

Abstract
The amino-terminal domain (ATD) of Saccharomyces cerevisiae mitochondrial RNA polymerase has been shown to provide a functional link between transcription and post-transcriptional events during mitochondrial gene expression. This connection is mediated in large part by its interactions with the matrix protein Nam1p and, based on genetic phenotypes, the mitochondrial membrane protein Sls1p. These observations led us to propose previously that mtRNA polymerase, Nam1p, and Sls1p work together to coordinate transcription and translation of mtDNA-encoded gene products. Here we demonstrate by specific labeling of mitochondrial gene products in vivo that Nam1p and Sls1p indeed work together in a pathway that is required globally for efficient mitochondrial translation. Likewise, mutations in the ATD result in similar global reductions in mitochondrial translation efficiency and sensitivity to the mitochondrial translation inhibitor erythromycin. These data, coupled with the observation that the ATD is required to co-purify Sls1p in association with mtDNA nucleoids, suggest that efficient expression of mtDNA-encoded genes in yeast involves a complex series of interactions that localize active transcription complexes to the inner membrane in order to coordinate translation with transcription.
The ∼80-kb Saccharomyces cerevisiae mitochondrial genome encodes seven oxidative phosphorylation subunits destined for the inner mitochondrial membrane, one ribosomal protein, two rRNAs, and a full complement of tRNAs (1). Transcription of yeast mtDNA is initiated at multiple promoters by a dedicated mtRNA polymerase that is encoded by the RPO41 gene (2) and homologous to the bacteriophage T7 family of RNA polymerases (3). Most mitochondrial transcripts are polycistronic and require extensive processing to release the mature RNA species for proper gene expression (4). In addition, introns are often present in certain messages that also must be excised. In the case of the COX1 and COB genes, this is a complex process that requires intron-encoded maturases that help catalyze splicing (5, 6). Once fully processed, mRNAs are translated within the matrix by ribosomes associated with the mitochondrial inner membrane (7, 8). Finally, the newly synthesized mtDNA-encoded proteins are assembled with the corresponding nucleus-encoded subunits to form the oxidative phosphorylation enzyme complexes in the inner membrane. The execution and coordination of these events are necessary for normal cellular respiration.
We previously examined the mechanism of mitochondrial gene expression in the budding yeast S. cerevisiae and elucidated a pathway of transcription-coupled events that involves an amino-terminal domain (ATD)1 of mtRNA polymerase (9–11). Over the course of these experiments, several mutations in the yeast mtRNA polymerase ATD have been characterized, including two deletion mutations, rpo41Δ2 and rpo41Δ3, and several single or double point mutations, which result in mitochondrial petite phenotypes and defects in mitochondrial RNA processing and/or mtDNA stability (10, 11). Specifically, the steady-state levels of mature mRNA from the intron-containing COB and COX1 genes are severely decreased in these strains, whereas those of an intronless gene, COX3, are minimally affected (10). These intron-processing defects suggest that the ATD affects translational efficiency (9) because studies using mitochondria-specific translation inhibitors cause the same defects in vivo (12). In addition, the minimal defects in COX3 transcript levels in the rpo41 ATD mutants, along with S1 nuclease protection assays of the ori5 promoter in an rpo41Δ3 strain (11), suggest that the catalytic activity and initiation properties of mtRNA polymerase are not drastically altered.
Analogous in some ways to the carboxyl-terminal domain of RNA polymerase II (13), the ATD of mtRNA polymerase is the binding site for at least one factor involved in post-transcriptional events. Specifically, it is the interaction point for Nam1p (10), a protein involved in RNA processing and translation (14–16). Two additional lines of evidence support a functional role for this interaction in coordinating mitochondrial translation. First, nam1Δ mutations result in similar, yet more severe, COX1 and CYTB intron-processing defects as ATD mutations (10). Second, overexpression of the mitochondrial membrane protein Sls1p suppresses the respiration-deficient petite phenotype of both nam1Δ and mtRNA polymerase ATD mutations, indicating that mtRNA polymerase, Nam1p, and Sls1p exist in an ordered genetic pathway required for normal mitochondrial gene expression (9). Based on these observations and on the proposed function of Sls1p in post-transcription events (17), we hypothesized that these three factors comprise important components of a mitochondrial RNA-handling pathway that targets newly synthesized transcripts to the inner membrane (9), where translation of mtDNA-encoded messages occurs (7, 8). Entirely consistent with this model is the recent identification of interactions between Nam1p and the COX-specific translational activator complex (18), implicating Nam1p as a potential envoy between mtRNA polymerase and the translation machinery. In this study we have demonstrated that Sls1p functions globally in mitochondrial translation, and we provide multiple new lines of evidence that the ATD of mtRNA polymerase is a focal point for critical interactions with Nam1p and Sls1p that act in concert to coordinate transcription and translation at the mitochondrial inner membrane.
Experimental Procedures
Media
Yeast were grown as described (19) in complete YPG (glycerol) medium, YPD (dextrose) medium, or synthetic dextrose (SD) medium with the necessary nutritional supplements as indicated. The bacto-yeast extract, dextrose, and yeast nitrogenous base (without amino acids) were obtained from Difco. Glycerol and peptone were obtained from Fisher.
