

Abstract
G protein coupled receptors can activate MAP kinase pathways via G protein-dependent and -independent mechanisms. However, the physiological outcomes correlated with the cellular signaling events are not as well characterized. Here we examine the involvement of G protein and β-arrestin 2 pathways in kappa opioid receptor (KOR)-induced, ERK1/2-mediated proliferation of both immortalized and primary astrocyte cultures. Since different agonists induce different cellular signaling pathways, we tested the prototypic kappa agonist, U69,593 as well as the structurally distinct, non-nitrogenous agonist, MOM-Salvinorin-B (MOM-Sal-B). In immortalized astrocytes, U69,593, activated ERK1/2 by a rapid (min) initial stimulation that was sustained over 2 hours and increased proliferation. Sequestration of activated Gβγ subunits attenuated U69,593 stimulation of ERK1/2 and suppressed proliferation in these cells. Furthermore, siRNA silencing of β-arrestin 2 diminished sustained ERK activation induced by U69,593. In contrast, MOM-Sal-B induced only the early phase of ERK1/2 phosphorylation and did not affect proliferation of immortalized astrocytes. In primary astrocytes, U69,593 produced the same effects as seen in immortalized astrocytes. MOM-Sal-B elicited sustained ERK1/2 activation which was correlated with increased primary astrocyte proliferation. Proliferative actions of both agonists were abolished by either inhibition of ERK1/2, Gβγ subunits or β-arrestin 2, suggesting that both G protein-dependent and - independent ERK pathways are required for this outcome.
Keywords: opioids, opioid receptors, astrocytes, ERK/MAP kinase, β-arrestins, G proteins
The binding of an agonist to a cell surface receptor has the potential to initiate numerous signaling cascades. In turn, diverse agonists are capable of initiating distinct pathways, and due to this functional selectivity such paths may be preferentially activated in certain cell types (for a review see Urban et al. 2007). A striking example of the multiplicity of signaling pathways is the recent finding that GPCRs activate the ERK1/2/MAPK phosphorylation cascade by G protein-dependent and β-arrestin-dependent mechanisms in the same cell (Ahn et al. 2004; Barnes et al. 2005; Lefkowitz and Shenoy 2005; Gesty-Palmer et al. 2006; Shenoy et al. 2006). In many of these studies, agonist induced ERK1/2 phosphorylation in receptor-transfected HEK293 cells was determined to be mediated via a G protein coupled mechanism involving PKA and/or PKC during the early phase of activation. The phosphoERK generated in this paradigm is then translocated to the nucleus to activate transcriptional substrates. In contrast, sustained ERK1/2 activation entails a G protein-, PKA- and PKC-independent pathway in which β-arrestins play a pivotal role (Tohgo et al. 2002). The phosphoERK produced by this mechanism is retained in the cytoplasm. Although these two independent ERK1/2 signaling pathways can have different effects on transcription, their impacts on outcomes of physiological regulation are not well characterized (Barnes et al. 2005; Lefkowitz et al. 2006).
The emerging status of astrocytes as equal partners in synaptic transmission has been supported by the recent discovery of a number of “gliotransmitters” (Diamond 2006; Seifert et al. 2006). These include D-serine, glutamate, leukemia inhibitory factor, ephrins and thrombospondins that are secreted by astrocytes and play dynamic roles in synaptic signaling, myelination and neurogenesis (Yang et al. 2003; Christopherson et al. 2005; Ishibashi et al. 2006; Panatier et al. 2006; Nishida and Okabe 2007; Jiao et al. 2008). Since astrocytes are a functionally and morphologically diverse group of cells, it is important to understand how cell division of each type is regulated to facilitate communication with neurons (Raff et al. 1983; Wang et al. 1994; Muroyama et al. 2005). MAPK phosphorylation cascades have been implicated in proliferation, differentiation and survival of primary astrocytes induced by various mitogens (Biesiada et al. 1996; Kurino et al. 1996; Lazarini et al. 1996; Riboni et al. 2000; Fanton et al. 2001; Lenz et al. 2001; Riboni et al. 2001). The opioid GPCRs have been demonstrated to regulate ERK1/2/MAPKs in immortalized and primary astrocytes, in some pathways by transactivation of growth factor receptors (Belcheva et al. 2003; Belcheva et al. 2005; Mahajan et al. 2005; Bruchas et al. 2006). Opioid receptor regulation of glial cell proliferation during CNS development and injury has also been well documented in vivo and in vitro (Hauser and Stiene-Martin 1991; Stiene-Martin et al. 1991; Barg et al. 1993; Barg et al. 1994; Hauser and Mangoura 1998; Xu et al. 2007).
The current study focuses on temporal and mechanistic aspects of κ-opioid receptor (KOR) activation of ERK1/2 and how this affects growth of astrocytes. Here we measure ERK1/2 activation by distinct κ-opioid agonists, MOM-Sal-B and U69,593 in rat cortical immortalized type-1 and primary (type-1 and type-2) astrocytes. We find that the two ligands promote different temporal signaling patterns with respect to ERK activation. This functional selectivity is dependent on the cellular environment as KOR ligand differences are only detected in immortalized cells. Furthermore, we find that KOR-mediated primary astrocyte proliferation involves both Gβγ- and β-arrestin 2-mediated ERK1/2 activation.
Experimental Procedures
Reagents
Chemicals were purchased from Sigma Chemical Co. (St. Louis, MO) with the following exceptions: U69,593 and norbinaltorphimine (NorBNI) were from NIDA Drug Supply (Research Triangle, NC); EGF and U0126 from Calbiochem (San Diego, CA); trypsin-EDTA solution from Gibco (Carlsbad, CA); pertussis toxin (PTX) from List Biological Laboratories, Inc. (Campbell, CA); Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) from ATCC (Manassas, VA); salvinorin A was isolated from Salvia divinorum and purified to >98% homogeneity; the salvinorin A derivative, MOM-Sal-B was a generous gift from Dr. David Y. W. Lee, McLean Hospital, Belmont, MA. Anti-phospho-ERK1/2 (directed against phospho Thr202/Tyr204) antibody (Ab) was from Cell Signaling Technology (Beverly, MA); anti-GFAP Ab was from ImmunoStar, Inc. (Hudson, WI, Catalog #: 22522); anti-β-arrestin 2 and anti ERK Ab were from Santa Cruz (Santa Cruz, CA); anti-TuJ 1 Ab was from Neuromics (Edina, MN); BrdU and the Abs for its detection from the BrdU Labeling and Detection Kit I (Roche, Basel, Switzerland), Alexa Fluor labeled secondary Abs and horse serum from Invitrogen - Molecular Probes (Carlsbad, CA); and VECTASHIELD Mounting Medium (Vector Laboratories, Inc., Burlingame, CA). CD8 and the Gβγ scavenger, CD8-β-adrenergic receptor (CD8-βARK-C) cDNA, were a generous gift from S. Gutkind (NIH). siRNA directed against the β-arrestin 2 gene and non-targeting control siRNA were purchased from Dharmacon RNA technologies (Lafayette, CO).
Primary astrocyte cultures
Postnatal day 1 Sprague-Dawley rat pups were euthanized and their cortical regions were dissected out, minced, suspended in 2.5 g/mL ice-cold PBS and trypsinized by incubation with an equal volume of 0.05% trypsin-EDTA solution at 37°C for 15 min. The tissue was pelleted (1000 g X 10 min), resuspended in 5 g/mL DMEM containing 5% FBS and 5% horse serum, triturated and plated onto poly-L-lysine (mol. wt. 30,000-70,000) coated tissue culture flasks as indicated.
After 7 days in culture, the poly-L-lysine coated flasks were shaken for at least 2 h, after which the unattached cells were removed and fresh culture medium was added (DMEM, 5% FBS and 5% horse serum EGF). For ERK1/2 assay, growth medium was replaced with DMEM without serum 24 h prior to ligand treatment. Of the total number of cells in primary cultures, 90% were GFAP positive and <1% were TuJ 1 (neuronal marker) positive. Generally, primary rat cortical astrocyte cultures were morphologically heterogeneous in that they contained both GFAP positive type-1 astrocytes that are flat polyhedral shaped cells and type-2 that are spindle shaped and possess two or more processes. Primary cultures from some tissues such as neonatal mouse spinal cord only contain type-2 astrocytes whereas primary neonatal mouse brain astrocyte cultures consist of predominantly type-1 cells (Raff et al. 1983; Hauser and Stiene-Martin 1991; Xu et al. 2007). Animals were handled in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Type-1 immortalized rat cortical astrocyte cultures
Rat cortical astrocytes (CTX TNA2, ATCC), were established from cultures of primary type-1 astrocytes from 1-day-old rat brain frontal cortex and grown as described (Radany et al. 1992; Belcheva et al. 2003; Belcheva et al. 2005).
