

Abstract
Growth factor binding to transmembrane protein receptors is generally understood to initiate cell signaling. Receptor binding of heparin-binding growth factors (HB-GFs), such as fibroblast growth factor-2 (FGF-2), is regulated by interactions with heparan sulfate proteoglycans. While there is some specificity for binding to heparan sulfate, overlap in sites for different growth factors may allow for cross regulation. Here we demonstrate, using experiments and computer simulations, that the HB-GFs FGF-2 and heparin-binding EGF-like growth factor (HB-EGF) can cross regulate receptor binding of the other despite having unique receptors. The ability of HSPG to stabilize HB-GF receptor binding is critical for competing growth factors to modulate receptor binding with both enhanced and reduced binding possible depending on this stabilization process. HSPG density and affinity for HB-GF are also critical factors for HB-GF cross regulation. Simulations further reveal that HB-GF can regulate receptor binding of non-HB-GFs such as EGF even when the two proteins share no binding sites when other HB-GF are present within the network. Proliferation studies demonstrate potentiation of HB-EGF-induced growth by FGF-2 indicating that competition networks can alter biological response. Exogenous manipulation of cellular responses to growth factors in complex living systems will require understanding the HSPG-controlled network.
Keywords: Receptors, Heparin, Fibroblast growth factor-2, Heparin-binding-EGF-like growth factor, Binding, Epidermal growth factor, Network, Growth factor
INTRODUCTION
Growth factors represent a large class of proteins that regulate cell proliferation, migration, differentiation, and survival/apoptosis. For most growth factors, signaling is mediated, in large part, via the binding and activation of cell surface receptor tyrosine kinases.11,13,26,33 In addition to binding to receptors, a large class of growth factors has been observed to also bind to heparan sulfate proteoglycans (HSPGs) on cell surfaces and within the extracellular matrix.4,15,25,32 The interaction of growth factors with HSPGs generally involves the binding to heparan sulfate (HS) chains, and in many instances these interactions have been demonstrated to modulate the cellular response to growth factors. For example, the ability of fibroblast growth factor-2 (FGF-2), the prototypical heparin-binding growth factor (HB-GF), to bind and activate its tyrosine kinase receptors is enhanced by HSPGs through the formation of stable high-affinity ternary complexes.23 Moreover, aspects of FGF-2 signaling have been demonstrated to be mediated by the HSPG syndecan-4 independent of FGF receptors.7,38,41 The cellular activity of other HB-GFs, such as heparin-binding epidermal growth factor-like growth factor (HB-EGF) and vascular endothelial growth factor (VEGF), has also been shown to be modulated by interactions with HSPGs.17,22 While it is well accepted that interactions with HSPGs play critical roles in mediating growth factor signaling to cells, it is not clear what defines the specificity of growth factor-HS interactions or how unique and exclusive those interactions are.25
Heparan sulfate represents a class of linear polysaccharides that are comprised of repeating 1-4 linked uronic acid-glucosamine disaccharide units that can be modified at several positions by sulfation, N-acetylation, and epimerization through a complex biosynthetic process.10,30,39 More than 200 extracellular proteins have been shown to bind with high-affinity to HS, and in many instances these interactions have been demonstrated to require distinct HS chemical characteristics.4,9 As one extreme example, the binding of antithrombin III to heparin and HS requires a specific pentasaccharide sequence containing a unique 3-O sulfate on the central glucosamine residue.1,2,27 However, the highly stringent requirements for antithrombin III binding to HS appears to be the exception, as most proteins show preferences for certain structures without clear requirements. Hence, most growth factor-HS interactions likely involve the binding to multiple structures within the array of HS chains presented on a particular cell.25 Moreover, while specific growth factors may have certain preferences for particular HS structures, they are also likely to show overlapping binding to sites containing characteristics that allow multiple growth factor interactions. Indeed, we have recently reported that FGF-2 and HB-EGF can compete for some but not all of the binding of one another to HSPG sites on vascular smooth muscle cells (SMCs).5 We noted that the majority of the HS binding sites, termed “common” binding sites, could bind both FGF-2 and HB-EGF. There were also HS-binding sites that were specific for either FGF-2 or HB-EGF, which were termed “unique” sites. The importance of these two classes of HS-binding sites with regard to signaling and receptor stabilization is unknown.
In the present study, we investigated the potential functions of “common” growth factor binding sites on HSPGs. Models of growth factor inter-competition were developed to evaluate the possibility that growth factors can influence one another even though they do not share tyrosine kinase receptors (Fig. 1). Specifically, models were established to describe the binding of FGF-2 and HB-EGF to their respective receptors as well as to “unique” and “common” HSPG sites using parameters determined with SMCs (Fig. 2). Simulations were run to investigate the influence of each growth factor on receptor binding of the other and the results were compared to experimental findings. Our results indicate that HB-GFs can influence the binding, activation, and biological response of other HB-GFs in a complex manner even when the growth factors do not share the same receptors. These studies indicate that the response of cells to a particular HB-GF is not only dependent on the concentration of the growth factor and the cellular expression level of its receptors, but also can be modulated by the presence of other HB-GFs as well as the particular array of HS structures present. We further show that in a complex environment when multiple factors may be present, HB-GFs can impact the binding of non-HB-GFs even when they do not share a common receptor. Accurate predictions of growth factor response within complex in vivo environments will require a detailed characterization of the levels of competing growth factors and an appreciation of their HS-binding properties.
FIGURE 1.
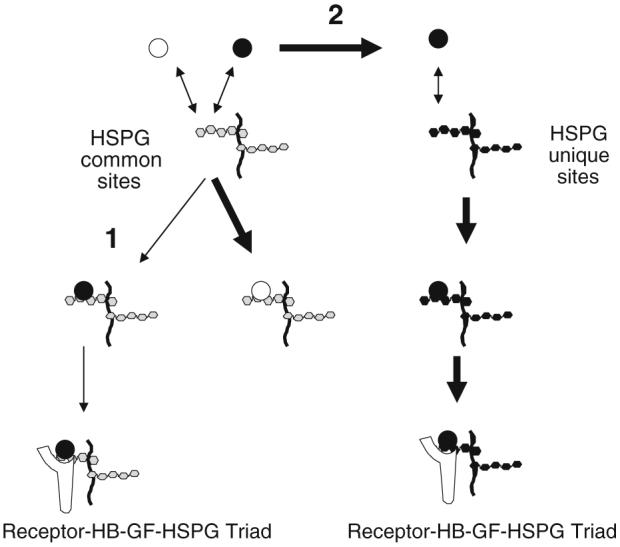
Competition model schematic. Binding of a heparin-binding growth factor (●) to its transmembrane protein receptor could be impacted by the presence of another heparin-binding growth factor (○) capable of competing for HSPG common sites. (1) Competition could result in binding of the competitor to the common sites (thick arrow) thus reducing ligand (●) binding to the common sites (thin arrow) and the subsequent ligand-mediated coupling with a receptor. (2) Competition could increase ligand (●) availability (thick arrow) for binding to HSPG unique sites and thus increase ligand-mediated coupling with receptors.
FIGURE 2.

Schematic diagram illustrating species within “non-receptor-coupling” and “receptor-coupling” models. Base model interactions show the ligand (i), receptor (R), ligand-receptor complex (C), unbound (P) and ligand bound (G) unique HSPG site, ternary complex (T), and ligand-receptor dimers (C2) with one (X) and two (T2) unique HSPG sites which are common to both models. Identical sets are included for each ligand i. In the “Non-receptor-coupling” model, the common HSPG sites (Pc) bind ligand (Gc) but do not interact with receptors. In the “receptor-coupling” model, the common HSPG (Pc) bind ligand (Gc) and form higher order complexes with receptors (Tc, Xc, , ). Specific reactions are shown in Table 1.
METHODS
Materials
Recombinant human FGF-2 was from Chiron, Inc. (Mountain View, CA, USA). Murine recombinant epidermal growth factor (EGF) was from InVitrogen Corp. (Carlsbad, CA, USA) and recombinant human HB-EGF was from R&D Systems (Minneapolis, MN, USA). 125I-FGF-2 and 125I-HB-EGF were prepared using a modified Bolton-Hunter procedure.6 125I-EGF was made using Iodobeads (Pierce Endogen; Rockford, IL, USA).40 [125I]-Bolton-Hunter and Na125I were from Perkin-Elmer (Boston, MA, USA). Heparinase III, from Flavobacterium heparinum, was a gift from Dr. E. Denholm at Biomarin Technologies (Montreal, Canada).
Cell Culture
Bovine aortic vascular SMCs were obtained from Coriell Cell Repositories (Camden, NJ, USA). For growth factor binding experiments, SMC (passages 6-15) were used at confluence and maintained as described previously.5-7,36,37 Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM-low glucose, Invitrogen), supplemented with 10% bovine calf serum (CS, Hyclone), penicillin (100 U/mL), streptomycin (100 μg/mL), and glutamine (2 mM). Cell number was determined with a Coulter Counter (Miami, FL, USA) and cell viability was determined by trypan blue exclusion.