Yeast Strains and Plasmid Construction
The yeast strains used in this study were derived from DBY2006 (α his3-Δ200 leu2-3, -112 ura3-52 trp1-Δ1 ade2). With the exception of MSR107 (GS122 + [YEp352-V5SLS1]) and MSR112 (GS125 + [YEp352-V5SLS1]), all yeast strains have been described previously (9, 10). The YEp352-V5SLS1 vector was created as follows. The SLS1 gene was PCR-amplified using a 5′-primer that annealed to a sequence 200 bp upstream of the SLS1 translation start site and a 3′-primer complementary to the 3′-end of the SLS1 open reading frame, which also contained the V5 epitope and a SacI site at its 5′-end. The PCR product was then ligated into the pGEM-T vector (Promega), from which the V5 epitope-SLS1 fusion allele was excised on a PstI-SacI fragment and ligated into YEp352 that was digested with the same restriction enzymes.
Radiolabeling of Mitochondrial Translation Products in Vivo
An overnight culture (2 ml) of each yeast strain was grown to saturation in YPG (or YPD for petite mutants) medium and used to inoculate a 15-ml SD culture supplemented with the appropriate amino acids. These cultures were then incubated at 30 °C for 2 h prior to overnight incubation at 37 °C. These cultures were then diluted to an A600 of 0.6 with SD medium. After incubation at 37 °C for 2.5 h, cycloheximide (Spectrum Laboratory Products, Inc.) was added to 250 μg/ml in 10 ml of each culture, and the incubation was continued for 5 min prior to the 15-min incubation with 100 μCi of [35S]methionine (PerkinElmer Life Sciences). Labeling was stopped by addition of 4 ml of 1 mM Na2SO4 and 1% casamino acids, followed by an additional 10-min incubation. The cells were harvested by centrifugation, and mitochondria were prepared as described (20). The procedure was identical for labeling done at 30 °C except that cycloheximide was added to a concentration of 150 μg/ml. Mitochondrial protein concentration was determined using the Bio-Rad Protein Assay kit. Mitochondrial protein (15 μg) was separated on a 17% SDS-PAGE gel. The gel was then treated with Enlightening (PerkinElmer Life Sciences), dried, and exposed to x-ray film for 40–72 h.
Erythromycin Sensitivity Assays
Yeast strains were grown at 30 °C to mid-log phase in 5 ml of liquid YPG media. Cultures were then diluted to an A600 of 0.4 and spotted onto solid YPG medium containing the either 0, 5, 10, 20, or 50 μg/ml of erythromycin (Sigma), using a 48-pin multiplex plating tool. The plates were then incubated at 36 or 30 °C for 6 days prior to the assessment of growth phenotypes. Complete lack of growth was scored as sensitivity to the drug.
Petite Induction Assays and mtDNA Southern Analysis
Yeast strains were set up and grown overnight at 37 °C and diluted to an A600 of 0.6 with SD medium as they were in the radiolabeling experiments. The cultures were then incubated at 37 °C for 3 h. These cultures were then diluted and plated for the petite induction assay, as described previously (21). Statistical analysis was applied to identify outlying data points (22). By using this criterion, one data point for rpo41Δ2 was omitted. Total cellular DNA was isolated from cultures grown in the same manner for Southern blot analysis. The total DNA was digested with NdeI and separated on a 0.8% agarose gel for Southern analysis. The Southern blots were hybridized sequentially with radiolabeled probes to mitochondrial encoded COX1 and COX3 and nuclear encoded ACT1 as a normalization control.
Western Immunoblot Analysis
Yeast cultures were grown and mitochondria prepared as they were for the in vivo translation assay described above, except that the cultures were not incubated with cycloheximide or [35S]methionine. Mitochondrial protein was separated by SDS-PAGE and transferred to nitrocellulose membranes by standard protocols (23), and Western analysis was performed as described in the ECL Western blotting detection kit protocol (Amersham Bio-sciences), using anti-Cox2p, anti-Cox1p, and anti-porin antibodies (Molecular Probes).
Translation in Organello and Isolation of Mitochondrial Nucleoids
Mitochondria competent for in organello translation were prepared and stored as described (24). Mitochondria (2 mg of mitochondrial protein/sample) were thawed, and in organello translation reactions were performed as described (24), except 20 mm HEPES was substituted for Tris in the protein synthesis medium. ATP, GTP, α-ketoglutarate, phosphoenolpyruvate, and pyruvate kinase (all obtained from Sigma) comprise the energy-regenerating system in the protein synthesis medium. Mitochondria were resuspended in 1 ml of ice-cold protein synthesis medium, and pyruvate kinase was added, and the sample was incubated at room temperature for 10 min prior to the addition of 400 μM of the cross-linking reagent dithiobis(succinimidyl propionate) (Pierce). After incubation for 20 min, Tris-Cl (pH 7.4) was added to a concentration of 20 mm, and mitochondria were pelleted by centrifugation (12,000 × g, 10 min) and resuspended in ice-cold NE2.5 (0.38 m sucrose, 20 mm Tris-HCl (pH 7.6), 2 mm EDTA, 0.8 mm spermidine) on ice. Mitochondria were then lysed by the addition of 12.5% Nonidet P-40 to a final concentration of 0.5%. After incubation on ice for 10 min, DNase I was added where indicated and then all samples were centrifuged (16,000 × g, 10 min). The supernatant was transferred to another tube and incubated on ice for 25 min. The supernatants were then layered on top of a 20/40/60% sucrose step gradient and centrifuged in an SW 41 rotor (111,000 × g for 75 min). The 40/60% interface was collected, diluted to 1 ml in NE2.5 buffer, and pelleted through 40% sucrose at 136,000 × g for 1 h in a TLA 100.3 rotor. The pellets were resuspended in NE2.5 buffer, diluted in 2× Laemmli buffer (23) with 10% β-mercaptoethanol, vortexed, and boiled for 5 min. Samples (10 μl) of each nucleoid fraction and of each lysate (supernatant) fraction (2 μl) were separated by 10% SDS-PAGE for silver staining. In parallel, a sample of each nucleoid fraction (40 μl) and each lysate sample (20 μl) were separated on an 8% SDS-PAGE gel and transferred to a membrane for Western analysis using an anti-V5 antibody (Invitrogen).