Transient transfections
Immortalized rat astrocytes were transiently transfected with pcDNA3 (for mock transfections) or KOR cDNA (pCMV-neo expression vector) using FuGENE 6 transfection reagent following the manufacturer's instructions and adding 1 μg of cDNA and 3 μl of transfection reagent. In some cases, cells were co-transfected with 1 μg CD8 (in pcDNAI AMP vector) or CD8-β-adrenergic receptor kinase (CD8-βARK-C in pcDNAIII vector) cDNA using FuGENE 6 as described (Belcheva et al. 1998). CD8-βARK-C expresses a membrane anchoring protein (CD8) fused to the βγ subunit-binding segment of the carboxyl terminus of βARK (Crespo et al. 1995).
siRNA preparation and transfection
siRNA targeting the rat β-arrestin 2 gene was designed and synthesized by Dharmacon RNA technologies (Lafayette, CO). The following siRNA preparations were used: siGENOME standard SMART pool to rat ARRB2 (Catalog # D-080157-00, target sequences: GGAGCUACCUUUCGUCCUA, GAUGAAGGAUGACGACUGU, GAGAAGACCUGGAUGUACU, GCAAAGAUCUGUUCAUCGC) and siCONTROL non-targeting siRNA pool (negative control that has been bioinformatically designed and validated to not have any known targets, Catalog # D-001206-13, target sequences: AUGAACGUGAAUUGCUCAA, UAAGGCUAUGAAGAGAUAC, AUGUAUUGGCCUGUAUUAG, UAGCGACUAAACACAUCAA). The siRNA preparations were resuspended in Dharmacon- provided siRNA buffer to a stock concentration of 20 μM. Immortalized and primary rat astrocytes were transiently transfected using the Amaxa Nucleofector electroporator (Amaxa Biosystems, Gaithersburg, MD). Briefly, cells were removed from flasks by treatment with 0.05% trypsin and 0.02% EDTA for 1 min at 37°C, washed with media and incubated for 2 h at 37°C in a 50 g/mL conical tube. Equal amount of cells, no more than 2.0x106, were distributed in tubes, harvested by centrifugation and resuspended in 100 μl Rat Astrocyte Nucleofector Solution (Amaxa). Two μg of KOR cDNA and 1 μM control or target siRNAs were added to each tube with cells and electroporation was performed using optimal Amaxa Nucleofector program T-20. This program was developed by Amaxa specifically for primary astrocytes. The transfection efficiency of Amaxa electroporation is much higher than the 9 +/- 1 % that we obtained with FuGENE 6 as determined by Gal expression measurements. The Amaxa estimate for rat astrocyte transfection is 70% using their T-20 program. To document this, cells were transfected with EGFP, along with GFAP and DAPI and their staining were compared. The majority of GFAP+ cells were labeled with EGFP with varying degrees of staining. Immediately following electroporation, cells were transferred to 6-well tissue culture plates or 8 well chambers containing growth media and cultured overnight at 37°C. Then cells were washed 3 times with media deprived of serum and cultured for an additional 24 h in fresh growth media. Cells were grown for 24 h in serum-deprived media prior to initiation of ERK1/2 activity or proliferation determinations. The efficiency of gene silencing was validated by immunoblotting with corresponding Abs.
ERK1/2 assays
ERK1/2 phosphorylation was measured by immunoblotting as described (Belcheva et al. 2001). Cells were treated first with either inhibitors or antagonist, and then with MOM-Sal-B or U69,593 as described in the Figure legends. Cells were then washed with PBS and lysed with buffer containing 20 mM HEPES, 10 mM EGTA, 40 mM β-glycerophosphate, 2.5 mM MgCl2, 2 mM sodium vanadate, 1% Nonidet-40, 1 mM PMSF, 20 μg/mL aprotinin and 20 μg/mL leupeptin. Cell lysates were centrifuged at 14,000 g for 20 min at 4°C and protein concentration of the supernatants was determined. Samples (10-20 μg protein/lane) were separated by 10% SDS-PAGE. Proteins were blotted on Immobilon P™ PVDF membranes. Nonspecific sites were blocked with 5% milk in Tris-buffered saline + 0.2% Tween 20 (TBST). Blots were then washed 3 times with TBST and incubated with anti-phospho-ERK1/2 Ab for at least 15 h at 4°C. After 3 washes with TBST, blots were incubated with 1:2000 diluted HRP-conjugated goat anti-mouse-IgG for 1 h at room temperature. For assurance of equivalent total ERK1/2 protein per lane, representative blots were stripped (0.2 M glycine, pH 2.5, 60 min at room temperature) and exposed to anti-ERK1/2 Ab (1:1000), followed by 1:20000 diluted HRP conjugated goat anti-rabbit-IgG. Bands were visualized using an ECL chemiluminescence detection system. Band intensities were determined by densitometry using photos taken with a Kodak DC120 digital camera, Kodak ds 1D version 3.0.2 (Scientific Imaging Systems) and analyzing them with NIH ImageJ version 1.32 software. ERK stimulation in opioid-treated cells is expressed as fold change over basal levels in untreated cells or % maximal expression.
Confocal imaging of HA-KOR and β-arrestin 2-GFP transfected HEK 293 cells
HEK 293 cells were cultured in minimum essential medium (MEM; Invitrogen, Carlsbad, California) containing 10% heat inactivated fetal bovine serum (FBS; Invitrogen) and 1% penicillin-streptomycin (Invitrogen) at 37°C under 5% CO2. The HEK 293 cells were transiently transfected with haemagglutinin (HA-N-terminus) tagged rat KOR (5 μg cDNA) and β-arrestin 2 tagged with green fluorescent protein (β-arr 2-GFP) (2μg cDNA). Live cell antibody staining of transfected HA-KOR was obtained by incubating cells in serum free MEM for 30 minutes with anti-HA-Alexa Fluor 594 conjugate (1:100; Molecular Probes/Invitrogen, Carlsbad, CA). Agonists were added and multiple cells were imaged per dish. HEK 293 cell transfection by electroporation and confocal imaging were performed as previously described (Bohn et al. 2004)
Immunocytochemical detection of BrdU incorporation and GFAP co-staining
Cells were grown in poly-L-lysine coated 8-well chamber slides until they were about 50% confluent in the case of primary or about 60% confluent for immortalized astrocytes and growth medium was replaced with DMEM without serum for 28 h. They were then treated with opioids and/or inhibitors for 24 h and were labeled with 10 μM BrdU for the final 4 h of treatment. Cells were fixed with 4% para-formaldehyde in PBS for 20 min at RT. After 3 X 5 min PBS washes, cells were incubated with 2N HCl/0.5% Triton X-100 in 0.1 X PBS for 1h at RT, washed once for 5 min with PBS pH 8.4 and 3 X 5 min with regular PBS, and blocked with PBS containing 0.5% BSA and 0.1% Tween 20 for 30 min. BrdU and the Abs for its detection were used following the manufacturer's directions. Cells were incubated with rabbit anti-GFAP Ab (1:1 dilution) in blocking solution overnight at 4°C and washed 3X 5 min with PBS. Then, cells were treated with mouse anti-BrdU monoclonal Ab, 1:10 dilution for 30 min at 37°C, washed 3 X 5 min with PBS and then incubated with both secondary Abs together in blocking solution (see above) for 1 h at RT (red fluorescence emitting Alexa Fluor 594 goat anti-rabbit IgG, highly cross-adsorbed Ab, 2 mg/mL, diluted 1:700 (for GFAP detection) and the green fluorescence emitting fluorescein-conjugated anti-mouse IgG Ab (1:10 dilution) from the kit for BrdU detection. After 3 PBS washes, slides were mounted using anti-fade VECTASHIELD mounting medium and covered with cover slips. Slides were examined for immunofluorescence with an OLYMPUS AH-3 microscope attached to a Soft Imaging Systems F-view II CCD digital camera that has simultaneous recording capability of dual fluorescence label images.
Protein assay and statistical analysis
Protein concentrations were determined by the Bradford method (Bradford 1976) with bovine serum albumin (1 mg/mL) as standard. In all experiments reported, each N value represents a result gained from one culture. In primary culture studies, cells from at least two different litters of rat pup brains were used for each final determination. Statistical determinations were made by Student t-test analysis using GraphPad Prism software. Data are expressed as the mean+/-SEM.