Dominant Negative FGFR1 Mutant
Dominant negative FGFR1 (cytoplasmic domain deletion) expressing SMC were generated using retroviral infection as described.7 Briefly, the pLNCX2 viral vector was used to create retrovirus containing truncated FGFR1 (Δ) or with no insert (X2) and SMC were infected in the presence of polybrene overnight. Infected SMC were trypsinized and grown in media supplemented with G418 (500 μg/mL) for 4 days. At day 4, SMC (X2 and Δ) were trypsinized and plated for experiments or frozen in liquid nitrogen. Frozen stocks of SMC (X2 and Δ) were thawed and used for experiments after one passage.
Growth Factor Cell Surface Equilibrium Binding
Equilibrium cell surface binding of 125I-labeled growth factors was conducted essentially as described.5,6,12,18,19,36,37 SMC were plated at 75,000 cells/2 cm2 (24-well plate) and cultured for 2 days in DMEM supplemented with 10% CS, penicillin (100 U/mL), streptomycin (100 μg/mL), and glutamine (2 mM). In some experiments, cells were treated with and without heparinase III (0.1 unit/mL) for 60 min at 37 °C prior to conducting growth factor binding assays. Following heparinase treatment, the cells were washed once with binding buffer to remove the HS degradation products. In preparation for growth factor binding analysis, cells were washed once with ice-cold binding buffer (DMEM-low glucose, 0.05% gelatin, 25 mM HEPES, pH 7.4), and incubated on ice for 10 min in fresh cold binding buffer (0.5 mL/well) to inhibit internalization. Unlabeled and radiolabeled growth factors were added directly to the binding buffer and the cells were incubated for 2.5 h at 4 °C. Unbound growth factor was removed by washing the cells with ice-cold binding buffer three times. To release HSPG-bound growth factor, cells were subjected to a brief (10 s) high salt, neutral pH extraction (2 M NaCl, 20 mM HEPES, pH 7.4) followed by a PBS rinse.12,29,31 To remove receptor-bound growth factor, a high salt, low pH extraction (2 M NaCl, 20 mM sodium acetate, pH 4) was conducted for 5 min followed by a PBS rinse. 125I-radiolabeled growth factor was quantitated in an Auto-Gamma Cobra II series gamma counter (Packard Instruments, Meriden, CT, USA).
Cell Growth Studies
The growth of SMC (X2 and Δ) was evaluated by plating cells (500 cells/well) into 96 well plates in DMEM, supplemented with 5% CS, penicillin (100 U/mL), streptomycin (100 μg/mL), and glutamine (2 mM) (200 μl/well). After an overnight incubation, the medium was changed to DMEM, 0.5% CS, penicillin (100 U/mL), streptomycin (100 μg/mL) and glutamine (2 mM) plus and minus the following growth factors (EGF, HB-EGF, and FGF2) in the indicated combinations. The cells were allowed to grow for 7 days and the number of cells in each well was determined by assaying for acid phosphatase activity.8
Model Development
In this paper, we propose two models with alternative mechanisms for regulation of growth factor binding by “common” HSPG (Fig. 1). Both models are based on our previous efforts looking at FGF-2 regulation14 but were modified to include additional competing and non-competing growth factors (Fig. 2, Table 1). The models are based on mass-action kinetics describing binding and cell surface trafficking interactions for HB-GFs such as FGF-2 and HB-EGF. Binding to growth factor receptors and both unique and common HSPG sites were included as well as dimerization of bound receptors and receptor-unique site HSPG-growth factor triads and triad dimers. Dimerization and triad formation occurs in the model via cell surface diffusion and coupling. Coupling rates were assumed to be constant, reflecting diffusion-limited binding and both coupling and uncoupling rates were equivalent for all species.22 The value used was similar to that obtained recently by Gavutis et al.16 using simultaneous reflectance interferometry and total internal reflection fluorescence spectroscopy along with FRET to analyze tethered extracellular receptor domain interactions on solid-supported lipid bilayers. No coupling independent of growth factor was included nor were mixed growth factor dimers permitted within the model. A 1:1 stoichiometry of growth factor to receptor or HSPG site was maintained in both models. Common site HSPG are unable to interact with receptors in the “non-receptor-coupling model” but are able to form higher order complexes in the “receptor-coupling model.” Each site is considered independent with no clustering of sites as might be found on large heparan sulfate glycosaminoglycan chains. Synthesis of both receptors and HSPG are included as are internalization of receptors and HSPG, both free and ligand bound. The models are composed of a set of nonlinear ordinary differential equations and these are included in the Appendix. Simulations were run in Matlab R2006b (The Mathworks, Inc., Natick, MA, USA) using the stiff ordinary differential equation solver ode15s with the backwards differentiation formulas option and an absolute tolerance criteria of 1 × 10-20. Parameter values are listed in Table 2. Values were used as reported in the literature with no attempt to adjust based on temperature.
TABLE 1. Model reactions.
| HB-EGF | FGF-2 | EGF |
|---|---|---|
| Non-receptor-coupling model | ||
| (Same receptors as HB-EGF) | ||
| (No HSPG binding) | ||
| RH + H ↔ CH | RF + F ↔ CF | RH + E ↔ CE |
| PH + H ↔ GH | PF + F ↔ GF | |
| CH + CH ↔ C2H | CF + CF ↔ C2F | CE + CE ↔ C2E |
| C2H + PH ↔ XH | CF + PF ↔ XF | |
| RH + GH ↔ TH | RF + GF ↔ TF | |
| PH + RH ↔ TH | PF + RF ↔ TF | |
| TH + TH ↔ T2H | TF + TF ↔ T2F | |
| XH + PH ↔ T2H | XF + PF ↔ T2F | |
| Receptor-coupling model (additions to non-receptor-coupling model) | ||
| (Same common sites HSPG) | (No HSPG binding) | |
H = HB-EGF, F = FGF-2, E = EGF, S = synthesis, R = receptors, C = ligand-receptor complexes, P = HSPG, T = ligand-receptor-HSPG complexes, C2 = dimers of C, X = C2 bound to P, T2 = dimers of T. Unique receptors, proteoglycans and their ligand complexes are distinguished with a subscript H or F. The common site HSPG and their resulting complexes are designated with a superscript c. Neither model includes mixed complexes of HB-EGF or FGF-2.
TABLE 2. Parameters.
| Symbol | Value | Meaning |
|---|---|---|
| RH (t = 0) | 16,000 #/cell | # of HB-EGF receptors per cell at time zero(set to experimental value for FGFR)14 |
| RF (t = 0) | 16,000 #/cell | # of FGF-2 receptors per cell at time zero14 |
| PH (t = 0) | 28,000 #/cell | # of unique HB-EGF binding HSPG sites per cell at time zero6 |
| PF (t = 0) | 118,000 #/cell | # of unique FGF-2 binding HSPG sites per cell at time zero6 |
| Pc (t = 0) | 1,560,000 #/cell | # of common HB-EGF and FGF-2 binding HSPG sites per cell at time zero6 |
| kiRH (t = 0) | Synthesis rate of HB-EGF receptors | |
| kiRF (t = 0) | Synthesis rate of FGF-2 receptors | |
| kiPH (t = 0) | Synthesis rate of unique HB-EGF-binding HSPG | |
| kiPF (t = 0) | Synthesis rate of unique FGF-2-binding HSPG | |
| kiPc (t = 0) | Synthesis rate of common HSPG | |
| Ki | 0.005 min-1 | Internalization rate14 |
| Ki* | 0.078 min-1 | Internalization rate of dimerized receptor complexes14 |
| 7.1 nM | Dissociation constant for HB-EGF binding to receptors24 | |
| 9.7 × 107 M-1 min -1; | Association/dissociation rate constant for HB-EGF binding to receptors (set to experimental value for EGF22) | |
| 2.5 × 108 M-1 min-1; 0.048 min-1 | Association/dissociation rate constant for FGF-2 binding to receptors14 | |
| 6.1 nM | Dissociation constant for HB-EGF binding to unique HSPG6 | |
| 44 nM | Dissociation constant for HB-EGF binding to common HSPG6 | |
| 0.6 nM | Dissociation constant for FGF-2 binding to unique HSPG6 | |
| 12 nM | Dissociation constant for FGF-2 binding to common HSPG6 | |
| 0.068 min-1 | Dissociation rate constant for growth factor binding to unique and common HSPG | |
| Association rate constant for HB-EGF binding to unique HSPG | ||
| Association rate constant for HB-EGF binding to common HSPG | ||
| Association rate constant for FGF-2 binding to unique HSPG | ||
| Association rate constant for FGF-2 binding to common HSPG | ||
| kc,kuc | 0.001 (#/cell)-1 min-1, 1 min-1 | Coupling and uncoupling rate constants22 |
Statistics
Figures show the mean ± standard error of the mean (SEM) from triplicate samples from a single experiment. Experiments were repeated at least three times and data from one representative experiment are shown. Differences between treatment means were identified by onetailed Student’s t-test assuming equal variance with p values less than 0.05 considered significant.