Results
Sls1p and Nam1p Collaborate in a Pathway Required for Optimal Mitochondrial Translation
We identified previously (9) SLS1 as a genetic suppressor of the mitochondrial petite phenotypes of mtRNA polymerase ATD and NAM1 null (nam1Δ) mutations. Because nam1Δ and ATD mutant strains all demonstrate a defect in intron processing of the COX1 and COB genes (10, 15) and Sls1p has been implicated to act in the same pathway of transcription-coupled events as Nam1p and mtRNA polymerase (9), we investigated whether an sls1Δ strain has a similar mitochondrial intron-processing defect as nam1Δ and rpo41 ATD mutant strains. Northern analysis of total mitochondrial RNA revealed that an sls1Δ strain expressed nearly wild-type levels of COX3 mRNA, a gene that does not contain introns, yet does not accumulate the mature mRNA from the intron-containing COX1 and COB genes (Fig. 1A). Therefore, the sls1Δ strain has the same intron-processing defects as nam1Δ and ATD mutant strains (10). Because mitochondrial intron-processing defects correlate with translation perturbation, these data suggested that Sls1p impacts mitochondrial translation efficiency. To test the hypothesis directly, mtDNA-encoded proteins in the sls1Δ strain were specifically labeled in vivo using [35S]methionine in the presence of the cytoplasmic translation inhibitor cycloheximide. Accordingly, the sls1Δ strain was severely deficient in labeling of all mtDNA-encoded products (Fig. 1B), indicating that Sls1p is, in fact, required globally for efficient translation in yeast mitochondria.
Fig. 1. Analysis of mitochondrial translation in nam1 and sls1 null mutant strains.
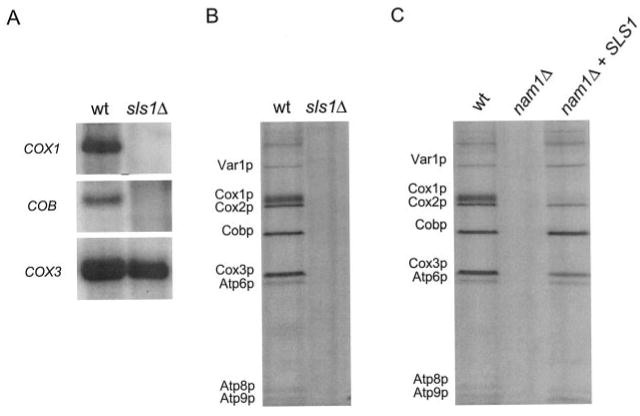
A, Northern analysis of mitochondrial transcripts. Mitochondrial RNA was extracted and prepared for Northern analysis as described (10). The strains analyzed have been described previously (9) and are indicated above the figure: CMW2 (wt) and CMW1 (sls1Δ). Signals for the mature mitochondrial RNA transcripts COB, COX1, and COX3 are indicated on the left. B, specific labeling of mitochondrial translation products in vivo. Shown is an autoradiogram of mtDNA-encoded proteins labeled with [35S]methionine at 30 °C in the presence of cycloheximide as described under “Experimental Procedures.” The strains analyzed are the same as those described in A. C, overexpression of Sls1p restores in vivo labeling of mitochondrial proteins in an nam1Δ strain. Shown is an autoradiogram of mtDNA-encoded translation products labeled in cultures at 30 °C. The strains analyzed have been described previously (9) and are indicated at the top of each lane: GS140 (wt), GS141 (nam1Δ), and ACB1 (nam1Δ + pRS316-SLS1). The identity of each labeled mtDNA-encoded protein is indicated on the left of the figure in both B and C.
Although sls1Δ strains exhibit a virtual absence of mitochondrial translation (Fig. 1B), nam1Δ mutants have been shown by others (14) to exhibit a moderate global decrease in mitochondrial translation and an apparent lack of labeling of Cox1p. Because we have previously placed Nam1p and Sls1p in the same genetic pathway (9), we analyzed the effect of deleting the NAM1 gene on mitochondrial translation in our strain background. Similar to the results obtained with the sls1Δ (Fig. 1B), in vivo labeling of mtDNA-encoded proteins revealed an extremely low if not complete absence of translation of all mtDNA-encoded proteins in this nam1Δ strain (Fig. 1C). We also demonstrated previously that overexpression of Sls1p suppresses the petite phenotype of the nam1Δ mutation in this genetic background (9). Therefore, we also examined whether Sls1p can suppress the observed translation defect in the nam1Δ strain when overexpressed. Labeling of translation products demonstrated that overexpression of Sls1p restored mitochondrial translation in our nam1Δ strain to close to wild-type levels at 30 °C (Fig. 1C), with the exception of Cox1p, which was only moderately increased. Similar results were obtained when in vivo labeling was performed at 37 °C (data not shown). The similar translation phenotypes of the nam1Δ and sls1Δ strains as well as the ability of increased levels of Sls1p to greatly restore translation in the nam1Δ strain provide strong evidence that these proteins act in the same pathway to determine the efficiency of mitochondrial translation.