Results
KOR activation of ERK1/2 in immortalized astrocytes
A time course study of the stimulation of ERK1/2 phosphorylation by salvinorin A, was conducted with type-1 immortalized rat cortical astrocytes. This nonnitrogenous, highly selective κ-opioid agonist stimulated ERK phosphorylation 3.6 fold at the 5 min interval (p<0.05 vs. control), but activation was not detected at 15 or 120 min. Salvinorin A is known to undergo a slow (∼20-30 min) hydrolysis of its ester bond at C-2 in vitro and as short as 8 min in primates in vivo (Roth et al. 2002; Yan and Roth 2004; Hooker et al. 2008). MOM-Sal-B is a salvinorin A analog that has a more stable methoxymethyl moiety at C(2), a high binding affinity for KOR, 7 fold greater potency in GTPγS binding assays and a longer half life in vitro than salvinorin A (Lee et al. 2005). Therefore, a time course study of the stimulation of ERK1/2 phosphorylation by MOM-Sal-B was conducted with type-1 immortalized rat cortical astrocytes (Fig. 1A). The κ opioid selective antagonist, NorBNI abolished ERK1/2 activation by MOM-Sal-B implicating KOR in the signaling pathway (Fig. 1B). The MOM-Sal-B results were compared to U69,593 data previously obtained for these same cells (Belcheva et al. 2003). While the κ opioid prototypic agonist, U69,593, induced a sustained activation of ERK1/2 lasting over 2 h, MOM-Sal-B elicited a short-lived increase in ERK1/2 phosphorylation (<30 min). Since MOM-Sal-B also stimulated ERK1/2 phosphorylation robustly but transiently, the waning effects on the extent of ERK1/2 phosphorylation were not likely to be attributable to the degradation of the agonist (Fig. 1A).
Fig. 1. Time course of MOM-Sal-B stimulation of ERK1/2 phosphorylation and antagonism by the selective KOR antagonist, NorBNI in type-1 immortalized astrocytes transiently transfected with KOR cDNA.
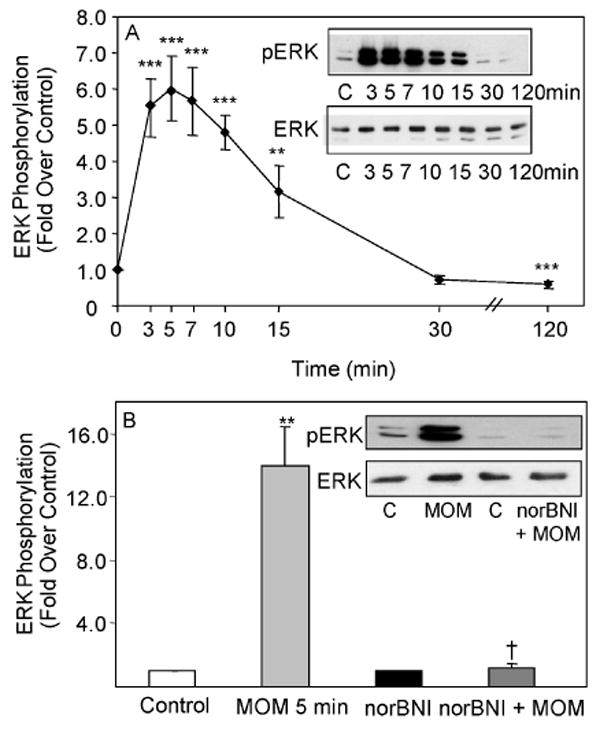
A. Cells were treated with 1 μM MOM-Sal-B and assayed at the indicated time intervals. N= 4-8. **p< 0.01, ***p< 0.001 vs. control. B. Cells were pre-treated with 1 μM NorBNI for 1 h, followed by a 5 min treatment with 1 μM MOM-Sal-B. N=4. **p< 0.01 vs. control, †p< 0.05 vs. agonist alone. The gels are representative immunoblots showing phosphorylated ERK1/2 and total ERK1/2 bands. The graph and curves show quantified ERK1/2 phosphorylation based on phosphoERK1/2 to ERK ratios.
Signaling mechanisms involved in κ opioid induced acute and sustained ERK1/2 phosphorylation
In immortalized astrocytes, U69,593 elicits both rapid (min) and sustained (h) activation of ERK1/2, while MOM-Sal-B induces only the early phase suggesting that multiple KOR-mediated ligand specific, pathways of ERK1/2 activation may exist in astrocytes. It has been previously demonstrated that the early phase of ERK1/2 phosphorylation may be mediated by a G protein-dependent mechanism that is distinct from a later sustained activation which involves β-arrestins (Ahn et al. 2004; Gesty-Palmer et al. 2006). Therefore, we used a series of inhibitor and siRNA knockdown studies to assess the nature of the rapid and sustained phases of ERK1/2 activation in response to the two ligands. An earlier study by our group suggested that the rapid phase of ERK1/2 phosphorylation is G protein-dependent and may be mediated by the Gβγ subunit of heterotrimeric G proteins (Belcheva et al. 2005). The role of the Gβγ subunit was assessed here by overexpressing CD8-βARK-C cDNA, a chimeric molecule comprised of the CD8 receptor and the carboxyl terminus of the beta-adrenergic receptor kinase that acts as a scavenger of Gβγ subunits (Crespo et al. 1995). As seen in figures 2A and 2B, CD8-βARK-C almost completely abolished the rapid phase and significantly attenuated the sustained phase of U69,593-induced ERK1/2 phosphorylation (see curve labeled G protein independent in Fig. 2B). Transfection of the CD8 cDNA construct had no effect on KOR mediated ERK activation.
Fig. 2. Selective inhibition of early and later phases of U69,593- and MOM-Sal-B-induced stimulation of ERK1/2 phosphorylation in type-1 immortalized astrocytes.
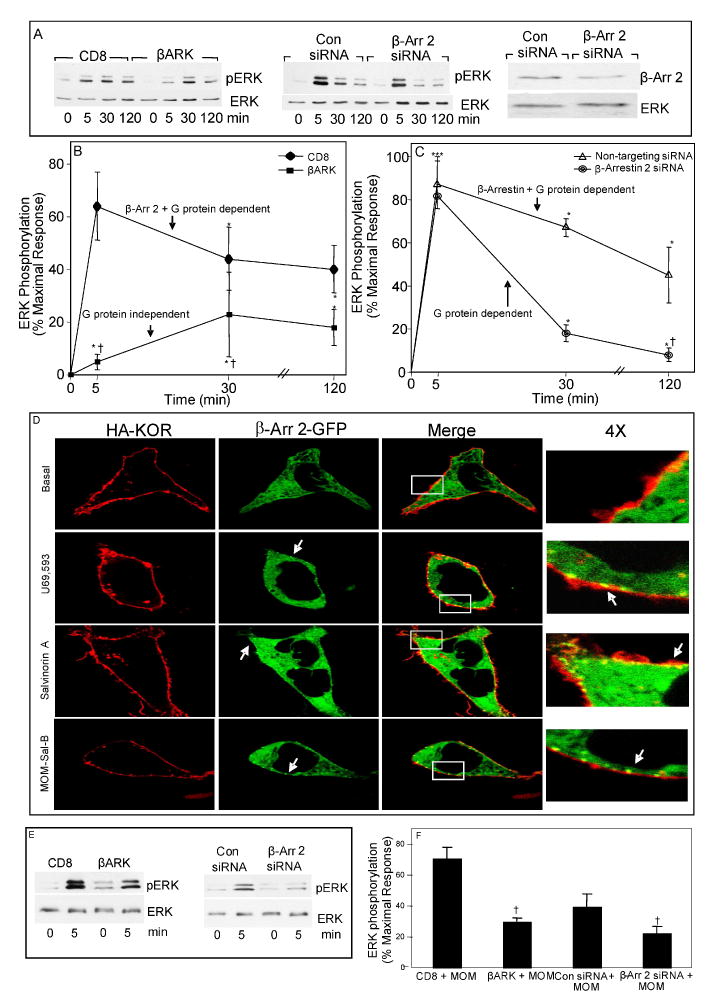
A. Effect of CD8-βARK-C and β-arrestin 2 siRNA on U69,593 signaling. Cells were transiently transfected with KOR +/- CD8 or CD8-βARK-C cDNA; or KOR cDNA +/- non-targeting control or β-arrestin 2 siRNA. After 24 h growth in serum-deprived media, cells were treated with 1 μM U69,593 for the indicated time intervals and ERK1/2 phosphorylation was assayed. The gels (left and center) are representative immunoblots showing phosphorylated ERK1/2. The gel on the right is a representative immunoblot showing β-arrestin 2 levels in cells transfected with non-targeting control or β-arrestin 2 siRNAs. B and C. Figures show curves of quantified % maximal response of ERK1/2 phosphorylation for each treatment in the experiment outlined in panel A. N=6-8. *p< 0.05, ***p< 0.001 vs. zero time control, †p< 0.05 vs. their respective control treatment (CD8 or non-targeting siRNA) at the same time point. D. Agonist-induced β-arrestin 2 translocation to KOR in living HEK 293 cells. HEK-293 cells were transiently transfected with HA-KOR and β-arr2-GFP. HA-KOR were labeled with anti-HA antibody conjugated to AlexaFluor 594. Cells were treated with MOM-Sal-B (100 nM), salvinorin A (100 nM, provided by Thomas E. Prisinzano) or U69,593 (100 nM) and images were taken at 5 minutes post-treatment. Representative cells are shown. β-arr 2-GFP puncta at cell surface are indicated by white arrows. E and F. Effect of CD8-βARK-C and β-arrestin 2 siRNA on MOM-Sal-B signaling. Cells were transiently transfected with KOR +/- CD8 or CD8-βARK-C cDNA; or KOR cDNA +/- non-targeting control or β-arrestin 2 siRNA, starved for 24-28 h, treated with 1 μM MOM-Sal-B for 5 min and ERK1/2 phosphorylation was assayed. The gels are representative immunoblots showing phosphorylated ERK1/2 and total ERK1/2 bands. The graph shows quantified % maximal response of ERK1/2 phosphorylation based on phosphoERK1/2 to ERK ratios. N=3. †p< 0.05 vs. MOM-Sal-B alone.