RESULTS
Inclusion of FGF-2 Reduces HB-EGF Receptor Binding and Vice Versa
Heparin-binding growth factors such as FGF-2 and HB-EGF are characterized by their ability to bind heparin and cellular HSPGs. Growth factors bound to HSPGs can generally be released via incubation with a high salt solution capable of disrupting the critical ionic interactions while release of receptor bound growth factors often requires exposure to a low pH solution. Elimination of HS moieties with heparinase III or by treating cells with chlorate has been shown to reduce FGF-2 and HB-EGF binding to both HSPGs (salt releasable) and receptors (pH releasable).6 These same treatments do not alter binding of non-HB-GFs such as EGF (Fig. 3). Previously we have shown that both FGF-2 and HB-EGF bind to “unique” HSPG sites (specific for each growth factor) and to “common” sites that are capable of binding either growth factor, albeit with different affinities.6 Given that receptor binding is critical for growth factor-mediated cell signaling, we were interested in how the presence of competing HSPG-binding growth factors would affect receptor interactions. Cellular binding studies at 4 °C with radiolabeled growth factors in the presence of unlabeled growth factors were performed and the quantity of labeled growth factor bound to receptors was quantified (Fig. 4). 125I-FGF-2 receptor binding on SMC was reduced in the presence of HB-EGF while, in contrast, EGF had negligible effect on 125I-FGF-2 binding to its receptors. HB-EGF and EGF, but not FGF-2, share affinity for the same transmembrane receptor.34 125I-HB-EGF receptor binding was inhibited by EGF as expected, but was also reduced by FGF-2. 125I-EGF receptor binding was reduced by HB-EGF but not FGF-2. Thus, FGF-2 and HB-EGF, despite binding to distinct receptors, influence the binding of one another to their respective receptors.
FIGURE 3.
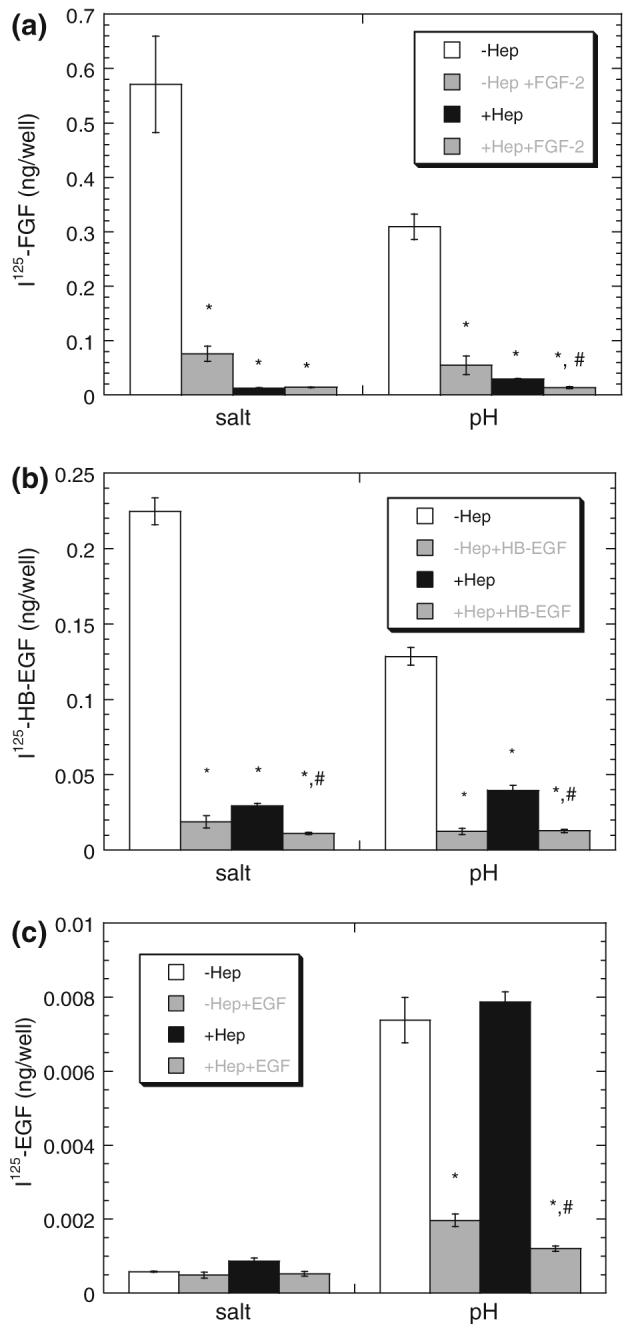
Heparinase reduces HSPG and receptor binding of HB-EGF and FGF-2 but not EGF. SMC were cultured, treated with heparinase (Hep) (0 (□) or (■) 0.1 units/mL), and incubated for 2.5 h with radiolabeled growth factor (0.28 nM 125I-FGF-2 (a), 0.42 nM 125I-HB-EGF (b), or 0.83 nM 125I-EGF (c)—corresponds to 5 ng/mL of each) as described in “Methods” section. Proteoglycan bound (‘salt’) and receptor-bound (‘pH’) growth factor were isolated using sequential washes. Radiolabeled growth factor bound in the presence of excess unlabeled growth factor (5 μg/mL) and released with respective wash is shown in the neighboring gray bar. Measurements (mean ± SE, n = 3) are representative of at least three independent experiments. *Treatment is significantly different than no heparinase (□) treatment within group (salt or pH).#Treatment is significantly different than heparinase (■) treatment (p < 0.05).
FIGURE 4.
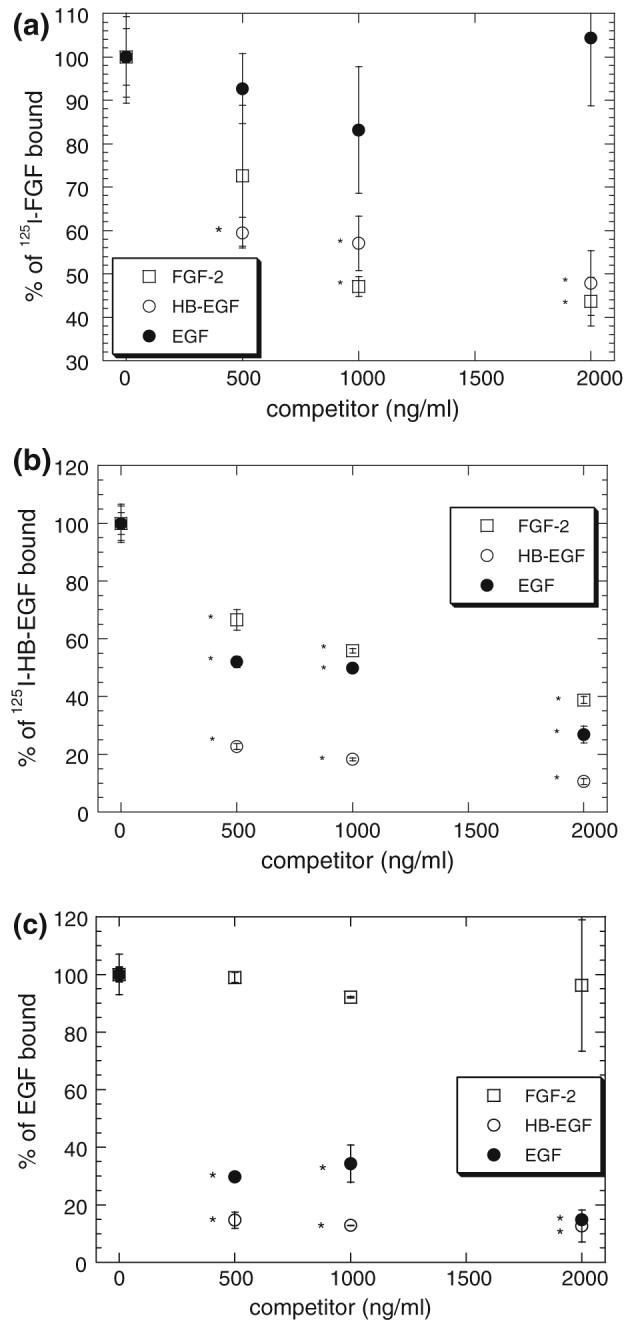
Effect of competitors on receptor binding. SMC Binding studies at 4 °C with 125I-FGF-2 (a), 125I-HB-EGF (b) and 125I-EGF (c) in the presence of FGF-2 (□), HB-EGF (○) and EGF (●) were performed as described in “Methods” section. Radiolabeled growth factor bound is plotted scaled to the amount of radiolabeled growth factor found in the absence of any competing unlabeled growth factor. All radiolabeled growth factors were used at a concentration of 5 ng/mL or 0.28 nM 125I-FGF-2 (a), 0.42 nM 125I-HB-EGF (b), or 0.83 nM 125I-EGF (c). Measurements (mean ± SE, n = 3) are representative of at least three independent experiments. *Significantly (p < 0.05) different from binding with no competitor.
