

Abstract
Background
Tests for resistance to HIV drugs are available for clinical use; however, their predictive value has not been fully assessed.
Objectives
To determine HIV-1 genotypic predictors of a virologic response to saquinavir–ritonavir therapy in patients in whom at least one previous protease inhibitor–containing regimen had failed and to compare the predictive value of baseline genotype with that of standard clinical evaluation.
Design
Retrospective clinical cohort study.
Setting
University-based HIV clinic.
Patients
54 HIV-1–infected adults treated with saquinavir–ritonavir who had experienced virologic failure while receiving a protease inhibitor–containing regimen for at least 3 months.
Measurements
HIV-1 reverse transcriptase and protease gene sequences, CD4 cell counts, clinical characteristics, detailed antiretroviral treatment history, and plasma HIV-1 RNA levels at baseline and at three follow-up time points (median, 4, 12, and 26 weeks). Virologic failure was defined as a plasma HIV RNA level greater than 1000 copies/mL.
Results
In 22 patients (41%), a plasma HIV-1 RNA level less than 500 copies/mL was achieved by week 12; in 15 patients (28%), this response was maintained through week 26. Clinical characteristics predicting a poorer response included a diagnosis of AIDS, lower CD4 cell count, and higher plasma HIV RNA level (P < 0.03). Number of previous nucleoside reverse transcriptase inhibitors, previous protease inhibitor therapy, and duration of previous protease inhibitor therapy were predictors of poorer response (P < 0.01). Multivariate regression models revealed that protease mutations present at the initiation of saquinavir-ritonavir therapy were the strongest predictors of virologic response. A model of clinical features explained up to 45% of the variation in virologic outcomes by week 12, whereas the explained variance was 71% when genotypic predictors were included.
Conclusions
In patients in whom protease inhibitor–containing antiretroviral therapy fails, HIV-1 genotype is predictive of virologic response to subsequent therapy. This predictive capacity adds to that of standard clinical evaluation.
Combination antiretroviral therapy for HIV-1 infection has resulted in profound control of HIV replication in vivo, improved immune function, and significant decreases in AIDS-related morbidity and mortality (1–9). For many persons, however, this therapy does not provide sustained viral suppression or durable clinical benefit (10, 11). Potential reasons for the loss of viral suppression include host immune defects, poor adherence to therapy, pharmacologic factors, and drug resistance (10–17). However, HIV-1 resistance to drug therapy is probably the central factor in the loss of viral suppression (18–22).
Mutations that result in reduced drug susceptibility have been demonstrated in vitro for all currently available antiretroviral agents, and some of these mutations have been associated with increasing plasma HIV-1 RNA levels and disease progression in clinical trials (19–30). Genotypic and phenotypic methods of measuring drug resistance are increasingly available to clinicians (31–37). However, the role of these tests in clinical practice has not been fully assessed. Many experts have been skeptical of resistance testing, although a recent consensus statement provides cautious support for testing in certain clinical circumstances (38–42).
Our objective was to determine the genotypic predictors of virologic response to saquinavir–ritonavir combination therapy in patients in whom therapy with at least one protease inhibitor–containing antiretroviral regimen had failed. We investigated whether HIV-1 reverse transcriptase and protease genotype predicts virologic response to saquinavir–ritonavir by week 12 and week 26 and compared those data with predictors from clinical and antiretroviral treatment history.
Methods
Patients
Two of the investigators treated patients in a university-based clinic that provides primary care for 500 HIV-infected adults. We identified 54 patients who received saquinavir–ritonavir between October 1996 and February 1998 after therapy with at least one protease inhibitor–containing antiretroviral regimen had failed. Treatment failure was defined as a greater than 0.5 log10 copies/mL (more than three-fold) increase in plasma HIV RNA level from a nadir value, an HIV RNA level greater than 10 000 copies/mL, or detectable HIV RNA after the level had been below the threshold of detection (<500 copies/mL) during a therapeutic regimen for more than 12 weeks.