To test the role of β-arrestin 2 in the agonist-induced KOR activation of ERK in our astrocytic model, we utilized a siRNA silencing approach. Immortalized astrocytes were transfected with siRNAs directed against β-arrestin 2 or a non-targeting control by electroporation. Figure 2A (right-last two lanes) demonstrates that the β-arrestin 2 protein content was reduced by 40 +/- 5%, p<0.05, N=4) by the siRNA in comparison to non-targeting control siRNA. U69,593-induced ERK1/2 phosphorylation was reduced by the knock-down of β-arrestin 2; however the temporal pattern differed greatly from that engendered by the Gβγ protein scavenger (Figs. 2A and 2C). While the late phase of U69,593-induced ERK1/2 phosphorylation was significantly diminished, the early phase was not affected upon β-arrestin 2 silencing (see curve labeled “G protein dependent” in figure 2C). These results suggest that U69,593 activation of KOR results in an early phase of ERK1/2 activation that is mainly due to a Gβγ-mediated pathway accompanied by a late phase which is driven predominantly by β-arrestin 2.
To demonstrate an agonist-induced interaction between KOR and β-arrestin 2, we utilized a β-arrestin 2-GFP (βarr2-GFP) translocation assay in HEK-293 cells expressing an HA-tagged KOR (Barak et al. 1997; Groer et al. 2007). In figure 2D, we demonstrate that U69,593, salvinorin A and MOM-Sal-B can induce β-arrestin 2 recruitment to the plasma membrane within 5 minutes of stimulation.
MOM-Sal-B stimulated only the early phase of ERK1/2 activation in the immortalized astrocytes and accordingly, CD8-βARK-C diminished MOM-Sal-B (5 min) induced ERK1/2 phosphorylation by 59% in these cells (Fig. 2E) demonstrating the importance of the Gβγ subunit in this early signaling event. However, siRNA targeting βarrestin-2 also attenuated the MOM-Sal-B activation of ERK at the early time point, suggesting that in response to this ligand, the early phase consists of additive G protein- and βarrestin-2-mediated ERK phosphorylation (Fig. 2F).
To further establish that the early phase of ERK phosphorylation is transduced by a G protein dependent mechanism, we treated the cells with the Gi/o inhibitor, PTX. Accordingly, PTX attenuated primarily the early phase of U69,593 induced ERK phosphorylation (Fig. 3A). Surprisingly, PTX did not affect MOM-Sal-B stimulation of ERK phosphorylation (Fig. 3B). Therefore, it appears that only U69,593 utilizes a PTX-sensitive Gi/o α protein. Since the Gβγ scavenger diminished MOM-Sal-B stimulated ERK phosphorylation (Fig. 2F), the data suggest that MOM-Sal-B is acting via a PTX insensitive G protein. There is precedent for opioid signaling to ERK via PTX insensitive G proteins (Belcheva et al. 2000; Bruchas et al. 2007). In addition, PTX-insensitive mediation by Gβγ subunits of serotonin signaling to phospholipase D has been reported (McGrew et al. 2002).
Fig. 3. Attenuation of κ opioid-induced stimulation of ERK1/2 phosphorylation by PTX in type-1 immortalized astrocytes.
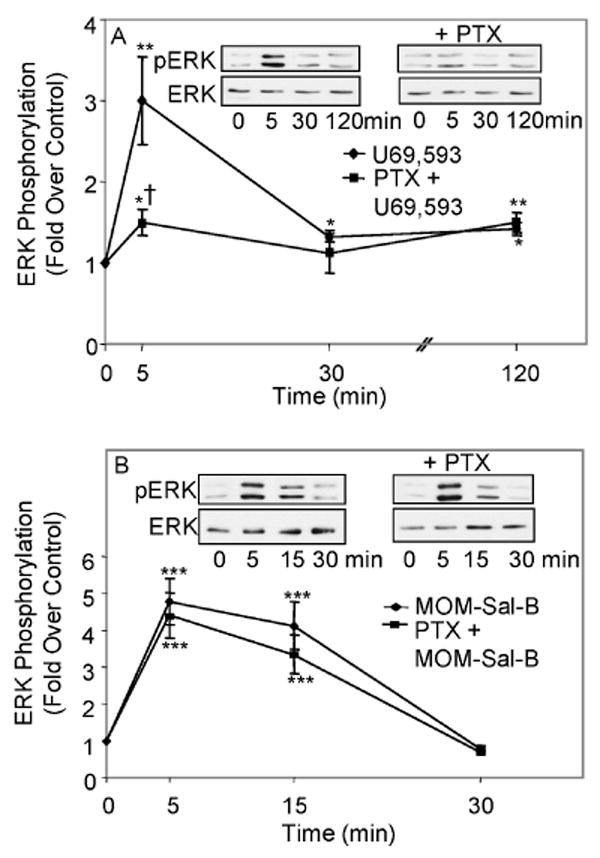
A. Effect of PTX on U69,593 signaling. Cells were transiently transfected with KOR cDNA, serum starved for 24 h in the presence of 100 ng/mL PTX and treated with 1 μM U69,593 for the indicated time intervals and ERK1/2 phosphorylation was assayed. N=4-10 *p< 0.05, **p< 0.01 vs. control; †p< 0.05, vs. U69,593 alone at the same time point. B. Lack of effect of PTX on MOM-Sal-B signaling. Cells were transiently transfected with KOR cDNA, serum starved for 24 h in the presence of 100 ng/mL PTX and treated with 1 μM MOM-Sal-B for the indicated time intervals and ERK1/2 phosphorylation was assayed. N=4-11. ***p< 0.001 vs. control.
Effects of κ opioids on immortalized astrocyte proliferation as measured by BrdU counting
Having obtained evidence for two mechanisms of ERK1/2 activation, the question arises as to the physiological outcome of each. Since opioid regulation of glial cell proliferation has been well established during CNS development and injury (Hauser and Stiene-Martin 1991; Stiene-Martin et al. 1991; Barg et al. 1993; Barg et al. 1994; Hauser and Mangoura 1998; Xu et al. 2007), we focused on the impact of the blockade of each of these pathways on astrocyte proliferation. When proliferation of type I immortalized astrocytes transfected with KOR was measured by BrdU counting after vehicle or U69,593 treatment, the κ agonist elicited a 1.6 fold increase in proliferation that was abolished by U0126 indicating the dependency of ERK1/2 (Fig. 4A). U69593-induced proliferation was also blocked by β-arrestin 2 targeting siRNA (Fig. 4B). CD8-βARK-C overexpression resulted in a 52% decrease in U69,593 stimulation of immortalized astrocyte proliferation (Fig. 4C). These results indicated that both ERK1/2-mediated pathways contribute to U69,593-induced immortalized astrocyte proliferation.