Models Predict Opposing Effects on Receptor Binding
The binding of most HB-GFs to their specific receptors is modulated by interactions with HSPG; thus, we proposed that HSPG common sites could serve as a control element through which HB-GFs regulate other growth factor receptor binding. Two possible mechanistic scenarios were examined (described in “Methods” section, Fig. 1). In both models, cells express FGF receptors and HB-EGF/EGF receptors as well as “common” HSPG sites (i.e., sites that bind both FGF-2 and HB-EGF) and “unique” HSPG sites (i.e., sites that bind either FGF-2 or HB-EGF specifically) (Fig. 2). Unique sites were able to interact with receptors via growth factor coupling to form higher-affinity complexes. Treatment of the common sites is what differentiates the models. In the “non-receptor-coupling” model, common sites were not able to interact with receptors to form a higher-affinity complex while in the “receptor-coupling” model they were. Both models showed a reduction in receptor bound growth factor under 4 °C conditions (i.e., no synthesis or degradation of receptors or HSPG and no internalization—corresponding parameters were set to zero) following cell treatment with heparinase III (i.e., removal of both unique and common HSPG sites) as found experimentally (Fig. 3). Only the “non-receptor-coupling” model results are shown in Fig. 5a since results for the “receptor-coupling” model are qualitatively similar exhibiting the same trends with regard to plus and minus heparinase. While the overall reduction in HB-EGF receptor binding in the simulations was similar to that found experimentally, the FGF-2 reduction was much smaller than that found experimentally. We hypothesized that the FGF-2 model might be more sensitive to parameter values and ran simulations over a range of FGF-2 concentrations. As shown in Fig. 5b, higher reductions were found when the FGF-2 concentration was reduced indicating that the model could capture the salient features of the experiment qualitatively if not exactly quantitatively.
FIGURE 5.
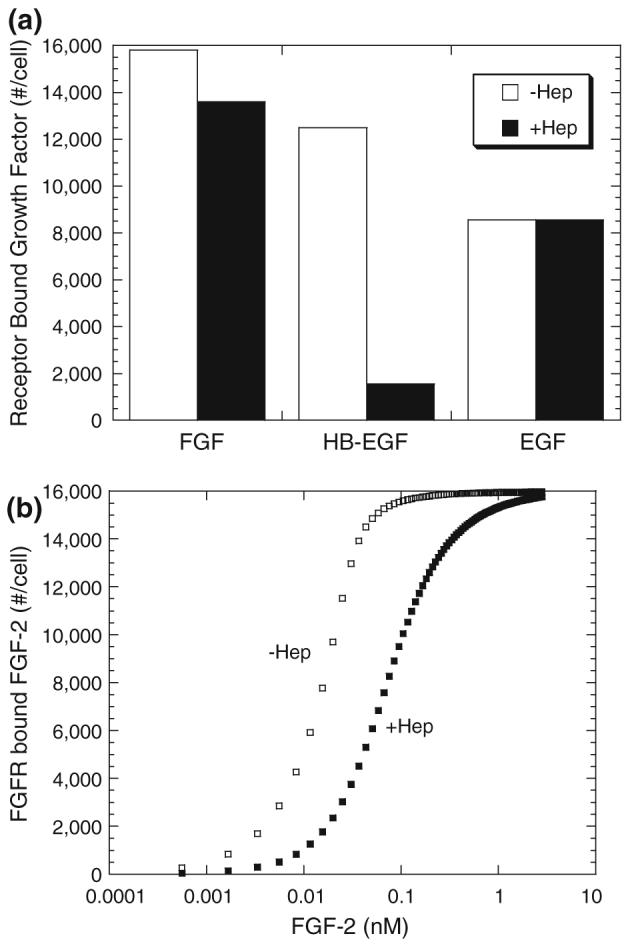
Heparinase Treatment reduces receptor binding for FGF-2 and HB-EGF (simulations). Simulations were performed using “non-receptor-coupling” model under 4 °C conditions (no synthesis or internalization of receptors or HSPG) and growth factor bound to receptor at 2.5 h under normal (□) and heparinase (■) conditions are shown. Simulations for (a) used 5 ng/mL or 0.28 nM FGF-2, 0.42 nM HB-EGF, and 0.83 nM EGF as used experimentally in Fig. 3. Receptor-bound FGF-2 as a function of FGF-2 treatment is shown for normal (□) and heparinase (■) conditions in (b).
We next investigated the effects of inclusion of FGF-2 on HB-EGF receptor binding for our two models. Simulations of HB-EGF binding at 4 °C were performed as a function of FGF-2 concentration using 5 ng/mL HB-EGF as in the experimental study (Fig. 4). With both models, inclusion of increasing concentrations of FGF-2 resulted in a decrease in HB-EGF bound solely to HSPG (Fig. 6). This would correspond experimentally to a decrease in the salt releasable HB-EGF and includes HB-EGF bound to both unique and common sites but not that bound to complexes including both HSPG and receptors. This decrease results from a reduction in HB-EGF binding to common sites due to competition from FGF-2. In contrast, the effect of FGF-2 on HB-EGF receptor binding differs markedly between the two models. With the “non-receptor-coupling” model, FGF-2 competition for common HSPG sites resulted in an increase in receptor-bound HB-EGF while a decrease was found with the “receptor-coupling” model. With the “non-receptor-coupling” model, only unique sites are able to interact with HB-EGF receptors thereby stabilizing the interaction. Common sites are at a high density, but lower affinity in our system and serve as a “sink” for HB-EGF in this model. The reduction in binding to the common sites due to FGF-2 competition facilitates binding of HB-EGF to the unique sites leading to receptor stabilization. With the “receptor-coupling” model, common sites are also able to interact with HB-EGF receptors and therefore FGF-2 competition for these sites leads to a decrease in HB-EGF bound receptors. Note however the difference in the scales and how the effects are more prominent with the receptor-coupling model. Using the raw simulation data from Fig. 6, with both models, more than 98% of the total HB-EGF bound to receptors is a triad or a triad dimer, a receptor-ligand complex stabilized via unique HSPG. With the “non-receptor-coupling” model, ∼53% of cell bound HB-EGF binds to HSPG alone in the absence of FGF-2 and this is reduced to ∼6.5% at high concentrations of FGF-2. In contrast, the “receptor-coupling” model has only ∼5% of cell bound HB-EGF bound to HSPG alone in the absence of FGF-2 and this drops to ∼3% at high concentrations of FGF-2. Similar effects of HB-EGF on FGF-2 binding were found for both models (data not shown) indicating that the phenomenon was not strictly dependent on the baseline set of parameters.
FIGURE 6.
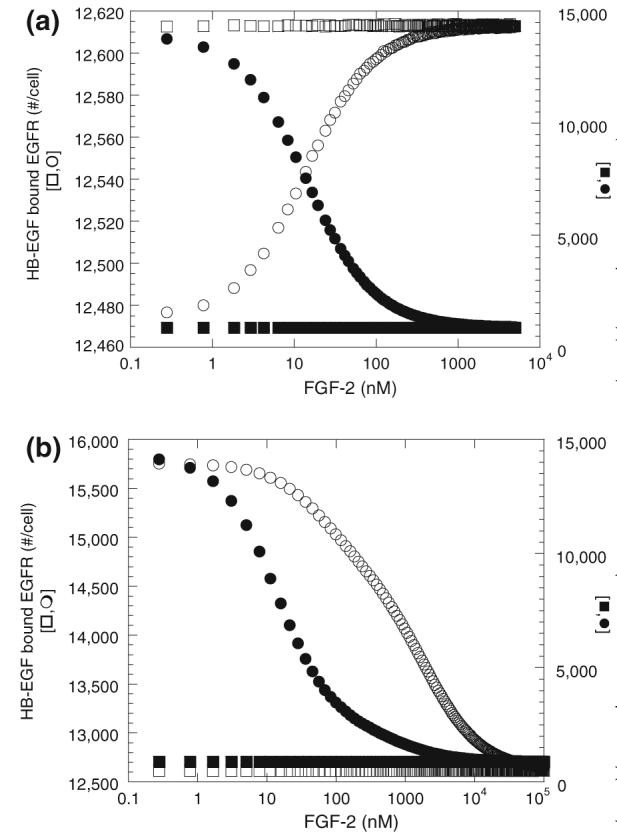
Effect of FGF-2 on HB-EGF binding (simulations)-HB-EGF (5 ng/mL or 0.42 nM) binding to SMC at 4 °C after 2.5 h incubation with FGF-2 for “non-receptor-coupling” model (a) and “receptor-coupling” model (b). HB-EGF bound to HSPG alone (salt releasable—●,■) and bound to EGF receptors (pH releasable—○,□) is shown. Results are from simulations with (circles) and without (squares) common HSPG sites.
HSPG Common Site Density Critical for Regulation of Receptor Binding
In order to maximize the effect of the competing growth factors on each other a lower concentration of HB-EGF (0.5 ng/mL) was selected for our parameter studies. We asked how the density of HSPG common sites affects HB-EGF-receptor binding. Low densities (<104 sites/cell) of common site, with a constant density of specific HSPG sites, had a negligible effect on HB-EGF receptor binding either in the presence or absence of FGF-2 (Fig. 7). At these levels, binding of HB-EGF to the common sites is not high enough to significantly impact receptor binding. With the “non-receptor-coupling” model, higher densities of common sites in the absence of FGF-2 resulted in decreased receptor binding due to sequestering of HB-EGF on HSPG sites not able to couple with receptors. Note that the experimentally measured value falls within this range.6 Addition of FGF-2 reduces HB-EGF binding to common sites and thus increases HB-EGF availability for binding to specific HB-EGF HSPG sites and receptors. In contrast, higher densities of common sites, which are capable of stabilizing HB-EGF receptor binding with the “receptor-coupling” model, results in increased receptor bound HB-EGF. FGF-2, by binding common sites, increases the HB-EGF available for HB-EGF specific sites and receptor binding but reduces the HSPG available for stabilized binding. With both models, FGF-2 counters the effect of increased common sites on HB-EGF receptor binding toward levels found in the absence of common sites. Again, the effects are more prominent for the “receptor-coupling” model as evident by the differences in scale. Similar effects are seen for FGF-2 receptor binding in the presence and absence of HB-EGF (data not shown). Specific receptors also altered the level of receptor-bound HB-EGF, particularly with the “non-receptor-coupling” model, but did not lead to differences in regulation by FGF-2 in either model than what has been shown at the baseline density (data not shown)
FIGURE 7.