Study patients received 400 to 600 mg of saquinavir in a hard-gel formulation (Invirase, Roche Laboratories, Nutley, New Jersey) and 300 to 400 mg of ritonavir in capsule form (Norvir, Abbott Laboratories, Abbott Park, Illinois) twice daily. In addition to the two protease inhibitors, 47 patients (87%) received two nucleoside reverse transcriptase inhibitors, 4 received three nucleoside reverse transcriptase inhibitors, 2 received two nucleoside reverse transcriptase inhibitors and either nevirapine or delavirdine, and one patient received lamivudine.
Clinical and demographic variables were abstracted from the medical records. Adherence, as recorded in the patient's record, was categorized by the self-reported number of missed doses in the month before evaluation and was classified as none, one to two, three to seven, or eight or more. Plasma levels of HIV-1 RNA were monitored on average every 4 to 6 weeks, and samples were stored at −70 °C.
The Stanford University Panel on Medical Human Subjects approved this study (#M1272).
HIV Genotyping
Baseline HIV-1 genotype was evaluated in plasma specimens that were stored within 1 month before initiation of saquinavir–ritonavir therapy and were obtained while patients were still receiving an ineffective antiretroviral regimen. Plasma HIV-1 RNA was extracted, and nested polymerase chain reaction (PCR) amplification generated a 1.3-kb fragment encompassing protease and the first 750 nucleotides of reverse transcriptase (43, 44). Direct bidirectional dideoxynucleotide terminator cycle sequencing of the PCR product was performed as described elsewhere (44). Sequencing reactions were analyzed by using an ABI 377 instrument (Perkin-Elmer, Foster City, California) and were manually proofread and edited. Sequences were compared to the HIV-1 clade B consensus sequence (Los Alamos database), and differences in amino acid sequence, including positions that contained a mixture of wild-type and mutant residues, were classified as mutations (45). Phylogenetic analysis of HIV-1 RNA sequence verified lack of cross-contamination (data not shown).
A priori, we decided to assess reverse transcriptase codons 41, 67, 69, 70, 74, 75, 151, 184, 210, 215, and 219 as predictors of virologic response. Mutations at these codons are known to be associated with resistance to one of the nucleoside reverse transcriptase inhibitors (25, 45). In the protease gene, mutations at codons 30, 46, 48, 54, 82, 84, and 90 were evaluated as potential predictors. We chose these “major” mutations a priori because they are associated with in vitro resistance to a protease inhibitor or occur commonly in patients in whom therapy with currently licensed protease inhibitors is failing. We also evaluated all other protease codons as predictors of response.
Virologic Outcomes
Virologic response to saquinavir–ritonavir was measured at two time points between 3 and 18 weeks and again around week 24 (range, 22 to 36 weeks); the median time points of the three follow-up evaluations were 4, 12, and 26 weeks. Levels of HIV-1 RNA were measured by using the Amplicor HIV Monitor Assay (Roche Molecular Systems, Alameda, California). Specimens with HIV RNA below the level of detection (<500 copies/mL) on this assay were retested by using the ultrasensitive modification with a lower limit of detection of less than 50 copies/mL (43).
Virologic response to saquinavir–ritonavir therapy was categorized on the basis of the larger response from baseline to the first or second evaluation (median, 4 and 12 weeks). The ordinal categories were 1) complete response if the plasma HIV-1 RNA level was less than 500 copies/mL, 2) partial response if the reduction from baseline RNA level was 0.5 log10 copies/mL or more but was not less than 500 copies/mL, and 3) nonresponse if reduction from baseline values was less than 0.5 log10 copies/mL.
Statistical Analysis
Demographic, clinical, and genotypic variables were analyzed as potential predictors of virologic response by using bivariate linear regression and multivariable linear regression. In the multivariable models, we included a subset of the reverse transcriptase mutations (listed above) identified through stepwise regression as significant (P < 0.05) predictors. This subset of mutations was included in the model as a signed-sum variable. For the protease mutations, we included the signed sum of the seven major mutations listed above and the signed sum of three additional mutations at codons 10, 19, and 71, which were found to be statistically significant bivariate predictors.