Fig. 4. Inhibition of κ opioid-induced stimulation of type-1 immortalized astrocyte proliferation.
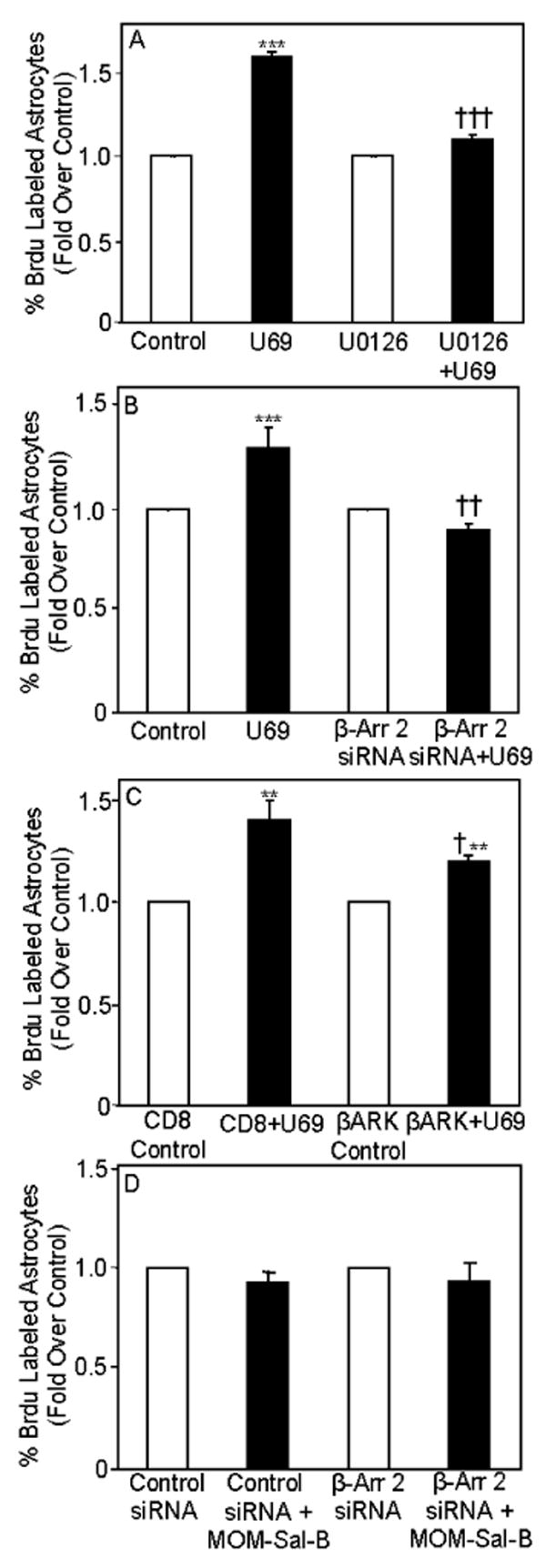
A. Effect of U0126 on U69,593 signaling. Cells were transiently transfected with KOR cDNA, starved for 28 h and treated for the last 24 h with 1 μM U69,593 and 1 μM U0126. BrdU was added for the last 20-30 min of treatment. In these studies >2500 cells were counted for each determination. N=4 ***p< 0.001 vs. control; †††p< 0.001 vs. U69,593 treated cells. B. Effect of β-arrestin 2 siRNA on U69,593 signaling. Cells were transiently transfected with KOR cDNA +/- non-targeting or β-arrestin 2 siRNA. After 24 h on growth medium, they were starved for 28 h and treated for the last 24 h with 1 μM U69,593. BrdU was added for the last 20-30 min of treatment. In these studies ≥850 cells were counted for each determination. N=3-5 ***p< 0.001, ††p< 0.01 vs. control; C. Effect of CD8-βARK-C on U69,593 signaling. Cells were transiently transfected with KOR +/- CD8 or CD8-βARK-C cDNA. After 24 h on growth medium, they were starved for 28 h and treated for the last 24 h with 1 μM U69,593. BrdU was added for the last 20-30 min of treatment. In these studies ≥850 cells were counted for each determination. N=3; **p<0.01 vs. control; †p< 0.01 vs. their CD8 control in the presence of U69,593. D. Effect of β-arrestin 2 siRNA on MOM-Sal-B signaling. Cells were transiently transfected with KOR cDNA and control, non-targeting or β-arrestin 2 siRNA). After 24 h on growth medium, they were starved for 28 h and treated for the last 24 h with 1 μM MOM-Sal-B. BrdU was added for the last 20-30 min of treatment. In these studies ≥3000 cells were counted for each determination. N=3-4.
Interestingly, MOM-Sal-B failed to stimulate proliferation of the immortalized astrocytes in the absence or presence of β-arrestin 2 siRNA suggesting that the late phase β-arrestin 2-dependent pathway may be required for KOR-induced proliferation in this cell line (Fig. 4D).
KOR stimulation of ERK and cell proliferation in primary rat astrocytes
Since the cellular environment can impact the receptor signaling potential as much as the ligand, we tested whether the signaling and physiological effects imparted by the KOR were conserved in primary astrocyte cultures. The use of primary cultures of astrocytes, may serve as a more physiologically relevant model system in which to study the mechanism of KOR-mediated effects on cell growth.
In primary astrocytes, both U69,593 and MOM-Sal-B, activated ERK1/2/MAPK via endogenous KOR by a rapid (min) initial stimulation that persisted up to 2 h (Fig. 5A & B). The long duration of MOM-Sal-B activation of ERK1/2 contrasted with its transient action in immortalized astrocytes (Fig. 1B). Cell type specific variations in the duration of GPCR ligand-induced ERK1/2 activation have been reported previously (Shah et al. 2003). The parallel comparison of the two astrocytic culture systems suggests that even two highly related cell types can undergo changes in their cellular environment sufficient to alter temporal aspects of the downstream signaling targets such as that seen with KOR-mediated ERK phosphorylation. The KOR-selective antagonist NorBNI also blocked MOM-Sal-B induced ERK1/2 activation in primary astrocytes (Fig. 5C) demonstrating the actions at KOR. However, unlike the results with immortalized astrocytes, norBNI did not completely abolish activity. Nevertheless, we did not observe a statistically significant change in ERK phosphorylation when using MOM-Sal-B in the presence of norBNI from the controls in the presence of norBNI alone (p<0.07) Although unlikely, this partial inhibition may reflect either a need for higher concentrations of norBNI to completely eliminate MOM-Sal-B activity when using primary astrocytes or there may be a small KOR-independent action of MOM-Sal-B.
Fig. 5. Time course of MOM-Sal-B and U69,593 modulation of ERK1/2 phosphorylation in type-1 and type-2 primary rat astrocytes.
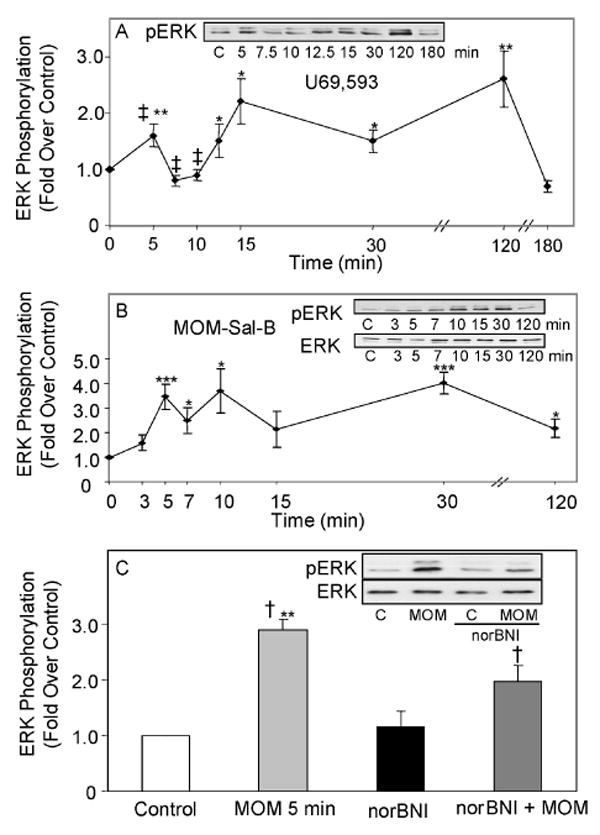
Cells were treated with 1 μM U69,593 (A) or 1 μM MOM-Sal-B (B) for specific time intervals. U69,593: N=7-12. MOM-Sal-B: N=5-6. *p < 0.05, **p< 0.01, ***p<0.001 vs. control, ‡p<0.05, the 5 min peak is significantly different from the 7.5 and 10 min points. C. Cells were pre-treated with 1 μM NorBNI for 1 h, followed by a 5 min treatment with 1 μM MOM-Sal-B. N=4. **p< 0.01 vs. control. †p< 0.05 vs. agonist alone. The gels are representative immunoblots showing phosphorylated ERK1/2 and total ERK1/2 bands. The graphs and curves show quantified ERK1/2 phosphorylation based on phosphoERK1/2 to ERK1/2 ratios.