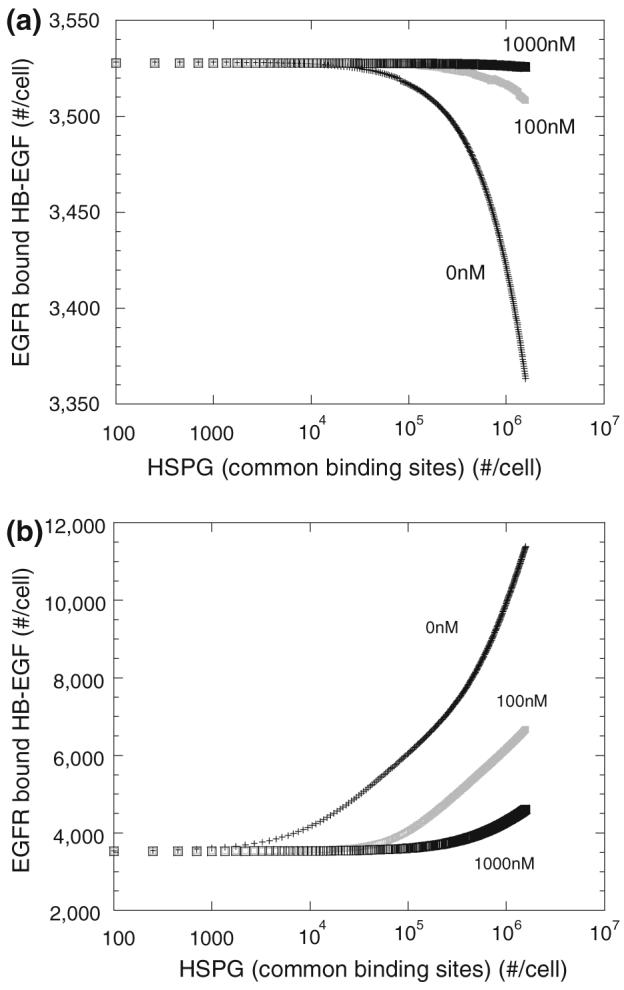
Effect of HSPG common sites density on HB-EGF receptor binding (simulations)—HB-EGF (0.5 ng/mL or 0.042 nM) bound to receptors after 2.5 h incubation at 4 °C with FGF-2 (0, 100, 1000 nM as listed on plot) for “non-receptor-coupling” (a) or “receptor-coupling” (b) model with varying density of HSPG common sites.
HSPG Affnity Ratio Impacts Regulation
Given the importance of the common site HSPG levels to FGF-2 regulation of HB-EGF receptor binding, we investigated how the relative affnity of the HSPG sites contributed to the competition. Maintaining the baseline receptor and HSPG common and unique sites densities, we varied the affnity of HB-EGF for the common sites. Note that the coupling rate was assumed to be independent of the species and this rate was maintained throughout all simulations. The effect of FGF-2 on HB-EGF binding is most pronounced when the affnity of the common sites is strong with only a minimal effect evident when the common site affnity for HB-EGF is an order of magnitude lower than that for the unique sites (i.e., ratio of KD (unique) to KD (common) is <0.1) (Fig. 8). This is particularly true with the “non-receptor-coupling” model where the effect of FGF-2 disappears as the affinity of HB-EGF for the common HSPG sites becomes small and hence binding to these sites becomes unlikely. The affinity ratio for HB-EGF to unique and common sites measured experimentally for our cell system was 0.14 putting it within the range for which a dose-dependent effect of FGF-2 should be evident. With the “receptor-coupling” model, the common sites can form high-affinity receptor complexes through coupling of an unbound common site with an HB-EGF bound receptor which is independent of the affinity of the growth factor for the HSPG site in our model (Table 2). Thus, even at essentially negligible affinity of HB-EGF for the common sites (i.e., ratio of KD(unique) to KD(common) approaching 0) the presence of FGF-2 impacts the receptor bound level of HB-EGF as the coupling rate within the model is independent of ligand affinity. We are, however, aware of no experimental evidence to either support or disprove this observation and do not suggest it is necessarily more than a function of our model structure at this extreme limit. We do note though that Shraga-Heled et al. recently showed that neuropilin modulate VEGF121 activity despite it not being a receptor for this isoform of VEGF suggesting receptor-coupling independent of ligand affinity can be biologically relevant.35
FIGURE 8.
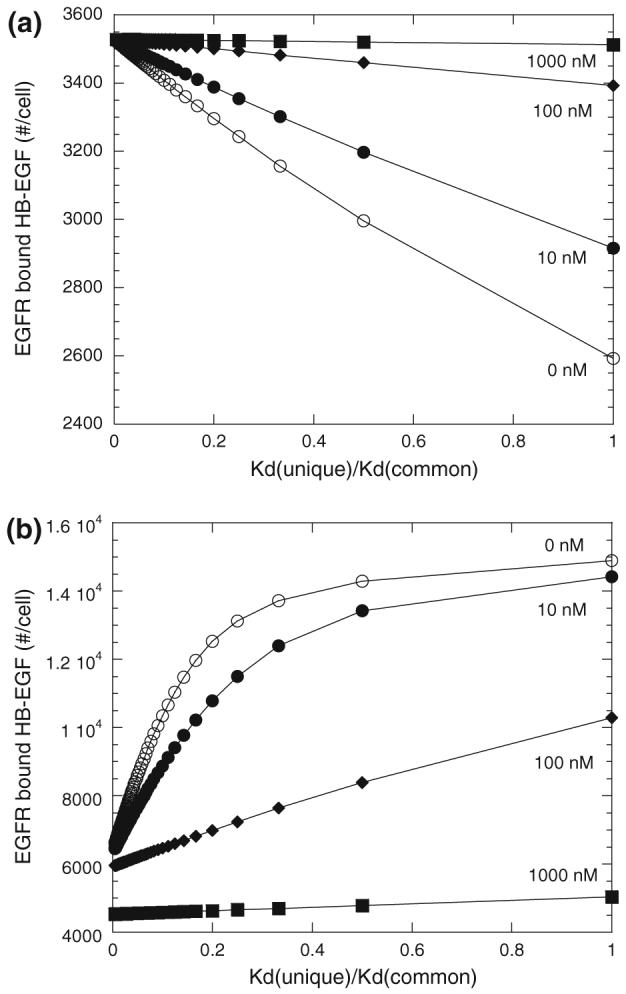
Effect of HB-EGF affinity for common site HSPG with FGF-2 added (simulations)—HB-EGF (0.5 ng/mL or 0.042 nM) bound to EGFR after 2.5 h incubation at 4 °C with FGF-2 (0 nM (○), 10 nM (●), 100 nM (◆), and 1000 nM (■) for “non-receptor-coupling” (a) or “receptor-coupling” (b) with varying affinity of HB-EGF for HSPG common sites. All other parameters as listed in Table 2.
FGF-2 Impacts EGF Binding in the Presence of HB-EGF
We next asked, using simulations, how FGF-2 might impact binding of EGF, a non-HB-GF, in the presence of HB-EGF, postulating that competition for common site HSPG would alter the availability of HB-EGF to compete for EGF receptors. In the absence of HB-EGF, FGF-2 had no effect on EGF binding with either model (Fig. 9), as would be expected. Moreover, in the absence of HSPG common sites, FGF-2 had no impact on EGF binding even in the presence of HB-EGF (data not shown). However, when model cells expressed HSPG common sites, FGF-2 impacted EGF binding in the presence of HB-EGF. With the “non-receptor-coupling” model, FGF-2 decreased EGF binding albeit the reduction was small compared to EGF binding in the presence of HB-EGF alone (∼3%). With the “receptor-coupling” model, FGF-2 increased EGF binding in the presence of HB-EGF and the effect was significant compared to binding in the presence of only HB-EGF (∼88%). With both models, HB-EGF at the concentration chosen (1 ng/mL or 0.083 nM) reduced EGF (1 ng/mL or 0.16 nM) receptor binding by ∼50% in the absence of HSPG common sites through direct competition of the two growth factors for the same receptor species (data not shown). Simulations performed at higher EGF and HB-EGF concentrations (5 ng/mL each) as used in earlier experimental figures yielded similar trends (∼99% and 77% reduction in EGF binding by HB-EGF for the “receptor-coupling” and “non-receptor-coupling” models, respectively, with high FGF-2 addition leading to an ∼80% reduction for both models).