The signed-sum variable is derived by a summation of the relevant mutations identified in the baseline sequence. A separate sum is derived from the seven major protease mutations, the three additional protease mutations, and the subset of statistically significant reverse transcriptase mutations (codons 69 and 210). In the signed-sum variable, mutations that are positively associated with virologic outcome (such as protease mutation D30N) are assigned a positive sign (+1) and mutations negatively associated with outcome are assigned a negative sign (−1).
We used the Cook distance to assess skewing of the ordinal outcome variable in the final multivariable model (model 5, Table 3). The value of 0.11 indicated no significant skewing; this result supports the use of linear regression models (46). We also evaluated the multivariate models for bias that would result from overfitting of the data. We used a bootstrap technique to estimate bias (“optimism”) in the explained variance values (R2) for the models presented and found minimal upward bias; for example, model 3 in Table 3 has a bias of approximately one fifth of the SE (data for other models not shown) (47). A bootstrap technique was used to provide the 95% CI estimates for the R2 values in Table 3. We selected 25 bootstrap samples of 54 with replacement from the original 54 patients to estimate the 95% CIs. The Wilcoxon rank test was used for comparisons between specific previous protease inhibitors in Table 1, and the F test was used for comparisons between models in Table 3. Two-sided P values are reported for all analyses. All analyses were conducted by using S-PLUS, version 4.0 (MathSoft, Seattle, Washington).
Table 3. Multivariable Linear Regression Models of Clinical, Antiretroviral Treatment History, and HIV-1 Genotypic Predictors of Virologic Response by Week 12 of Saquinavir–Ritonavir Therapy*.
| Model | Model 1: Clinical and Treatment History |
Model 2: Major Protease Mutations |
Model 3: Major and Minor Protease Mutations |
Model 4: Protease and Reverse Transcriptase Mutations |
Model 5: Comprehensive Model 1 |
Model 6: Comprehensive Model 2 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Regression Coefficient |
P Value | Regression Coefficient |
P Value | Regression Coefficient |
P Value | Regression Coefficient |
P Value | Regression Coefficient |
P Value | Regression Coefficient |
P Value | |
| Clinical and treatment history | ||||||||||||
| Plasma HIV RNA level at baseline | −0.312 | <0.01 | – | – | – | – | – | – | – | – | −0.10 | >0.2 |
| Duration of previous therapy with nucleoside reverse transcriptase inhibitors | −0.002 | <0.05 | – | – | – | – | – | – | – | – | −0.001 | 0.18 |
| Number of previous protease inhibitors | −0.388 | <0.01 | – | – | – | – | – | – | −0.17 | 0.07 | −0.11 | >0.2 |
| Previous nelfinavir treatment | 0.491 | <0.03 | – | – | – | – | – | – | – | – | −.051 | >0.2 |
| Genotypic variables | ||||||||||||
| Signed sum of the major protease mutations† | – | – | 0.413 | <0.01 | 0.24 | <0.01 | 0.20 | <0.01 | 0.22 | <0.01 | 0.21 | <0.01 |
| Signed sum of the minor protease mutations‡ | – | – | – | – | 0.45 | <0.01 | 0.42 | <0.01 | 0.43 | <0.01 | 0.38 | <0.01 |
| Signed sum of the reverse transcriptase mutations§ | – | – | – | – | – | – | −0.08 | 0.5 | – | – | – | – |
| Explained variance of model, R2 value (95% CI) | 0.45 (0.31– 0.59) | 0.59 (0.45– 0.73) | 0.66 (0.52– 0.80)‖ | 0.67 (0.53– 0.81)¶ | 0.69 (0.57–0.81)** | 0.71 (0.59–0.83) | ||||||
Based on ordinal response categories and complete, partial, and nonresponse to therapy (see Methods section).