Mutations in the ATD of mtRNA Polymerase Cause a Global Decrease in Mitochondrial Translation
To test the hypothesis that, like sls1Δ and nam1Δ strains, the documented intron-processing defects observed in rpo41 ATD mutant strains (10) result from impaired mitochondrial translation, labeling experiments were performed in these strains. All ATD mutations tested resulted in decreased incorporation of radiolabel into mtDNA-encoded proteins, indicative of a global decrease in translation efficiency (Fig. 2A). The rpo41-N152A/Y154A mutation resulted in the most severe global translation defect (Fig. 2A), requiring extended exposures to reveal the extremely low amount of labeling (data not shown). Additional experiments revealed that labeling of translation products did not diminish when the chase time was increased to 1 h in any of the rpo41 ATD mutants, demonstrating that the defect is not a result of a decrease in stability of the translation products during the labeling period (data not shown).
Fig. 2. Mitochondrial RNA polymerase ATD mutations result in global mitochondrial translation defects.

A, specific labeling of mitochondrial translation products in vivo. Shown is an autoradiogram of mtDNA-encoded proteins labeled with [35S]methionine in the presence of cycloheximide at 37 °C as described under “Experimental Procedures.” The ATD mutation in the RPO41 gene (encoding mtRNA polymerase) harbored by each strain tested is indicated above the figure as follows: RPO41 (wt); rpo41Δ2, deletion of amino acids 27–117 (Δ2); rpo41Δ3, deletion of amino acids 27–212 (Δ3); rpo41-E119A-C121A (119–121); and rpo41-N152A-Y154A (152-154). The identity of each labeled protein is indicated to the left of the figure. B, Southern analysis of mtDNA isolated from wild-type (wt) and rpo41-N152A-Y154A (152–154) mutant strains after growth under the conditions used for in vivo labeling in A. Radiolabeled probes were hybridized to mtDNA (COX1 and COX3 genes) as well as nuclear DNA (ACT1 gene) as indicated to the left of the figure.
The [35S]-labeling assays require that the cultures be grown in synthetic glucose medium, a condition that allows growth of cells that have lost wild-type (rho+) mtDNA. Because mtRNA polymerase is required for rho+ mtDNA maintenance (1, 2), in principle, the translation defects observed in the in vivo labeling assays could result from a decrease in the number of rho+ genomes. To assess the degree of mtDNA instability in the ATD mutant strains under these conditions, cultures were grown in the same manner as the [35S] incorporation assays and quantitated for the amount of spontaneous petite formation (Table I), which is largely a measure of rho− genome production (1). The deletion mutants, rpo41Δ2 and rpo41Δ3, produce spontaneous petite mutants at a rate similar to the wild-type strain, indicating that their mtDNA is as stable as in the wild-type strain under these conditions. The rpo41-E119A/C121A mutation produced more spontaneous petite mutants than either of the deletion mutants (Table I), yet consistently incorporated more radiolabel in the mitochondrial translation assay (Fig. 2A), indicating that the translation defect observed in the deletion mutants is likely not the result of increased rho− genome production. The strain that exhibited the most severe translation defect, rpo41-N152A/Y154A (Fig. 2A), was also the strain that exhibited the largest amount of spontaneous petite formation (Table I). However, the ∼30% instability observed presumably cannot account for the very small percentage of total translation activity that was observed in the in vivo labeling experiments (Fig. 2A). Furthermore, Southern analysis of mtDNA from the rpo41-N152A/Y154A strain using two probes that hybridize to mtDNA sequences physically separated on the genome (COX1 and COX3) also revealed that mtDNA is largely intact in this strain under these conditions (Fig. 2B). Altogether, these data indicate that the translation defects measured in all of the rpo41 ATD mutant strains using the in vivo labeling approach, with the possible exception of the rpo41-E119A/C121A strain (see “Discussion”), are not the result of mtDNA instability at the time of the assay.
Table I. Petite induction in ATD mutant strains.
| ATD mutant | Percent respiratory competent |
|---|---|
| Wild-type | 88.9 ± 10.6 |
| rpo41Δ2 | 99.1 ± 2.6 |
| rpo41Δ3 | 95.2 ± 5.0 |
| rpo41-E119A/C121A | 80.1 ± 6.3 |
| rpo41-N152A/Y154A | 71.3 ± 8.2 |
To confirm that the observed translation defect results in decreased steady-state levels of mtDNA-encoded proteins in the rpo41 ATD mutant strains, Western blot analysis was performed on mtDNA-encoded Cox1p and Cox2p. Analysis of total mitochondrial protein revealed that all rpo41 ATD mutants accumulated reduced steady-state levels of both proteins, whereas the amount of the nucleus-encoded mitochondrial protein porin was apparently unaffected (Fig. 3A). These data demonstrate that mutations in the ATD perturb mitochondrial translation, resulting in accumulation of less mtDNA-encoded protein as expected.
Fig. 3. ATD mutations result in reduced steady-state accumulation of mtDNA-encoded proteins and hypersensitivity to the mitochondrial translation inhibitor erythromycin.