FIGURE 9.
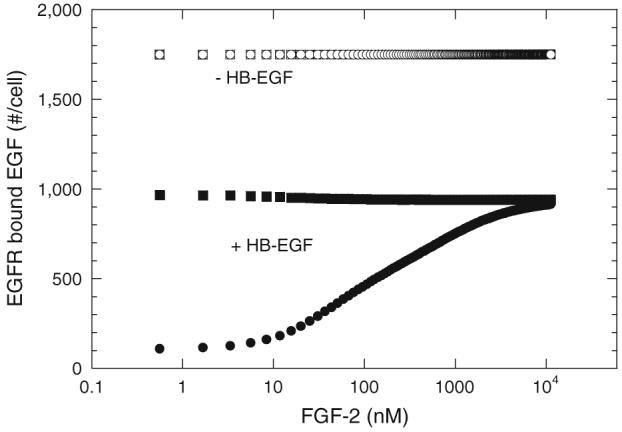
Effect of FGF on EGF binding in the presence of HB-EGF. EGF (1 ng/mL or 0.16 nM) bound to EGFR after 2.5 h incubation at 4 °C with (●, ■) and without (○, □) HB-EGF (1 ng/mL or 0.083 nM) for “non-receptor-coupling” (□, ■) and “receptor-coupling” (○,●) models. All other parameters as listed in Table 2. Note direct overlap of binding of EGF in the absence of HB-EGF for both models (□,○).
FGF-2 Enhances HB-EGF-induced Proliferation
Given the reduction in HB-EGF receptor binding found experimentally in the presence of FGF-2 (Fig. 3B), we hypothesized that the presence of FGF-2 would reduce HB-EGF-induced proliferation. This, however, was not the case (Fig. 10). Control SMCs (SMC-X2) or cells that have been engineered to express the dominant negative FGF receptor (SMC-Delta) were cultured in the presence of EGF, HB-EGF, and FGF-2 or combinations of these growth factors for 7 days and the proliferative effects evaluated. SMC-X2 responded to all three growth factors (FGF-2, EGF, and HB-EGF) with increased proliferation compared to control and an additive effect was evident when cells were cultured with FGF-2 in the presence of EGF or HB-EGF. In contrast, the growth of SMC-Delta cells was increased in response to EGF and HB-EGF but not to FGF-2 confirming the specific effect of the dominant negative FGF receptor expression on proliferation. Treatment of SMC-Delta cells with both EGF and FGF-2 showed no increased response compared to EGF alone while HB-EGF-induced proliferation was enhanced by the presence of FGF-2. While these data do not directly correlate with the ability of FGF-2 to reduce HB-EGF binding, they do indicate an ability of FGF-2 to modulate HB-EGF response possibly through HSPG competition.
FIGURE 10.
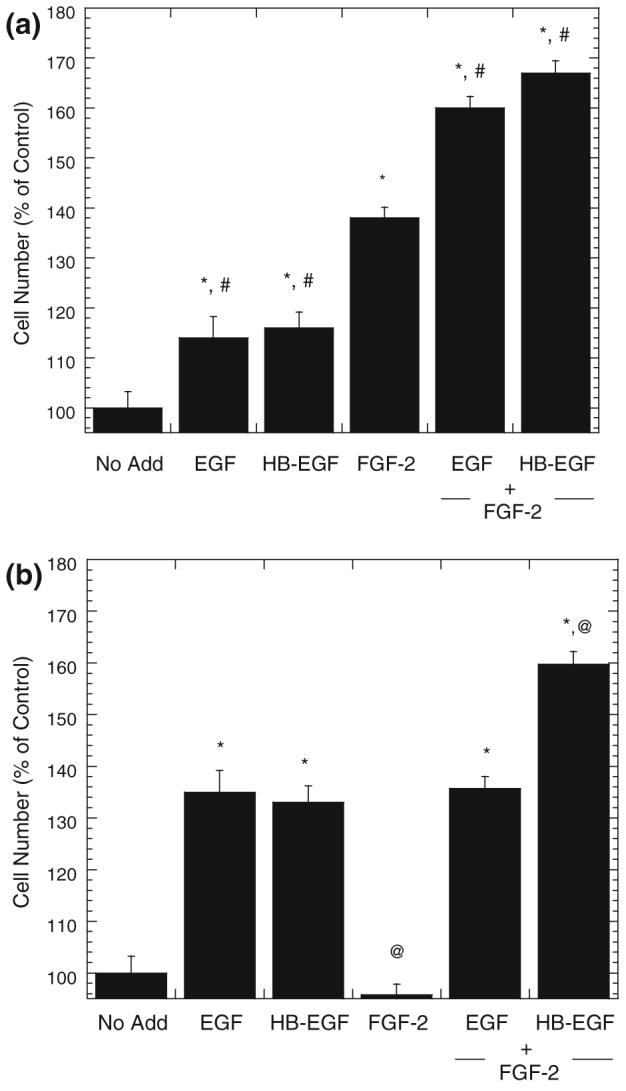
FGF-2 potentiates HB-EGF mitogenesis on FGF receptor-negative cells (SMC-Delta). SMC X2 (a) or SMC-Delta (b) were grown in DMEM, 4% CS for 7 days in the presence of EGF (2.5 nM), HB-EGF (2.5 nM), FGF-2 (60 nM) or combinations. Cell number was determined by acid phosphatase and is related to the no growth factor control (100%). Measurements (mean ± SE, n = 3) are representative of at least three independent experiments. *Significantly different than “no add”. #Significantly different than FGF-2 alone. @Significantly different than HB-EGF alone.
Impact of FGF-2 on HB-EGF Receptor Binding with Active Receptor Traffcking
Simulations from the “receptor-coupling” model agreed well with the experimental 4 °C growth factor binding data (Fig. 3B), yet reduced receptor binding would not generally predict the enhanced HB-EGF-mediated proliferation observed in the presence of FGF-2 at 37 °C (Fig. 10). Thus, simulations were performed including synthesis and internalization. It is possible that these cellular trafficking events could impact ligand, receptor, and HSPG site availability and hence alter regulation by FGF-2. All other parameter values were maintained at their baseline values. The levels of HB-EGF receptor dimers as a function of time with and without FGF-2 inclusion were evaluated (Fig. 11). We focused on HB-EGF receptor dimers since EGF receptor signal transduction is generally accepted to require dimerization.3 At high HB-EGF concentration (>1.0 nM), peak levels of HB-EGF receptor dimers occurred relatively quickly (≤10 min) with this level being reduced to ∼25% of peak level in <40 min (data not shown). FGF-2 had negligible effects on the overall process with minimal changes evident over the time frame studied. At high HB-EGF concentrations, there are sufficient HB-EGF molecules present such that competition for common sites by FGF-2 was not critical with regard to receptor binding and dimerization. At low HB-EGF concentration (≤0.1 nM or 1.2 ng/mL), the peak level of HB-EGF receptor dimers was reduced compared to that seen when 1.0 nM HB-EGF was added and the time to reach the peak concentration was delayed (>25 min) (data not shown). The addition of FGF-2 reduced both peak HB-EGF receptor dimer levels as well as the overall level of bound dimers over the entire time course (150 min). However, at the intermediate HB-EGF concentration (0.25 nM or 3 ng/mL), an interesting effect of FGF-2 on HB-EGF dimerization was found. FGF-2 addition muted the peak level of bound HB-EGF as it had for the lower HB-EGF concentration, however there was a significant period of time (40-120 min) where the number of surface HB-EGF receptor dimers was greater in the presence of FGF-2 than in the absence (Fig. 11). While it is not clear how, or even if, these levels translate to the proliferative response, the observation that there are some regimes where the presence of FGF-2 results in increased HB-EGF receptor dimers, despite inhibiting steady-state HB-EGF bound receptor levels at 4 °C, indicates that the “receptor-coupling” model might be able to accommodate conditions whereby signaling through receptors is enhanced at 37 °C. We also noted a small increase in peak HB-EGF-receptor dimers with FGF-2 when simulations at 37 °C were performed using the “non-receptor-coupling” model (data not shown).
FIGURE 11.