We then utilized the BrdU incorporation assay to assess cell growth in the primary cultures. Due to the presence of both type-1 and type-2 cells in the cultures generated under our growth conditions, GFAP immunocytochemical studies were performed on cells labeled with BrdU. As seen in figure 6A (far right panel), the primary astrocyte cultures were heterogeneous in that they contained GFAP positive type-1 astrocytes that are flat polyhedral shaped cells and type-2 that are spindle shaped and possess two or more processes (Raff et al. 1983). A small number of stellate shaped GFAP positive cells that contain many processes were also observed. Type-1 and type-2 cells were counted on the basis of their differences in morphology. The ratios of type-1 and 2 cells were generally about 1-3:1 in the P1 rat cortical astrocytes under our experimental conditions.
Fig. 6. Inhibition of κ opioid-induced stimulation of type-1 and type-2 primary astrocyte proliferation.
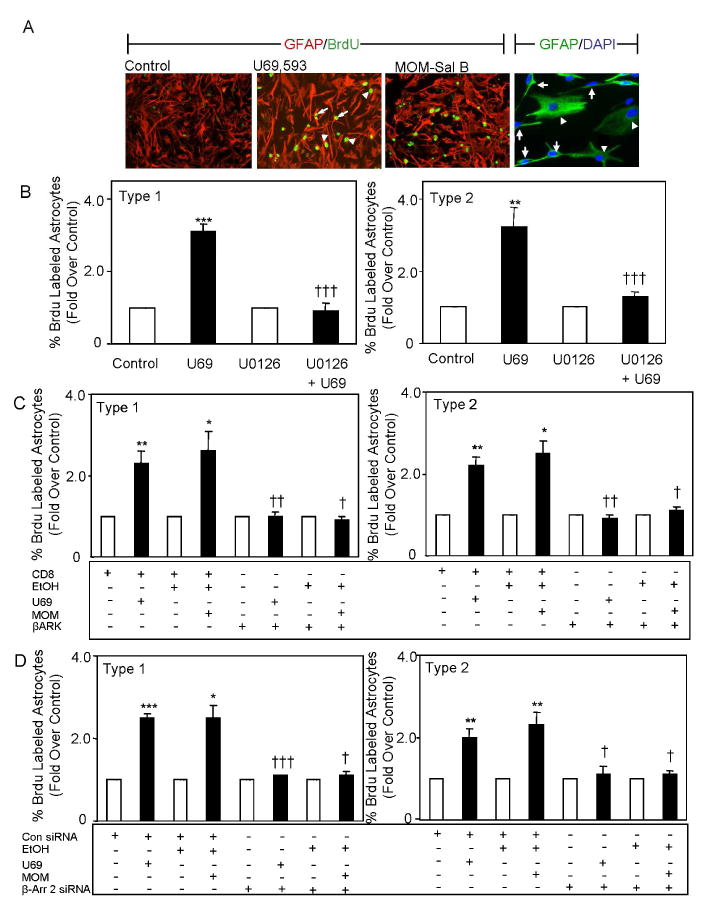
A. Micrograph of GFAP and BrdU stained astrocytes. Cells were starved for 28 h, treated for the last 24 h with 1 μM U69,593 or 1 μM MOM-Sal-B and BrdU was added for the last 4 h of treatment. Primary Abs: mouse anti-BrdU monoclonal Ab (1:10 dilution) and rabbit anti-GFAP polyclonal Ab (1:1 dilution). Secondary Ab: Green fluorescence emitting fluorescein-conjugated anti-mouse IgG Ab (1:10 dilution) and Alexa Fluor 594 goat anti-rabbit IgG (1:700 dilution). Cells were designated type-1 or type-2 based on their morphology. Arrowheads show type-1 and arrows show type-2 astrocytes in the far right panel. B. Effect of U0126 on U69,593 signaling. Cells were starved 28 h, treated for the last 24 h with 1 μM U69,593 ± 1 μM U0126 and BrdU was added for the last 4 h of treatment. In these studies ≥1100 cells were counted for each determination. N=5. **p<0.01, ***p<0.001 vs. control. †††p<0.001 vs. U69,593 without inhibitor. C. Effect of CD8-βARK-C on U69,593 and MOM-Sal-B signaling. Cells were transfected with CD8 or CD8-βARK-C cDNA, starved 28 h and treated for the last 24 h with 1 μM U69,593 or 1 μM MOM-Sal-B. BrdU was added for the last 4 h of treatment. In these studies ≥650 cells were counted for each determination. N=3. *p<0.05, **p<0.01 vs. control. †p<0.05, ††p<0.01 vs. their CD8 control in the presence of U69,593. D. Effect of β-arrestin 2 siRNA on U69,593 and MOM-Sal-B signaling. Cells were transfected with KOR cDNA +/- non-targeting or β-arrestin 2 siRNA). After 24 h on growth medium, they were starved for 28 h and treated for the last 28 h with 1 μM U69,593 or 1 μM MOM-Sal-B. In these studies ≥390cells were counted for each determination. N=3. *p<0.05, **p<0.01, ***p<0.001 vs. control. †p≤0.05, †††p<0.001 vs. non-targeting siRNA control in the presence of U69,593.
BrdU labeling studies revealed that both U69,593 and MOM-Sal-B stimulate proliferation in primary type-1 and type-2 astrocytes by 2-3 fold (Fig. 6B&C). Also the κ agonists elicited a greater increase in proliferation of primary type-1 and type-2 astrocytes than in immortalized type-1 astrocytes. The MEK1 inhibitor U0126 essentially abolished the proliferative actions of both κ agonists indicating that ERK1/2 mediates their activity. Both κ agonists elicit persistent ERK1/2 activation in these cells (Fig. 5A&B), consistent with the notion that sustained ERK1/2 activation is required for type-1 and 2 primary astrocyte proliferation.
CD8-βARK-C overexpression resulted in a complete inhibition of U69,593 and MOM-Sal-B stimulation of primary type-1 and type-2 astrocyte proliferation (Fig. 6C). These results implicate Gβγ in the mechanism of κ agonist-induced proliferation of primary astrocytes. Furthermore, siRNA targeting β-arrestin 2 also abolished MOM-Sal-B elicited proliferation (Fig. 6D). Taken together, our findings suggest that the G protein and β-arrestin mediated signaling pathways are likely not additive and may be distinct.
Discussion
Here we found that both κ opioid agonists, U69,593 and MOM-Sal-B, activated ERK/MAPK via KOR in immortalized and primary astrocytes. Time course studies of ERK1/2 phosphorylation, revealed that U69,593 initiated a rapid (min) activation that was sustained for at least 2 h in immortalized and primary astrocytes. In contrast, MOM-Sal-B activated ERK1/2 transiently (<30 min) in immortalized astrocytes but persistently in primary astrocytes. Nevertheless, both U69,593- and MOM-Sal-B-induced ERK1/2 phosphorylation was inhibited by a Gβγ scavenger and β-arrestin 2 targeting siRNA. PTX inhibition of the early phase of ERK activation by U69,593 supports the data gained with the Gβγ scavenger. In addition, the confocal microscopy data provides evidence that the agonists can induce β-arrestin 2-receptor interactions and together with the siRNA knockdown of β-arrestins 2 lends evidence to support β-arrestin 2-mediated signaling by KOR. Therefore, a multiplicity of κ opioid signaling pathways to ERK may exist in astrocytes.
Since U69,593 differs from MOM-Sal-B in its ability to induce sustained ERK activation in immortalized astrocytes, our results are also consistent with the notion of functional selectivity (Mukhopadhyay and Howlett 2005; for a review see Urban et al. 2007). Furthermore, the non-nitrogenous ligand is capable of selectively stimulating PTX-insensitive G protein and a transient β-arrestin 2 dependent signaling to ERK in immortalized astrocytes but it activates both G protein and sustained β-arrestin 2 dependent ERK signaling in primary astrocytes. However, in this instance cellular environment may be a more important factor as MOM-Sal-B appears to be more functionally selective in immortalized than in primary astrocytes. This is keeping with recent signaling data demonstating unique binding properties of members of the salvinorin family (Groer et al. 2007; Vortherms et al. 2007; Tidgewell et al. 2008).
