CT) (~35 Å),21 a pathologically active agent secreted by the bacterium Vibrio cholerae. The inhibition of CT binding by G6 was evaluated via competitive ELISA (enzyme-linked immunosorbent assay) via previously reported methods.22 CT inhibition curves are shown in Figure 3 for G6 and the monovalent galactose. IC50 values (the concentration of inhibitor, on a saccharide basis, that inhibits 50% of CT binding) were calculated via nonlinear regression methods.23 IC50 values of ~350 μM for G6 and ~68 mM for galactose were determined, indicating a 200-fold improvement in inhibition upon multivalent presentation of the galactose on the protein polymer. Control samples preincubated with B6 were also measured and showed only a small level of inhibition, indicating that the protein polymer is not the primary cause of the improvements in inhibition. The improvement in IC50 values afforded by G6 is similar to those previously observed for the inhibition of other binding events by various linear glycopolymers,24-27 despite the short length of the linker arm that may reduce the binding of the G6 polymer-linked saccharides to the CT receptor.22,26 The production of an expanded series of well-defined glycopolymeric structures, in which the saccharide linker arm, saccharide density, and protein polymer backbone have been modified to manipulate avidity and perhaps tailor bioactivity,8,27,28 is currently under investigation.
Figure 3.
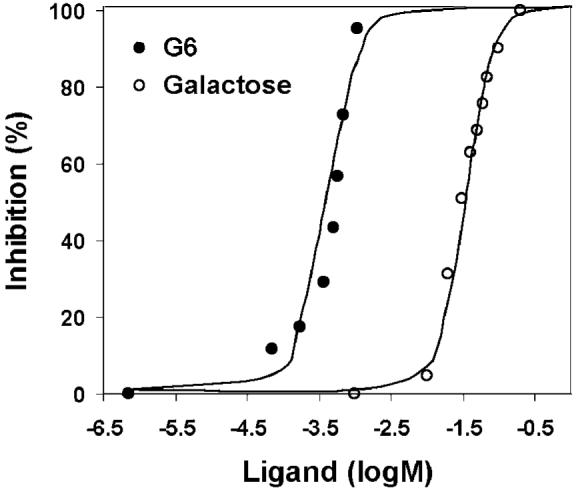
CT inhibition as a function of inhibitor (ligand) concentration, as determined via competitive ELISA. Ligand concentration is reported as the concentration of saccharide.
In conclusion, a combination of biological and chemical methods has been employed for the production of a homogeneous protein-based glycopolymer with saccharide residues precisely placed along the polymer chain. The glycopolymer exhibits controlled conformational behavior and efficient binding to CT. The employed chemical modification strategies are widely applicable to a variety of amine-functionalized saccharides and carboxylic acid-decorated protein polymers and, therefore, provide a route to a range of monodisperse glycosylated polymers with controlled sequences and conformations. The reported methods offer unprecedented opportunities to explore and control the roles of saccharide number, saccharide spacing, and polymer conformation on multivalent binding events mediated by linear polymeric materials.
Acknowledgment
This work has been supported in part by the Camille and Henry Dreyfus Foundation, the National Institutes of Health (1-P20-RR17716-01), the University of Delaware Research Foundation, and the Army Research Office (DAAD 119-02-1-0173). Robin Farmer and Brian Polizzotti are thanked for expression plasmid preparation and helpful discussions.