Major protease codons were 30, 46, 48, 54, 82, 84, and 90. Mutation D30N was positively associated with a virologic outcome; mutations at all other codons were negatively associated with such an outcome.
Minor protease codons were 10, 19, and 71. Mutation at codon 19 was positively associated with virologic outcome; mutations at condons 10 and 71 were negatively associated with such an outcome.
Reverse transcriptase codons were 69 and 210. Mutations at these codons were negatively associated with virologic outcome.
P < 0.002 (model 3 compared with model 2; F test).
P > 0.2 (model 4 compared with model 3; F test).
P = 0.07 (model 5 compared with model 3; F test).
Table 1. Baseline Demographics, Clinical Characteristics, and Antiretroviral Treatment History as Predictors of Virologic Response to Saquinavir–Ritonavir Therapy.
| Characteristic | Overall | Complete Response* | Partial Response† | Nonresponse‡ | P Value§ |
|---|---|---|---|---|---|
| Demographic characteristic | |||||
| Patients, n (%) | 54 | 22 (41) | 14 (26) | 18 (33) | |
| Median age, y | 41 | 39 | 42.5 | 38.5 | >0.2 |
| Men, n (%) | 51 (94) | 20 (90) | 13 (93) | 18 (100) | >0.2 |
| White, non-Hispanic ethnicity, n (%) | 39 (72) | 14 (64) | 12 (86) | 13 (72) | >0.2 |
| Baseline clinical characteristics | |||||
| AIDS diagnosis, n (%)‖ | 20 (37) | 5 (23) | 5 (36) | 10 (56) | 0.03 |
| Median CD4 count (range), ×109/L | 245 (5–1060) | 295 (20–1060) | 220 (5–460) | 155 (9–470) | <0.01 |
| Median log10 plasma HIV-1 RNA level (range), copies/mL | 5.0 (3–5.9) | 4.0 (3.0 –5.9) | 5.0 (4.1–5.9) | 5.0 (4.0 –5.9) | <0.01 |
| Antiretroviral treatment history | |||||
| Median nucleoside reverse transcriptase inhibitors (range), n | 4 (2–5) | 3 (2–5) | 4 (3–5) | 4 (2–5) | 0.01 |
| Median duration of prior nucleoside reverse transcriptase inhibitor therapy (range), wk | 188 (30–472) | 118 (30–346) | 226 (64–408) | 334 (75–472) | <0.01 |
| Patients with previous non-nucleoside reverse transcriptase inhibitor therapy, n (%) | 16 (29) | 8 (36) | 5 (36) | 3 (17) | 0.19 |
| Median previous protease inhibitors (range), n | 2 (1–4) | 1 (1–3) | 2 (1–3) | 2 (1– 4) | 0.01 |
| Median duration of protease inhibitor thearapy (range), wk | 48 (12–216) | 38 (12–104) | 48 (32–216) | 58 (24–156) | 0.08 |
| Patients who received new nucleoside reverse transcriptase inhibitors, n (%) | 13 (24) | 7 (32) | 3 (16) | 3 (17) | >0.2 |
| Previous protease inhibitors, n | |||||
| Indinavir | 14 | 5 | 4 | 5 | –¶ |
| Nelfinavir | 5 | 5 | 0 | 0 | – |
| Saquinavir then indinavir | 14 | 4 | 4 | 6 | – |
| Saquinavir then nelfinavir | 9 | 6 | 2 | 1 | – |
| Indinavir then nelfinavir | 1 | 1 | – | ||
| Ritonavir then indinavir | 2 | 0 | 1 | 1 | – |
| Saquinavir, nelfinavir, and indinavir | 7 | 1 | 2 | 4 | – |
| Saquinavir, ritonavir, and indinavir | 1 | 0 | 1 | 0 | – |
| All four protease inhibitors | 1 | 0 | 0 | 1 | – |
Defined as ≤500 copies/mL at 4 or 12 weeks.