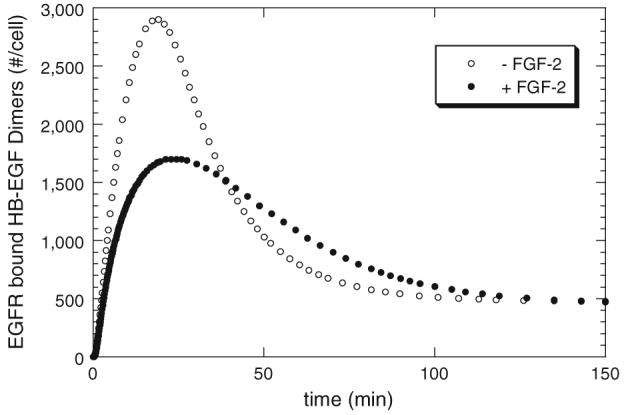
Effect of FGF-2 on HB-EGF binding at 37 °C (simulations)—cell surface dimers of HB-EGF bound EGFR as a function of time when HB-EGF (0.25 nM) is with (●) and without (○) FGF-2 (60 nM) using “receptor-coupling” model. All parameters are as listed in Table 2 except that there are more FGFR (1.6 × 105 receptors/cell) reflecting an increased receptor levels due to expression of the dominant negative receptors in the SMC-Delta cells.7,14
DISCUSSION
Binding and dimerization of cell surface tyrosine kinase receptors is the paradigm by which growth factors initiate signal transduction. With HB-GFs, this situation is complicated by the importance of HSPGs in regulating receptor binding, ligand availability, and, in some cases, direct signaling.7,38,41 In this paper, we analyze how the presence of competing HB-GFs can impact cell surface interactions and show, using simulations, that the ability of “common” HSPG binding sites to couple with receptors to stabilize binding determines whether the competing growth factors increase or decrease receptor binding. While the computational simulations used parameters for FGF-2 and HB-EGF, the model of competition by which accessory proteins impact receptor binding and dimerization could be applicable to any number of growth factor systems. Further, our simulations demonstrate how receptor binding of non-HB-GFs may also be impacted by HB-GFs which do not share a common receptor. Our results suggest that “common” binding sites such as HSPGs hold the key to enhanced or diminished receptor binding in growth factor networked systems.
Certainly agreement with the experimental data is desirable but given the variability in glycosaminoglycan structure, the inherent diffculty with experimental parameter measurement as well as the simplifying assumptions, quantitative agreement is unlikely. The structural modifications to the critical glycosaminoglycan sidechains provide the very moieties that dictate growth factor binding and specificity, but the forces that regulate these modifications are not well understood and not easily controlled. Models, such as we present here, describe subsets of these populations that act in like manner and increase our knowledge of the dynamic role of proteoglycans on cell surfaces and in extracellular matrices. As our abilities to control glycosaminoglycan expression become more advanced, more complex analyses can be incorporated to understand the individual contributions of each moiety. This information will add to the details of the model and undoubtedly result in greater correlation with experimental data but will not likely undermine the fundamental strategy of competitive binding described within it.
Our simulations were based on simple models wherein the pivotal difference was in the ability of these HSPG “common” sites to facilitate receptor coupling (Fig. 2). When common sites were not allowed to couple with receptors to form high-affinity complexes, the addition of FGF-2 and its competitive binding for common sites resulted in increased availability of HB-EGF for binding to both unique HSPG sites and specific receptors (Fig. 6). When common HSPG sites were able to couple with receptors to form high-affinity sites, addition of FGF-2 and its competitive binding of common sites resulted in increased availability of HB-EGF for direct binding to both unique HSPG sites and specific receptors, but the overall receptor binding level was reduced due to a loss of high-affinity common site-mediated receptor complex formation. While the overall density and affinity of the common sites was important (Figs. 7 and 8), it was the ability to couple and stabilize growth factor-receptor interactions that ultimately led to the difference in responses. These findings suggest a very different response regulation by ECM versus cellular HSPG as well as indicating that “fencing” or “patching” of HSPG in cell surface domains inaccessible to receptors or vice versa would be quite different from freely accessible interactions.
We have assumed that all surface coupling reactions occur at the same rate in our model. Differences in the coupling rate via diffusion or affnity differences could also impact the resulting effect. Ligand-independent coupling was not included for either FGF-2 or HB-EGF as our simulations previously showed a negligible effect on FGF-2 binding distribution.20 Recent work from Mac Gabhan and Popel28 looking at dimerization of VEGF receptors (VEGFR1 and VEGFR2) in the presence of VEGF and placental growth factor (PlGF) suggest though that competition for binding to VEGFR1 and VEGFR2 and pre-coupling of these receptors can impact the homo- and heterodimerization process and likely impact signal generation. This suggests that dimerization and its ligand independent and dependent rates maybe require future attention for this more complex system. Further experimental and computational studies investigating coupling and cell localization of binding sites would provide valuable insight into how these factors can influence growth factor responsiveness.
In contrast to the inhibition of HB-EGF binding by FGF-2 (Fig. 3), enhanced HB-EGF-induced proliferation by FGF-2 was found (Fig. 10). These findings indicate that steady-state binding levels at 4 °C are not necessarily indicative of proliferation in our cell system. It is important to note that the proliferation studies were performed at 37 °C when membrane trafficking and cell activity are not negligible. Further, proliferation is a complex downstream response induced by growth factor binding that is dependent on a complex series of steps that remain incompletely understood for HB-EGF. For example, in our cell system we have previously shown that FGF-2 activates MAPK, but not proliferation, through HSPG interactions that suggests that HB-EGF could also generate signals through cellular HSPGs.7 We were, however, intrigued by the simulation results suggesting that there are conditions, based on time and dose of FGF-2, where HB-EGF receptor surface dimers are enhanced in the presence of FGF-2 compared to the absence of FGF-2 (Fig. 11). Of course this does not account for endocytic dimers which have been demonstrated to be capable of signaling in some growth factor systems.21 Clearly there is need for more experimental measurements in this area in order to reveal the underlying controlling elements that dictate the proliferative response to growth factors. In this regard, the use of computational models in conjunction with experimental studies can help focus the proposed studies.
Our most intriguing result may be the prediction that HB-GFs are able to impact the binding of non-HB-GFs even when they do not share a common receptor (Fig. 9). For example, FGF-2 does not directly impact EGF receptor binding (Fig. 4) however our simulations suggest that FGF-2 can impact EGF binding when HB-EGF is present via competition for HSPG “common” sites. When common sites are unable to couple to receptors (“non-receptor-coupling” model), FGF-2 competition with HB-EGF for HSPG “common” sites increases the available HB-EGF for binding to both unique HSPG sites and receptors leading to reduced binding of EGF to these same receptors. When receptor-coupling is also included (“receptor-coupling” model), FGF-2 competition with HB-EGF for HSPG “common” sites reduces the probability of HB-EGF forming high-affinity receptor complexes and, thus, increases EGF receptor binding. For our parameter values, this effect was particularly significant for the “receptor-coupling” model where almost four times as much EGF is predicted to be bound to receptors when FGF-2 and HB-EGF are present compared to that bound when only HB-EGF is there. This certainly suggests that care must be used in interpreting complex system results as well as in vivo studies where unknown components may directly or indirectly alter results. Heparin-binding and non-binding growth factors and their receptors may be intimately connected through cell and ECM HSPGs forming a regulatory signaling network.
ACKNOWLEDGMENT
This study was supported by grants from NIH (HL56200 and HL86644).
APPENDIX
We developed two related models for this work and the equations are listed below. For HB-EGF binding in the “non-receptor-coupling” model:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
Nine additional equations identical in structure are written for FGF-2 binding with H replaced by F
For HSPG common sites:
| (10) |
| (11) |
| (12) |
In the “receptor-coupling” model, HSPG common sites are able to interact with receptors to form higher order complexes. Hence Eqs. (10-12) are replaced by Eqs. (13-15)
| (13) |
| (14) |
| (15) |
Plus the addition of:
| (16) |
| (17) |
| (18) |
| (19) |
and four additional identical equations for FGF-2 with H replaced by F.
Also, note that additional terms reflecting the interactions of the HSPG common sites with HB-EGF and FGF-2 receptors were added to Eqs. (1-9) and the accompanying FGF-2 equations to balance the system.
The parameter symbols and values are given in Table 2. Unbound receptors (RH, RF), ligand bound receptors (CH, CF), receptor-ligand dimers (C2H, C2F), unbound ligand-specific proteoglycans (PH, PF), unbound common site proteoglycan (Pc), ligand bound proteoglycans (GH, GF, , ), receptor-ligand-proteoglycan complexes (TH, TF, , ), receptor-ligand dimers bound to a proteoglycan site (XH, XF, , ), dimers of receptor-ligand-proteoglycan complexes (T2H, T2F, THH, , , ), and ligand concentration (H, F) form the variables in the system with subscripts H and F representing HB-EGF and FGF-2, respectively. The superscript c indicates HSPG common site.