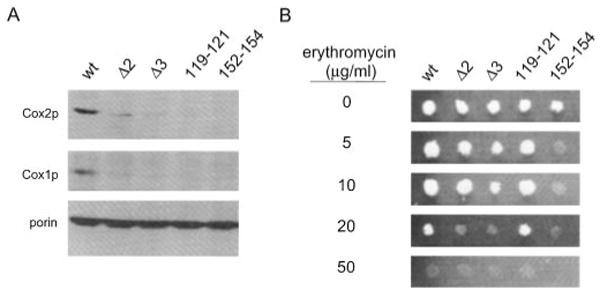
A, Western analysis of mitochondrial Cox1p and Cox2p. Cultures and mitochondria were prepared in the same manner as described for the in vivo labeling experiments except that cycloheximide was omitted, and the cells were not labeled with [35S]methionine. Blots of total mitochondrial protein (40 μg/lane) were sequentially probed using antibodies against mtDNA-encoded Cox2p and Cox1p and porin, a nucleus-encoded mitochondrial outer membrane protein. The mutation in RPO41 harbored by each strain tested is indicated as described in Fig. 2A. B, erythromycin sensitivity assays. A small volume of a liquid culture containing the same number of cells of the indicated yeast strains were spotted onto YPG medium without (0 μg/ml) and with the indicated amounts (5, 10, 20, and 50 μg/ml) of erythromycin after growth as described under “Experimental Procedures.” The plates were then incubated at 36 °C for 6 days. Complete lack of growth was scored as sensitivity to the drug at the given concentration. The yeast strains analyzed are indicated at the top of the figure in the same manner as in Fig. 2A.
Finally, to assess independently whether the rpo41 ATD mutations affect mitochondrial translation, their sensitivity to the mitochondrial translation inhibitor erythromycin was measured. The yeast cultures used in this assay were maintained on glycerol medium throughout the experiment, selecting for maintenance of rho+ genomes. With the exception of the rpo41-E119A-C121A mutation, all of the rpo41 ATD mutants are hypersensitive to the mitochondrial translation inhibitor erythromycin at 36 °C (Fig. 3B). Consistent with results of the in vivo labeling experiments (Fig. 2A) and the steady-state analysis (Fig. 3A), the rpo41-N152A/Y154A strain was by far the most sensitive to the drug.
Sls1p Co-purifies with Mitochondrial Nucleoids in an mtRNA Polymerase ATD-dependent Fashion
The ability of Sls1p to suppress nam1Δ and ATD mutations (9) (Fig. 1C) led us to investigate the possibility that Sls1p interacts in some manner with mtRNA polymerase. We hypothesized that if Sls1p interacts with the ATD, either directly or as a complex with other proteins, it may do so in the context of the mitochondrial nucleoid, where mtRNA polymerase is predicted to reside. To test this possibility, nucleoids in actively translating mitochondria were isolated from wild-type and rpo41Δ3 mutant strains expressing a functional V5-tagged version of Sls1p. Western analysis of nucleoids isolated from wild-type strains confirmed that mtRNA polymerase is present in the nucleoid fraction in a DNA-dependent manner (data not shown), demonstrating that mitochondrial nucleoids are being isolated. Western analysis of mitochondrial lysates using an anti-V5 antibody revealed that expression of the tagged Sls1p was similar in both strains (Fig. 4). Analysis of the corresponding nucleoid fraction from the wild-type strain revealed the presence of Sls1p in the nucleoid that was dependent on the presence of mtDNA (Fig. 4), indicating that Sls1p is not simply a contaminant in the fraction. In addition, its nucleoid association was dependent on the presence of an energy-regenerating system that is needed for optimal in organello translation under these conditions (24). Furthermore, in the rpo41Δ3 strain, a mutant in which the ATD is deleted and does not interact with Nam1p (10), Sls1p no longer associated with the nucleoid fraction (Fig. 4). These data indicate that Sls1p interacts with the ATD, either directly or in a complex with other proteins, and this interaction is necessary for the localization of Sls1p to the nucleoid. Because Sls1p is an integral membrane protein, these data also suggest that mtDNA is associated with the inner membrane during active gene expression. It is noted that Nam1p was not detected in the nucleoid fraction under these conditions (data not shown), consistent with the results of Kaufman et al. (25).
Fig. 4. Sls1p is a component of the mitochondrial nucleoid.
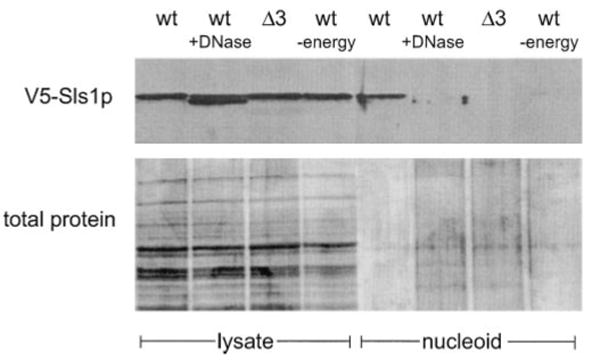
Mitochondrial lysates and nucleoid fractions were prepared as described under “Experimental Procedures.” V5-tagged Sls1p was detected by Western analysis using an anti-V5 antibody (top panel). A separate gel was run and silver-stained to visualize the amount of protein loaded (bottom panel). Lanes 1–4 are the mitochondrial lysates prior to fractionation, and lanes 5–8 are the purified nucleoid fractions. The sample analyzed is indicated at the top of each lane: MSR107 (wt); MSR107 treated with DNase I (wt +DNase); MSR110(Δ3) and MSR107 without the energy regenerating system (wt −energy).