Footnotes
Supporting Information Available: Materials and methods for protein polymer and glycopolymer synthesis. Amino acid analysis, MALDI-MS, NMR spectra, periodate assays, and circular dichroic spectra. Detailed methods for ELISA. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Lee RT, Lee YC. Glycoconjugate J. 2000;17:543–551. doi: 10.1023/a:1011070425430. [DOI] [PubMed] [Google Scholar]
- (2).Mammen M, Chio SK, Whitesides GM. Angew. Chem., Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- (3).Bertozzi CR, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- (4).Kiessling LL, Strong LE, Gestwicki JE. Annu. Rep. Med. Chem. 2000;35:321–330. [Google Scholar]
- (5).Mammen M, Dahmann G, Whitesides GM. J. Med. Chem. 1995;38:4179–4190. doi: 10.1021/jm00021a007. [DOI] [PubMed] [Google Scholar]
- (6).Glunz PW, Hintermann S, Williams LJ, Schwarz JB, Kuduk SD, Kudryashov V, Lloyd KO, Danishefsky SJ. J. Am. Chem. Soc. 2000;122:7273–7279. [Google Scholar]
- (7).Ambrosi M, Batsanov AS, Cameron NR, Davis BG, Howard JAK, Hunter R. J. Chem. Soc., Perkin Trans. 1. 2002;1:45–52. [Google Scholar]
- (8).Wolfenden ML, Cloninger MJ. J. Am. Chem. Soc. 2005;127:12168–12169. doi: 10.1021/ja053008n. [DOI] [PubMed] [Google Scholar]
- (9).Gomez-Garcia M, Benito JM, Rodriguez-Lucena D, Yu J-X, Chmurski K, Ortiz MC, Gutierrez GR, Maestre A, Defaye J, Garcia-Fernandez JM. J. Am. Chem. Soc. 2005;127:7970–7971. doi: 10.1021/ja050934t. [DOI] [PubMed] [Google Scholar]
- (10).Reuter JD, Myc A, Hayes MM, Gan Z, Roy R, Yin R, Piehler LT, Esfand R, Tomalia DA, Baker JR., Jr. Bioconjugate Chem. 1999;10:271–178. doi: 10.1021/bc980099n. [DOI] [PubMed] [Google Scholar]
- (11).Dam TK, Oscarson S, Roy R, Das SK, Page D, Macaluso F, Brewer CF. J. Biol. Chem. 2005;280:8640–8646. doi: 10.1074/jbc.M412827200. [DOI] [PubMed] [Google Scholar]
- (12).Baird EJ, Holowka D, Coates GW, Baird B. Biochemistry. 2003;42:12739–12748. doi: 10.1021/bi034884l. [DOI] [PubMed] [Google Scholar]
- (13).Gordon EJ, Sanders WJ, Kiessling LL. Nature. 1998;392:30–31. doi: 10.1038/32073. [DOI] [PubMed] [Google Scholar]
- (14).Roy R. Trends Glycosci. Glycotechnol. 2003;15:291–310. [Google Scholar]
- (15).Kiick KL. In: The Encyclopedia of Polymer Science and Technology. 3rd ed. Kroschwitz JI, editor. John Wiley and Sons; New York: 2004. pp. 145–197. [Google Scholar]
- (16).Farmer RS, Kiick KL. Biomacromolecules. 2005;6:1531–1539. doi: 10.1021/bm049216+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Farmer RS, Argust LM, Sharp JD, Kiick KL. Submitted.
- (18).Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; New York: 1986. p. 17. [Google Scholar]
- (19).Pierce Glycoprotein Carbohydrate Estimation Kit. 2004 [Google Scholar]
- (20).Miller JS, Kennedy RJ, Kemp DS. J. Am. Chem. Soc. 2002;124:945–962. doi: 10.1021/ja011726d. [DOI] [PubMed] [Google Scholar]
- (21).Pickens JC, Mitchell DD, Liu J, Tan X, Zhang Z, Verlinde CLMJ, Hol WGJ, Fan E. Chem. Biol. 2004;11:1205–1215. doi: 10.1016/j.chembiol.2004.06.008. [DOI] [PubMed] [Google Scholar]
- (22).Minke WE, Roach C, Hol WGJ, Verlinde CLMJ. Biochemistry. 1999;38:5684–5692. doi: 10.1021/bi982649a. [DOI] [PubMed] [Google Scholar]
- (23).Bowen WP, Jerman JC. TIPS. 1995;16:413–417. doi: 10.1016/s0165-6147(00)89091-4. [DOI] [PubMed] [Google Scholar]
- (24).Ohta T, Miura N, Fujitani N, Nakajima F, Niikura K, Sadamoto R, Guo C-T, Suzuki T, Suzuki Y, Monde K, Nishimura S-I. Angew. Chem., Int. Ed. 2003;42:5186–5189. doi: 10.1002/anie.200351640. [DOI] [PubMed] [Google Scholar]
- (25).Rendle PM, Seger A, Rodrigues J, Oldham NJ, Bott RB, Jones JB, Cowan MM, Davis BG. J. Am. Chem. Soc. 2004;126:4750–4751. doi: 10.1021/ja031698u. [DOI] [PubMed] [Google Scholar]
- (26).Vrasidas I, de Mol NJ, Liskamp RMJ, Pieters RJ. Eur. J. Chem. 2001:4685–4692. [Google Scholar]
- (27).Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. J. Am. Chem. Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- (28).Cairo CW, Gestwicki JE, Kanai M, Kiessling LL. J. Am. Chem. Soc. 2002;124:1615–1619. doi: 10.1021/ja016727k. [DOI] [PubMed] [Google Scholar]