Defined as a >0.5 log10 or greater reduction in plasma HIV-1 RNA level but not <500 copies/mL at 4 or 12 weeks.
<0.5 log10 reduction at 4 or 12 weeks.
Linear regression.
Excludes CD4 cell count criterion.
No statistical test as a bivariate predictor.
Results
Virologic Response to Saquinavir–Ritonavir Therapy
Of the 54 study patients, 22 (41%) achieved a “complete” response, with plasma HIV-1 RNA levels less than 500 copies/mL by the second follow-up evaluation (at a median of 12 weeks). Of these 22 patients, 10 (18.5% of the entire cohort) achieved a plasma HIV-1 RNA level less than 50 copies/mL. Fourteen patients (26%) had a partial response to saquinavir–ritonavir, and 18 (33%) were nonresponders (Table 1). The virologic response to saquinavir–ritonavir is shown by initial response category in Figure 1. The response waned somewhat in the partial and complete response groups by week 26: The HIV RNA level remained below 500 copies/mL in 15 patients (28%) and below 50 copies/mL in 10 patients (19%).
Figure 1. Virologic response to saquinavir plus ritonavir through week 26 based on response by week 12.
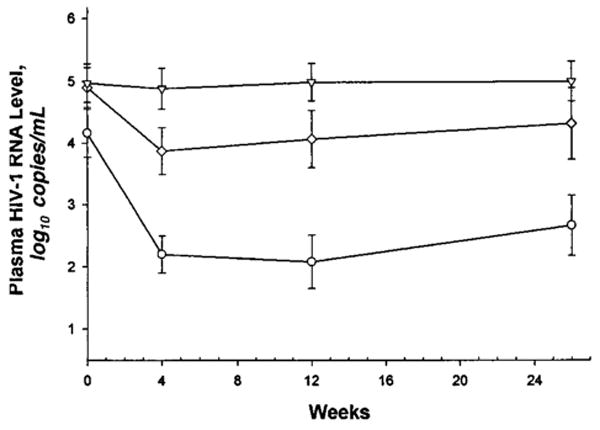
See the Methods section for further explanation. Triangles represent nonresponders; diamonds represent partial responders; circles represent complete responders.
Predictors of Virologic Response from the Clinical and Antiretroviral Treatment History
Table 1 delineates baseline characteristics of the cohort as predictors of virologic response to saquinavir–ritonavir therapy by week 12. Plasma levels of the HIV-1 RNA and CD4 count within 4 weeks of the start of combination therapy and a history of an AIDS-defining opportunistic infection or malignant disease were predictors of response.
The cohort was heavily pretreated with nucleoside reverse transcriptase inhibitors. Only 24% of patients received a new nucleoside reverse transcriptase inhibitor along with saquinavir–ritonavir. Sixteen patients (29%) had received a non-nucleoside reverse transcriptase inhibitor (15 received nevirapine and 1 received delavirdine) before saquinavir–ritonavir but only 2 patients received a non-nucleoside inhibitor as part of saquinavir–ritonavir therapy. Before saquinavir–ritonavir therapy, the median number of protease inhibitors taken was two and the median duration of protease inhibitor therapy was 48 weeks. Previous protease inhibitor therapies are listed in Table 1.
The number and duration of nucleoside reverse transcriptase inhibitors and the number of protease inhibitors taken before saquinavir–ritonavir therapy were predictive of virologic response, and a trend was seen for duration of previous protease inhibitor therapy as a predictor. Patients who had had unsuccessful nelfinavir therapy as their sole previous protease inhibitor had a better response to saquinavir–ritonavir than did patients who had had unsuccessful indinavir therapy as their sole previous protease inhibitor (P < 0.01). No other statistically significant differences were seen among the nine previous protease inhibitor treatment regimens listed in Table 1.