Overexpression of Sls1p Increases the Steady-state Levels of mtDNA-encoded Proteins in rpo41 ATD Mutant Strains Independently of Its Role in Translation
Because Sls1p overexpression suppresses the translation defect in the nam1Δ strain (Fig. 1C) and partially rescues the petite phenotypes of all the ATD mutant strains (9), we next investigated whether the same conditions would restore translation in the rpo41 ATD mutants. Perhaps unexpectedly, overexpression of Sls1p did not result in increased labeling of mtDNA-encoded proteins in any of the rpo41 ATD mutant strains (data not shown). However, Western analysis of Cox1p and Cox2p revealed that over-expression of Sls1p did result in a moderate increase in steady-state levels of both mtDNA-encoded proteins in the ATD mutant strains (Fig. 5). We note that the effect on Cox1p was not as dramatic as Cox2p; nonetheless, this difference was reproducible. These data indicate that Sls1p suppresses the rpo41 ATD mutants by increasing the steady-state level of mtDNA-encoded proteins presumably in a manner largely independent of its effects on translation.
Fig. 5. Overexpression of Sls1p increases the steady-state levels of mtDNA-encoded proteins in ATD mutant strains.

Western analysis of ATD mutants with and without a plasmid that overexpresses Sls1p. Blots of total mitochondrial protein (40 μg/lane) were sequentially probed with antibodies to mtDNA-encoded Cox2p, Cox1p, and the nucleus-encoded porin. The mutation in the RPO41 gene harbored by each strain tested is indicated at the top of the figure in the same manner as in Fig. 2A. The 1st to 5th lanes are the strains without the SLS1 plasmid as a control, and the 6th to the 10th lanes (bracketed) are the strains expressing SLS1 from pRMS5-6 (9), indicated as (+SLS1).
Discussion
A large body of evidence now indicates that mitochondrial translation is primarily, if not exclusively, a membrane-associated process (7, 8, 26–29). In yeast, this is a complex process that involves not only membrane-associated ribosomes but also mRNA-specific translational activators and numerous other proteins involved in processing and stability of mRNA that are directly or indirectly associated with the inner membrane (4, 7). Our previous work (9–11) in this area suggested that mtRNA polymerase is also intricately involved in the efficiency of translation not only directly, through synthesizing the requisite RNA species, but also via functions of the ATD in coordinating transcription with post-transcriptional events. In this report, we have elucidated a role for Sls1p in mitochondrial translation, and we provide additional new lines of evidence that support a model in which one function for the ATD of mtRNA polymerase is to nucleate a series of interactions involving Nam1p and Sls1p that are ultimately required to link mtRNA polymerase to the inner membrane to facilitate efficient mitochondrial protein synthesis. The data supporting these conclusions are discussed below.
A recent key observation that led to our initial proposal that the ATD of mtRNA polymerase is involved in coupling transcription to membrane-associated events is the ability of the membrane protein Sls1p to rescue the petite phenotype of nam1Δ and ATD mutations when overexpressed (9). Because Sls1p is required for proper assembly of the respiratory chain, but not for transcription or mtDNA maintenance per se, it has been postulated to be involved in post-transcriptional steps of mitochondrial gene expression (17). Our results (Fig. 1), which clearly demonstrate that our sls1Δ strain has the same intron-processing defect we reported previously for nam1Δ and ATD mutations (10) and are globally deficient in labeling of mtDNA-encoded proteins in vivo, establish a function for Sls1p in mitochondrial protein synthesis.
Next, we investigated whether the ability of Sls1p to rescue a nam1Δ phenotype (9) was linked to its role in mitochondrial translation identified here. Nam1p was shown by others (14) to be involved in overall mitochondrial translation efficiency, accompanied by a severe defect in translation of Cox1p. We confirmed a role for Nam1p in overall mitochondrial translation (Fig. 1C) and went on to show that overexpression of Sls1p re-established a nearly wild-type mitochondrial labeling pattern in an nam1Δ strain (Fig. 1C), supporting a critical role for Sls1p in mitochondrial translational efficiency. Interestingly, overexpression of Sls1p in our nam1Δ strain resulted in a Cox1p-specific translation defect (Fig. 1C) that is virtually identical to that originally reported by Asher et al. (14), suggesting that the differences observed between these two strains may involve strain-dependent differences in endogenous expression of SLS1.
The elucidation that Sls1p is involved in translation in cooperation with Nam1p (Fig. 1) suggested to us that one function of the pathway of gene expression events involving Nam1p, Sls1p and the ATD that we have described previously (9, 10) is to facilitate delivery of transcripts to the translation machinery at the inner mitochondrial membrane. Two additional lines of evidence provided by this study support this conclusion. First, all mtRNA polymerase ATD mutations examined here resulted in translation-related defects, including a significant reduction in labeling of all mtDNA-encoded proteins in vivo (Fig. 2A), reduced steady-state levels of the mtDNA-encoded proteins Cox1p and Cox2p (Fig. 3A), and, with the exception of the rpo41-E119A-C121A mutation (discussed later), an increased sensitivity to the mitochondrial translation inhibitor erythromycin (Fig. 3B). That the three ATD mutations that have translation defects in all three assays (rpo41Δ2, rpo41Δ3, and rpo41-N152A/Y154A) are those that were shown previously to negatively impact Nam1p binding (10) strongly suggests that the observed defects result from disruption of the proposed RNA-handeling pathway. Second, Sls1p is found associated with nucleoids in an ATD-dependent and energy-dependent manner (Fig. 4), strongly supporting the notion that efficient mitochondrial translation is occurring when Sls1p is found in a complex with those mtRNA polymerase molecules that are bound to mtDNA and presumably actively engaged in transcription of the mitochondrial genome.