There is also precedent for μ opioid signaling to ERK via PTX insensitive G proteins that is cell type specific (Belcheva et al. 2000). In COS-7 cells, DAMGO stimulates ERK phosphorylation via a PTX-insensitive mechanism, possibly entailing G(z) and G(12). In contrast, we have found that U69,593 stimulation of ERK phosphorylation in an astrocytoma model (C6 glioma cells) is PTX sensitive whereas morphine is PTX insensitive (Barg et al. 1994; Bohn et al. 2000). This effect of cellular environment may be attributed to differences of ligand signaling in astrocytes versus neurons and in cells from different brain regions. In recent studies of κ opioid stimulation of JNK kinase phosphorylation in HEK293 cells, it was found that the elevation of phosphoJNK kinase by U50,488 was PTX sensitive (Bruchas et al. 2007). The κ antagonist, norBNI also increased JNK kinase phosphorylation, but by a PTX insensitive mechanism. As the authors point out, there is precedent for functional selectivity in which a ligand can display agonist and antagonist properties (see also Urban et. al. 2007). However, the norBNI stimulation of JNK kinase phosphorylation was detected in both HEK293 cells and in vivo, thereby arguing against cell type specificity. Nevertheless, both our results and the norBNI data can be explained by G protein selectivity. The data shown here raise yet another parameter that must be considered in our attempts to understand functional selectivity, i.e. temporal aspects. The action of norBNI on JNK kinase phosphorylation was detected after 1 h treatment with the antagonist. It would be of interest to investigate the temporal profile of norBNI activation of JNK kinase. Moreover, in our studies and those of others, full understanding of functional selectivity will also depend upon delineation of all the features of the individual pathways leading to activation of MAP kinases. Differences in intermediary signaling components and the presence of endogenous receptor ligands in cells may impact on feedback mechanisms and ultimate outcomes.
Cortical astrogliosis has been found to be associated with neurodegenerative and other diseases such as autism, muscular dystrophy, HIV and dementia in drug users and Alzheimer's Disease (Bell et.al. 1998; Terai et al. 2001; Yang et al. 2007; Buffo et al. 2008; Fatemi et al. 2008). This proliferative action may be triggered for the purpose of structural reorganization as proposed for spinal cord injury (Xu et al. 2007). Astrogliosis may be beneficial or it may be a maladaptive feature ensuing under pathophysiological conditions. Another factor that contributes to the complexity of the role of astrogliosis in injury and disease is the observed increase in dynorphin expression that also has neuroprotective as well as maladaptive roles (Hauser et al. 2005). This endogenous KOR ligand can promote DNA synthesis in developing brain at a time when astrocytes are being synthesized (Gorodinsky et al. 1995). Moreover, the astrogliosis seen after partial sciatic nerve lesion is not detected in dynorphin null mice (Xu et al. 2007). Therefore, the integration of the multiple-input ERK1/2 signaling pathways that contribute to cell proliferation is an important question to address.
The total abolishment of primary astrocyte proliferation driven by U69,593 and MOM-Sal-B in the presence of U0126, CD8-βARK-C or β-arrestin 2 targeting siRNA raises the issue of redundancy (Fig. 6). However, the considerable redundancies recently seen in receptor tyrosine kinase and PI3K signaling pathways in gliomas result in only partial blockade of viability and colony number (Stommel et al. 2007). We have implicated EGFR and PI3K in immortalized astrocytes (Belcheva et al. 2003 and Belcheva et al. 2005) and partial inhibition of proliferation by CD8-βARK-C is evidenced in figure 5C. Since under similar conditions, CD8-βARK-C abolishes proliferation of primary astrocytes, it cannot be ruled out that both G protein and sustained β-arrestin 2 signaling play specific, integral roles required for the regulation of primary cell division. A recent report suggests that as many as 408 genes are involved in normal human fibroblast cell division (Bar-Joseph et al. 2008). Experiments are underway to delineate the mechanism further and to determine whether each pathway may affect an essential aspect of the complex process of primary astrocyte division.
Acknowledgments
Supported in part by grants from the National Institutes of Health DA-05412 (CJC) and DA-18860 (LMB). We thank Ciara C. Coleman and Jannie S. Serna for their excellent technical assistance.
The abbreviations used are
- Ab
antibody
- BrdU
5′-bromo-2′-deoxy-uridine
- DAMGO
[D-ala2,mephe4,gly-ol5] enkephalin
- DMEM
Dulbecco's modified Eagle's medium
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ERK1/2
extracellular signal-regulated kinase
- G-protein
GTP binding regulatory protein
- GPCR
G protein-coupled receptor
- HRP
horse radish peroxidase
- KOR
κ opioid receptor
- MAPK
mitogen activated protein kinase
- MOM-Sal-B
C(2)-methoxymethyl salvinorin B
- OR
opioid receptor
- PTX
pertussis toxin
- TBST
Tris-buffered saline + 0.2% Tween 20
References
- Ahn SK, Shenoy SK, Wei HJ, Lefkowitz RJ. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Bar-Joseph Z, Siegfried Z, Brandeis M, Brors B, Lu Y, Eils R, Dynlacht BD, Simon I. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc Natl Acad Sci USA. 2008;105:955–960. doi: 10.1073/pnas.0704723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak LS, Ferguson SSG, Zhang J, Caron MG. A β-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- Barg J, Belcheva MM, Rowinski J, Coscia CJ. κ-opioid agonist modulation of [3H]thymidine incorporation into DNA: evidence for the involvement of pertussis toxin-sensitive G protein-coupled phosphoinositide turnover. J Neurochem. 1993;60:1505–1511. doi: 10.1111/j.1471-4159.1993.tb03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg J, Belcheva MM, Zimlichman R, Levy R, Saya D, McHale RJ, Johnson FE, Coscia CJ, Vogel Z. Opioids inhibit endothelin-mediated DNA synthesis, phosphoinositide turnover, and Ca2+ mobilization in rat C6 glioma cells. J Neurosci. 1994;14:5858–5864. doi: 10.1523/JNEUROSCI.14-10-05858.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, Lefkowitz RJ. β-arrestin 1 and Gαq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem. 2005;280:8041–8050. doi: 10.1074/jbc.M412924200. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. Opioid modulation of extracellular signal-regulated protein kinase activity is Ras-dependent and involves G(βγ) subunits. J Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcheva MM, Wong YH, Coscia CJ. Evidence for transduction of μ but not κ opioid modulation of extracellular signal-regulated kinase activity by G(z) and G(12) proteins. Cell Signal. 2000;12:481–489. doi: 10.1016/s0898-6568(00)00095-4. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Szucs M, Wang DX, Sadee W, Coscia CJ. μ-Opioid receptor-mediated Erk activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Tan Y, Heaton VM, Clark AL, Coscia CJ. μ-Opioid transactivation and down-regulation of the epidermal growth factor receptor in astrocytes: implications for mitogen-activated protein kinase signaling. Mol Pharmacol. 2003;64:1391–1401. doi: 10.1124/mol.64.6.1391. [DOI] [PubMed] [Google Scholar]
- Belcheva MM, Clark AL, Haas PD, Serna JS, Hahn JW, Kiss A, Coscia CJ. μ and κ opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J Biol Chem. 2005;280:27662–27669. doi: 10.1074/jbc.M502593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesiada E, Razandi M, Levin ER. Egr-1 activates basic fibroblast growth factor transcription. Mechanistic implications for astrocyte proliferation. J Biol Chem. 1996;271:18576–18581. doi: 10.1074/jbc.271.31.18576. [DOI] [PubMed] [Google Scholar]
- Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS. Effect of neocortical involvement. Brain. 1998;121:2043–2052. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Belcheva MM, Coscia CJ. Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: Evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. κ Opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Yang T, Schreiber S, DeFino M, Kwan SC, Li S, Chavkin C. Long-Acting {kappa} Opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Crespo P, Cachero TG, Xu N, Gutkind JS. Dual effect of β-adrenergic receptors on mitogen-activated protein kinase. Evidence for a βγ-dependent activation and a Gαs-cAMP-mediated inhibition. J Biol Chem. 1995;270:25259–25265. doi: 10.1074/jbc.270.42.25259. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Astrocytes put down the broom and pick up the baton. Cell. 2006;125:639–641. doi: 10.1016/j.cell.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Fanton CP, McMahon M, Pieper RO. Dual growth arrest pathways in astrocytes and astrocytic tumors in response to Raf-1 activation. J Biol Chem. 2001;276:18871–18877. doi: 10.1074/jbc.M011514200. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Lee S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse. 2008;62:501–507. doi: 10.1002/syn.20519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ. Distinct β-Arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- Gorodinsky A, Barg J, Belcheva MM, Levy R, McHale RJ, Vogel Z, Coscia CJ. Dynorphins modulate DNA synthesis in fetal brain cell aggregates. J Neurochem. 1995;65:1481–1486. doi: 10.1046/j.1471-4159.1995.65041481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce μ opioid receptor--arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Stiene-Martin A. Characterization of opioid-dependent glial development in dissociated and organotypic cultures of mouse central nervous system: critical periods and target specificity. Brain Res Dev Brain Res. 1991;62:245–255. doi: 10.1016/0165-3806(91)90172-f. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Aldrich JV, Anderson KJ, Bakalkin G, Christie MJ, Hall ED, Knapp PE, Scheff SW, Singh IN, Vissel B, Woods AS, Yakovleva T, Shippenberg TS. Pathobiology of dynorphins in trauma and disease. Front Biosci. 2005;10:216–235. doi: 10.2741/1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Mangoura D. Diversity of the endogenous opioid system in development. Novel signal transduction translates multiple extracellular signals into neural cell growth and differentiation. Perspect Dev Neurobiol. 1998;5:437–449. [PubMed] [Google Scholar]
- Hooker JM, Xu Y, Schiffer W, Shea C, Carter P, Fowler JS. Pharmacokinetics of the potent hallucinogen, salvinorin A in primates parallels the rapid onset and short duration of effects in humans. Neuroimage. 2008;41:1044–1050. doi: 10.1016/j.neuroimage.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Jw, Feldheim DA, Chen DF. Ephrins as negative regulators of adult neurogenesis in diverse regions of the central nervous system. Proc Natl Acad Sci USA. 2008;105:8778–8783. doi: 10.1073/pnas.0708861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Cyclic AMP inhibits activation of mitogen-activated protein kinase and cell proliferation in response to growth factors in cultured rat cortical astrocytes. J Neurochem. 1996;67:2246–2255. doi: 10.1046/j.1471-4159.1996.67062246.x. [DOI] [PubMed] [Google Scholar]
- Lazarini F, Strosberg AD, Couraud PO, Cazaubon SM. Coupling of ETB endothelin receptor to mitogen-activated protein kinase stimulation and DNA synthesis in primary cultures of rat astrocytes. J Neurochem. 1996;66:459–465. doi: 10.1046/j.1471-4159.1996.66020459.x. [DOI] [PubMed] [Google Scholar]
- Lee DY, Karnati VV, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon WA, Jr, et al. Synthesis and in vitro pharmacological studies of new C(2) modified salvinorin A analogues. Bioorg Med Chem Lett. 2005;15:3744–3747. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for β-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Lenz G, Goncalves D, Luo ZJ, Avruch J, Rodnight R, Neary JT. Extracellular ATP stimulates an inhibitory pathway towards growth factor-induced cRaf-1 and MEKK activation in astrocyte cultures. J Neurochem. 2001;77:1001–1009. doi: 10.1046/j.1471-4159.2001.00299.x. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MP. Morphine modulates chemokine gene regulation in normal human astrocytes. Clin Immunol. 2005;115:323–332. doi: 10.1016/j.clim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- McGrew L, Chang MSS, Sanders-Bush E. Phospholipase D activation by endogenous 5-hydroxytryptamine 2C receptors is mediated by Gα13 and pertussis toxin-insensitive Gβγ subunits. Mol Pharmacol. 2002;62:1339–1343. doi: 10.1124/mol.62.6.1339. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Howlett AC. Chemically distinct ligands promote differential CB1 cannabinoid receptor-Gi protein interactions. Mol Pharmacol. 2005;67:2016–2024. doi: 10.1124/mol.104.003558. [DOI] [PubMed] [Google Scholar]
- Muroyama Y, Fujiwara Y, Orkin SH, Rowitch DH. Specification of astrocytes by bHLH protein SCL in a restricted region of the neural tube. Nature. 2005;438:360–363. doi: 10.1038/nature04139. [DOI] [PubMed] [Google Scholar]
- Nishida H, Okabe S. Direct astrocytic contacts regulate local maturation of dendritic spines. J Neurosci. 2007;27:331–340. doi: 10.1523/JNEUROSCI.4466-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Radany EH, Brenner M, Besnard F, Bigornia V, Bishop JM, Deschepper CF. Directed establishment of rat brain cell lines with the phenotypic characteristics of type 1 astrocytes. Proc Natl Acad Sci USA. 1992;89:6467–6471. doi: 10.1073/pnas.89.14.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Abney ER, Cohen J, Lindsay R, Noble M. Two types of astrocytes in cultures of developing rat white matter: differences in morphology, surface gangliosides, and growth characteristics. J Neurosci. 1983;3:1289–1300. doi: 10.1523/JNEUROSCI.03-06-01289.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riboni L, Viani P, Bassi R, Stabilini A, Tettamanti G. Biomodulatory role of ceramide in basic fibroblast growth factor-induced proliferation of cerebellar astrocytes in primary culture. GLIA. 2000;32:137–145. doi: 10.1002/1098-1136(200011)32:2<137::aid-glia30>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Riboni L, Viani P, Bassi R, Giussani P, Tettamanti G. Basic fibroblast growth factor-induced proliferation of primary astrocytes- evidence for the involvement of sphingomyelin biosynthesis. J Biol Chem. 2001;276:12797–12804. doi: 10.1074/jbc.M011570200. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Shah BH, Farshori MP, Jambusaria A, Catt KJ. Roles of Src and epidermal growth factor receptor transactivation in transient and sustained Erk1/2 responses to gonadotropin-releasing hormone receptor activation. J Biol Chem. 2003;278:19118–19126. doi: 10.1074/jbc.M212932200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Stiene-Martin A, Gurwell JA, Hauser KF. Morphine alters astrocyte growth in primary cultures of mouse glial cells: evidence for a direct effect of opiates on neural maturation. Brain Res Dev Brain Res. 1991;60:1–7. doi: 10.1016/0165-3806(91)90149-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- Terai K, Iwai A, Kawabata S, Sasamata M, Miyata K, Yamaguchi T. Apolipoprotein E deposition and astrogliosis are associated with maturation of beta-amyloid plaques in betaAPPswe transgenic mouse: Implications for the pathogenesis of Alzheimer's disease. Brain Res. 2001;900:48–56. doi: 10.1016/s0006-8993(01)02202-8. [DOI] [PubMed] [Google Scholar]
- Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, Hiemstra J, Partilla JS, Dersch CM, Rothman RB, Bohn LM, Prisinzano TE. Herkinorin analogues with differential beta-arrestin-2 interactions. J Med Chem. 2008;51:2421–2431. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo AK, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. β-arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277:9429–9436. doi: 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional Selectivity and Classical Concepts of Quantitative Pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Vortherms TA, Mosier PD, Westkaemper RB, Roth BL. Differential helical orientations among related G protein-coupled receptors provide a novel mechanism for selectivity: studies with salvinorin A and the κ-opioid receptor. J Biol Chem. 2007;282:3146–3156. doi: 10.1074/jbc.M609264200. [DOI] [PubMed] [Google Scholar]
- Wang LC, Baird DH, Hatten ME, Mason CA. Astroglial differentiation is required for support of neurite outgrowth. J Neurosci. 1994;14:3195–3207. doi: 10.1523/JNEUROSCI.14-05-03195.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by κ opioid activation of p38 mitogen-activated protein Kinase. J Neurosci. 2007;27:2570–2581. doi: 10.1523/JNEUROSCI.3728-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Roth BL. Salvinorin A: a novel and highly selective κ-opioid receptor agonist. Life Sci. 2004;75:2615–2619. doi: 10.1016/j.lfs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci USA. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang P, Xiong Y, Li X, Qi Y, Hu H. Ectopia of meningeal fibroblasts and reactive gliosis in the cerebral cortex of the mouse model of muscle-eye-brain disease. J Comp Neurol. 2007;505:459–477. doi: 10.1002/cne.21474. [DOI] [PubMed] [Google Scholar]