REFERENCES
- 1.Atha DH, Lormeau JC, Petitou M, Rosenberg RD, Choay J. Contribution of 3-O- and 6-O-sulfated glucosamine residues in the heparin-induced conformational change in antithrombin III. Biochemistry. 1987;26:6454–6461. doi: 10.1021/bi00394a024. doi:10.1021/bi00394a024. [DOI] [PubMed] [Google Scholar]
- 2.Atha DH, Stephens AW, Rosenberg RD. Evaluation of critical groups required for the binding of heparin to antithrombin. Proc. Natl. Acad. Sci. U.S.A. 1984;81:1030–1034. doi: 10.1073/pnas.81.4.1030. doi:10.1073/pnas.81.4.1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bublil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007;19:124–134. doi: 10.1016/j.ceb.2007.02.008. doi:10.1016/j.ceb. 2007.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem. Int. Ed. Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. doi:10.1002/1521-3773(20020201)41:3<390::AID-ANIE390>3.0.CO; 2-B. [DOI] [PubMed] [Google Scholar]
- 5.Chu CL, Buczek-Thomas JA, Nugent MA. Heparan sulphate proteoglycans modulate fibroblast growth factor-2 binding through a lipid raft-mediated mechanism. Biochem. J. 2004;379:331–341. doi: 10.1042/BJ20031082. doi:10.1042/ BJ20031082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu CL, Goerges AL, Nugent MA. Identification of common and specific growth factor binding sites in heparan sulfate proteoglycans. Biochemistry. 2005;44:12203–12213. doi: 10.1021/bi050241p. doi:10.1021/bi050241p. [DOI] [PubMed] [Google Scholar]
- 7.Chua CC, Rahimi N, Forsten-Williams K, Nugent MA. Heparan sulfate proteoglycans function as receptors for fibroblast growth factor-2 activation of extracellular signal-regulated kinases 1 and 2. Circ. Res. 2004;94:316–323. doi: 10.1161/01.RES.0000112965.70691.AC. doi:10.1161/01.RES.0000112965.70691.AC. [DOI] [PubMed] [Google Scholar]
- 8.Connolly DT, Knight MB, Harakas NK, Wittwer AJ, Feder J. Determination of the number of endothelial cells in culture using an acid phosphatase assay. Anal. Biochem. 1986;152:136–140. doi: 10.1016/0003-2697(86)90131-4. doi:10.1016/0003-2697(86)90131-4. [DOI] [PubMed] [Google Scholar]
- 9.Conrad HE. Heparin-binding proteins. Academic Press; San Diego: 1998. [Google Scholar]
- 10.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. doi:10.1146/annurev.biochem. 71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 11.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–149. doi: 10.1016/j.cytogfr.2005.01.001. doi:10.1016/j.cytogfr. 2005.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Fannon M, Forsten KE, Nugent MA. Potentiation and inhibition of bFGF binding by heparin: a model for regulation of cellular response. Biochemistry. 2000;39:1434–1445. doi: 10.1021/bi991895z. doi:10.1021/bi991895z. [DOI] [PubMed] [Google Scholar]
- 13.Fantl WJ, Johnson DE, Williams LT. Signalling by receptor tyrosine kinases. Annu. Rev. Biochem. 1993;62:453–481. doi: 10.1146/annurev.bi.62.070193.002321. [DOI] [PubMed] [Google Scholar]
- 14.Forsten-Williams K, Chua CC, Nugent MA. The kinetics of FGF-2 binding to heparan sulfate proteoglycans and MAP kinase signaling. J. Theor. Biol. 2005;233:483–499. doi: 10.1016/j.jtbi.2004.10.020. doi:10.1016/j.jtbi.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 15.Gallagher JT. Heparan sulfate: growth control with a restricted sequence menu. J. Clin. Invest. 2001;108:357–361. doi: 10.1172/JCI13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gavutis M, Jaks E, Lamken P, Piehler J. Determination of the two-dimensional interaction rate constants of a cytokine receptor complex. Biophys. J. 2006;90:3345–3355. doi: 10.1529/biophysj.105.072546. doi:10.1529/biophysj.105.072546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J. Biol. Chem. 1992;267:6093–6098. [PubMed] [Google Scholar]
- 18.Goerges AL, Nugent MA. Regulation of vascular endothelial growth factor binding and activity by extracellular pH. J. Biol. Chem. 2003;278:19518–19525. doi: 10.1074/jbc.M211208200. doi:10.1074/jbc.M211208200. [DOI] [PubMed] [Google Scholar]
- 19.Goerges AL, Nugent MA. pH regulates vascular endothelial growth factor binding to fibronectin: a mechanism for control of extracellular matrix storage and release. J. Biol. Chem. 2004;279:2307–2315. doi: 10.1074/jbc.M308482200. doi:10.1074/jbc.M308482200. [DOI] [PubMed] [Google Scholar]
- 20.Gopalakrishnan M, Forsten-Williams K, Tauber UC. Ligand-induced coupling versus receptor pre-association: cellular automaton simulations of FGF-2 binding. J. Theo. Biol. 2004;227:239–251. doi: 10.1016/j.jtbi.2003.11.004. doi:10.1016/j.jtbi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Haugh JM, Huang AC, Wiley HS, Wells A, Lauffenburger DA. Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts. J. Biol. Chem. 1999;274:34350–34360. doi: 10.1074/jbc.274.48.34350. doi:10.1074/jbc.274.48.34350. [DOI] [PubMed] [Google Scholar]
- 22.Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: distribution of homo- and heterodimers depends on relative HER2 levels. J. Biol. Chem. 2003;278:23343–23351. doi: 10.1074/jbc.M300477200. doi:10.1074/jbc.M300477200. [DOI] [PubMed] [Google Scholar]
- 23.Higashiyama S, Abraham JA, Klagsbrun M. Heparin-binding EGF-like growth factor stimulation of smooth muscle cell migration: dependence on interactions with cell surface heparan sulfate. J. Cell Biol. 1993;122:933–940. doi: 10.1083/jcb.122.4.933. doi:10.1083/jcb.122.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones JT, Akita RW, Sliwkowski MX. Binding specificities and affinities of egf domains for ErbB receptors. FEBS Let. 1999;447:227–231. doi: 10.1016/s0014-5793(99)00283-5. doi:10.1016/S0014-5793(99)00283-5. [DOI] [PubMed] [Google Scholar]
- 25.Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J. Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. doi:10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemmon MA, Schlessinger J. Transmembrane signaling by receptor oligomerization. Methods Mol. Biol. 1998;84:49–71. doi: 10.1385/0-89603-488-7:49. [DOI] [PubMed] [Google Scholar]
- 27.Lindahl U, Backstrom G, Thunberg L, Leder IG. Evidence for a 3-O-sulfated d-glucosamine residue in the antithrombin-binding sequence of heparin. Proc. Natl. Acad. Sci. U.S.A. 1980;77:6551–6555. doi: 10.1073/pnas.77.11.6551. doi:10.1073/pnas.77.11.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mac Grabhann F, Popel AS. Dimerization of VEGF receptors and implications for signal transduction: a computational study. Biophys. Chem. 2007;128:125–139. doi: 10.1016/j.bpc.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moscatelli D. High and low affinity binding sites for basic fibroblast growth factor on cultured cells: absence of a role for low affinity binding in the stimulation of plasminogen activator production by bovine capillary endothelial cells. J. Cell. Physiol. 1987;131:123–130. doi: 10.1002/jcp.1041310118. doi:10.1002/jcp.1041310118. [DOI] [PubMed] [Google Scholar]
- 30.Nugent MA. Heparin sequencing brings structure to the function of complex oligosaccharides. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10301–10303. doi: 10.1073/pnas.97.19.10301. doi:10.1073/pnas.97.19.10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nugent MA, Edelman ER. Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: a mechanism for cooperativity. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. doi:10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- 32.Nugent MA, Forsten-Williams K, Karnovsky MJ, Edelman ER. Mechanisms of cell growth regulation by heparin and heparan sulfate. In: Garg HG, editor. Chemistry and Biology of Heparin and Heparan Sulfate. Elsevier Ltd.; 2005. pp. 533–570. [Google Scholar]
- 33.Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin. Transl. Oncol. 2006;8:77–82. doi: 10.1007/s12094-006-0162-1. doi:10.1007/s12094-006-0162-1. [DOI] [PubMed] [Google Scholar]
- 34.Raab G, Klagsbrun M. Heparin-binding EGF-like growth factor. Biochim. Biophys. Acta. 1997;1333:F179–199. doi: 10.1016/s0304-419x(97)00024-3. [DOI] [PubMed] [Google Scholar]
- 35.Shraga-Heled N, Kessler O, Prahst C, Kroll J, Augustin H, Neufeld G. Neuropilin-1 and neuropilin-2 enhance VEGF121 stimulated signal transduction by the VEGFR-2 receptor. FASEB J. 2007;211:915–926. doi: 10.1096/fj.06-6277com. doi:10.1096/fj.06-6277com. [DOI] [PubMed] [Google Scholar]
- 36.Sperinde GV, Nugent MA. Heparan sulfate proteoglycans control bFGF processing in vascular smooth muscle cells. Biochemistry. 1998;37:13153–13164. doi: 10.1021/bi980600z. doi:10.1021/bi980600z. [DOI] [PubMed] [Google Scholar]
- 37.Sperinde GV, Nugent MA. Mechanisms of FGF-2 intracellular processing: A kinetic analysis of the role of heparan sulfate proteoglycans. Biochemistry. 2000;39:3788–3796. doi: 10.1021/bi992243d. doi:10.1021/bi992243d. [DOI] [PubMed] [Google Scholar]
- 38.Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ. Rec. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. doi:10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 39.Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. doi:10.1016/S0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- 40.Wouters-Ballman P, Donnay I, Devleeschouwer N, Verstegen J. Iodination of mouse EGF with chloramine T at 4 degrees C: characterization of the iodinated peptide and comparison with other labelling methods. J. Recept. Signal. Transduct. Res. 1995;15:737–746. doi: 10.3109/10799899509079903. doi:10.3109/10799899509079903. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Li J, Partovian C, Sellke FW, Simons M. Syndecan-4 modulates basic fibroblast growth factor 2 signaling in vivo. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H2078–H2082. doi: 10.1152/ajpheart.00942.2001. [DOI] [PubMed] [Google Scholar]