Based on these data, we propose a revised model for mitochondrial gene expression involving these factors (Fig. 6). In this model, Nam1p is predicted to bind to the ATD of mtRNA polymerase to facilitate the interaction of a transcriptionally active, nucleoid-associated mtRNA polymerase with Sls1p at the inner mitochondrial membrane. Once this connection is established between mtRNA polymerase and Sls1p (which may involve additional factors), translation of the mRNA is accomplished in a transcription-coupled manner. The precise functions of Nam1p and Sls1p remain to be elucidated. However, based on the recent report by Fox and colleagues (18), Nam1p may facilitate interactions between the nascent mRNA and COX-specific translation activators. If this were the case, then it is tempting to speculate that, once a functionally coupled transcription/translation complex is fully established, Nam1p would dissociate in order to locate another template-bound mtRNA polymerase that has yet to be membrane-coupled. A transient nature to the Nam1p interactions is postulated based on the fact that Nam1p is not found as a nucleoid component (see Ref.25; this study, data not shown) and is localized primarily to the matrix in mitochondrial fractionation studies (30). At present we speculate that the most likely function for Sls1p in this regard is to serve as part of a membrane-anchoring point for mtRNA polymerase during active gene expression.
Fig. 6. Model describing critical interactions required to coordinate transcription and translation at the inner mitochondrial membrane.

The mitochondrial inner membrane is shown with associated gene-expression components (e.g. RNA processing factors, ribosomes, and other translation components) indicated as large shaded rectangles. Nam1p is shown binding to the ATD of mtRNA polymerase (10) in order to facilitate the interaction of mtDNA-bound (i.e. transcription-competent) mtRNA polymerase with Sls1p at the inner membrane. Based on its ATD-dependent localization to mtDNA nucleoids (Fig. 4), Sls1p is shown in association with the ATD of mtRNA polymerase that is engaged in transcription. However, it is noted that whether such an interaction is direct or mediated by other proteins is currently not known. In this model, Sls1p is postulated to function as part of an interaction point for the ATD, perhaps to localize mtRNA polymerase and its associated nascent transcript to the inner membrane to facilitate RNA interactions with the translational activators. The nascent transcript emerging from mtRNA polymerase represents one of the cytochrome oxidase subunit mRNAs (e.g. COX1, COX2, or COX3) and is shown interacting with the membrane-associated COX translational-activator complex. Based on the recent identification of the interactions between Nam1p and members of this complex (18), we show Nam1p binding here as well, perhaps to help establish binding of the 5′-untranslated region of the mRNA with its cognate translational activator. After a membrane-associated mtRNA polymerase complex is established, Nam1p is postulated to dissociate, perhaps to locate the next mtRNA polymerase to be coupled to the membrane.
Whereas the vast majority of data presented herein are consistent with this model, it is clear that mutations in the ATD can cause multiple defects in mitochondrial gene expression that are not as easily explained. First, although we were successful at eliminating mtDNA instability as an explanation for the observed overall translation defects seen in most of ATD mutant strains tested (Table I), the rpo41-E119A-C121A is a notable exception. This mutation resulted in a relatively large defect in steady-state accumulation of Cox1p and Cox2p (Fig. 3A). In fact, its steady-state defect was comparable with that of the rpo41-N152A/Y154A strain (Fig. 3A), despite the fact that its in vivo labeling capacity was substantially greater (Fig. 2A), and it was not hypersensitive to erthyromycin (Fig. 3B). This mutation also does not appear to disrupt Nam1p binding (10). Altogether, these results suggest that the rpo41-E119A-C121A defect is not due to disruption of the proposed translation-coordination function of the ATD but rather its mtDNA instability phenotype (Table I). Second, overexpression of Sls1p is unable to rescue the in vivo labeling defect in the rpo41 ATD mutant strains (data not shown) but does moderately increase the steady-state level of Cox1p and Cox2p in the ATD mutants (Fig. 5), suggesting that Sls1p has a role in assembly of the oxidative phosphorylation complexes as originally postulated by others (17). These data indicate the ability to Sls1p to partially rescue the ATD mutant phenotypes (9) is most likely through this second function and not it ability to reestablish normal membrane coupling of mtRNA polymerase through the ATD. One possibility is that overexpression of Sls1p may allow increased numbers of membrane complexes to form in the ATD mutant strains and, even though they are not “wild-type” complexes, facilitates assembly (and hence stability) of the proteins that do manage to get translated, thus partially rescuing the mutant phenotype. In the case of nam1Δ strains, a similar scenario is envisioned where the ability of Sls1p to increase the number of membrane sites would, in principle, increase the probability that normally coupled complexes would form because the ATD is intact in these strains, thus providing an explanation for the nearly full rescue of the nam1Δ phenotype by Sls1p overexpression (9). This interpretation is entirely consistent with the proposal that Nam1p functions to facilitate formation of the membrane-coupled mtRNA polymerase complex involving Sls1p (Fig. 6), but itself is not an active member of the complex once it is formed. Finally, the rpo41-R129D mutation results in only a moderate reduction in mitochondrial translation (data not shown), yet causes the most severe glycerol growth phenotype (10), suggesting that this mutation in the ATD of mtRNA polymerase can compromise additional cellular functions.
In conclusion, we have elucidated important new aspects of how mitochondrial gene expression is accomplished in yeast. Our results indicate that the primary mechanism involves a complex series of interactions that ensure coordination of transcription and translation of mitochondrial transcripts at the inner mitochondrial membrane that is mediated through the ATD of mtRNA polymerase. It will be of great general importance to determine whether a similar mechanism is operating in human cells, where defects in mitochondrial gene expression can cause and exacerbate disease states and impact the aging process.
Footnotes
The abbreviation used is: ATD, amino-terminal domain.
This work was supported by National Institutes of Health Grant HL-59655 (to G. S. S.).
References
- 1.Shadel GS. Am J Hum Genet. 1999;65:1230–1237. doi: 10.1086/302630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenleaf AL, Kelly JL, Lehman IR. Proc Natl Acad Sci U S A. 1986;83:3391–3394. doi: 10.1073/pnas.83.10.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masters BS, Stohl LL, Clayton DA. Cell. 1987;51:89–99. doi: 10.1016/0092-8674(87)90013-4. [DOI] [PubMed] [Google Scholar]
- 4.Dieckmann CL, Staples RR. Int Rev Cytol. 1994;152:145–181. doi: 10.1016/s0074-7696(08)62556-5. [DOI] [PubMed] [Google Scholar]
- 5.Lazowska J, Jacq C, Slonimski PP. Cell. 1980;22:333–348. doi: 10.1016/0092-8674(80)90344-x. [DOI] [PubMed] [Google Scholar]
- 6.Pel HJ, Grivell LA. Mol Biol Rep. 1993;18:1–13. doi: 10.1007/BF01006890. [DOI] [PubMed] [Google Scholar]
- 7.Fox TD. Experientia (Basel) 1996;52:1130–1135. doi: 10.1007/BF01952112. [DOI] [PubMed] [Google Scholar]
- 8.Sanchirico ME, Fox TD, Mason TL. EMBO J. 1998;17:5796–5804. doi: 10.1093/emboj/17.19.5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryan AC, Rodeheffer MS, Wearn CM, Shadel GS. Genetics. 2002;160:75–82. doi: 10.1093/genetics/160.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodeheffer MS, Boone BE, Bryan AC, Shadel GS. J Biol Chem. 2001;276:8616–8622. doi: 10.1074/jbc.M009901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Shadel GS. Proc Natl Acad Sci U S A. 1999;96:8046–8051. doi: 10.1073/pnas.96.14.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Bell A, Perlman PS, Leibowitz MJ. RNA (New York) 2000;6:937–951. doi: 10.1017/s1355838200991726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Proudfoot NJ, Furger A, Dye MJ. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 14.Asher EB, Groudinsky O, Dujardin G, Altamura N, Kermorgant M, Slonimski PP. Mol Gen Genet. 1989;215:517–528. doi: 10.1007/BF00427051. [DOI] [PubMed] [Google Scholar]
- 15.Groudinsky O, Bousquet I, Wallis MG, Slonimski PP, Dujardin G. Mol Gen Genet. 1993;240:419–427. doi: 10.1007/BF00280396. [DOI] [PubMed] [Google Scholar]
- 16.Lisowsky T. Mol Gen Genet. 1990;220:186–190. doi: 10.1007/BF00260480. [DOI] [PubMed] [Google Scholar]
- 17.Rouillard JM, Dufour ME, Theunissen B, Mandart E, Dujardin G, Lacroute F. Mol Gen Genet. 1996;252:700–708. doi: 10.1007/BF02173976. [DOI] [PubMed] [Google Scholar]
- 18.Naithani S, Saracco S, Butler C, Fox T. Mol Biol Cell. 2002;14:324–333. doi: 10.1091/mbc.E02-08-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherman F. Methods Enzymol. 1991;194:3–21. doi: 10.1016/0076-6879(91)94004-v. [DOI] [PubMed] [Google Scholar]
- 20.Fox TD, Folley LS, Mulero JJ, McMullin TW, Thorsness PE, Hedin LO, Costanzo MC. Methods Enzymol. 1991;194:149–165. doi: 10.1016/0076-6879(91)94013-3. [DOI] [PubMed] [Google Scholar]
- 21.O'Rourke TW, Doudican NA, Mackereth MD, Doetsch PW, Shadel GS. Mol Cell Biol. 2002;22:4086–4093. doi: 10.1128/MCB.22.12.4086-4093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grubbs FE. Technometrics. 1969;11:1–21. [Google Scholar]
- 23.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. pp. A8.40–A8.55. [Google Scholar]
- 24.Poyton RO, Bellus G, McKee EE, Sevarino KA, Goehring B. Methods Enzymol. 1996;264:36–42. doi: 10.1016/s0076-6879(96)64006-3. [DOI] [PubMed] [Google Scholar]
- 25.Kaufman BA, Newman SM, Hallberg RL, Slaughter CA, Perlman PS, Butow RA. Proc Natl Acad Sci U S A. 2000;97:7772–7777. doi: 10.1073/pnas.140063197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuriyama Y, Luck DJ. J Cell Biol. 1973;59:776–784. doi: 10.1083/jcb.59.3.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu M, Spremulli L. J Biol Chem. 2000;275:29400–29406. doi: 10.1074/jbc.M002173200. [DOI] [PubMed] [Google Scholar]
- 28.Marzuki S, Hibbs AR. Biochim Biophys Acta. 1986;866:120–124. doi: 10.1016/0167-4781(86)90108-9. [DOI] [PubMed] [Google Scholar]
- 29.Spithill TW, Trembath MK, Lukins HB, Linnane AW. Mol Gen Genet. 1978;164:155–162. doi: 10.1007/BF00267380. [DOI] [PubMed] [Google Scholar]
- 30.Wallis MG, Groudinsky O, Slonimski PP, Dujardin G. Eur J Biochem. 1994;222:27–32. doi: 10.1111/j.1432-1033.1994.tb18837.x. [DOI] [PubMed] [Google Scholar]