Adherence to Saquinavir–Ritonavir Therapy
Thirty-nine (81%) of the 48 patients with recorded self-reported adherence measures missed two or fewer doses of saquinavir–ritonavir in the month before the first and second evaluations. Although self-report of adherence was lower in the nonresponse group, the differences between response groups did not reach statistical significance. Furthermore, this measure of adherence did not add to the multivariate prediction models described below (data not shown).
HIV-1 Genotype as Predictor of Virologic Response by Week 12
Mutations in the protease gene were strongly associated with virologic outcome. Each of the seven major mutations defined a priori, except for I84V, were predictors of virologic response (P < 0.01) (Figure 2). In addition to the seven “major” mutations, the “minor” mutations at codons 10, 19, and 71 were associated with virologic response (P < 0.01). All protease mutations evaluated except for L19Q/I and D30N were associated with a poor virologic response.
Figure 2. Frequencies of protease and reverse transcriptase mutations by virologic response at week 12.
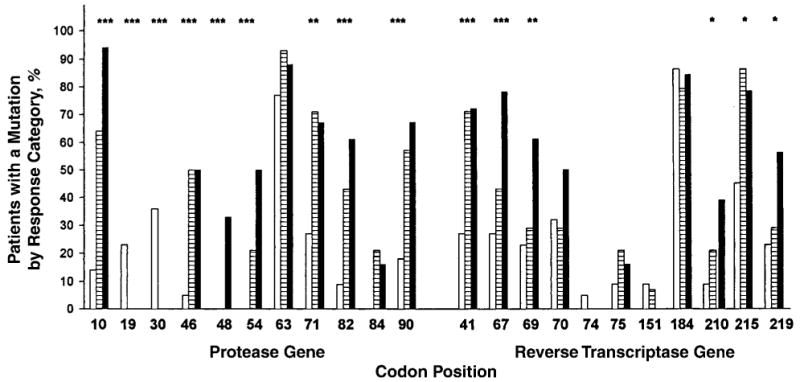
Mutation frequencies at baseline in complete responders (white bars), partial responders (striped bars), and nonresponders (solid bars) are shown. *P < 0.05; **P < 0.01; ***P < 0.001 (as bivariate predictors).
Linear regression models demonstrated a strong relation between the number of major mutations in the protease gene at baseline and the virologic response to saquinavir–ritonavir therapy by week 12 (P < 0.001). In Table 2, the relation between the seven major protease mutations present at the start of saquinavir–ritonavir therapy and the virologic response is shown. Patients without major protease mutations or with a D30N mutation had approximately a 100-fold (2 log10 copies/mL) reduction in plasma HIV-1 RNA levels at weeks 4 and 12 and an increase in CD4 count of 55 to 60 × 109 cells/L by week 12. Of the 6 patients with the L90M mutation, 2 had a complete response, 2 had a partial response, and 2 were nonresponders. Among the 10 patients with two major mutations, 2 had a complete response, 6 had a partial response, and 2 were nonresponders by week 12. In contrast, none of the 17 patients with three or more major protease mutations had a complete response, and only 4 (24%) had a partial response.
Table 2. Protease Mutation Patterns at Baseline and Response to Saquinavir–Ritonavir Combination Therapy*.
| Major Protease Mutations at Baseline† | Patients | Median Additional Baseline Mutations (Range) | Median Baseline CD4 Count | CD4 Count at 12 Weeks: Median Change from Baseline | Median Baseline Plasma HIV RNA Level | Follow-up Plasma HIV RNA Level during Saquinavir–Ritonavir Therapy | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Protease | Reverse Transcriptase | Change from Baseline at 4 Weeks | Change from Baseline at 12 Weeks | <500 copies/mL | <50 copies/mL | |||||
| ×109 cells/L | n (%) | |||||||||
| None | 11 | 5 (1–7) | 10 (0–17) | 240 | 60 | 4.7 | −1.8 | −2.4 | 9 (82) | 6 (55) |
| D30N | 7 | 4 (3–9) | 11 (9–14) | 500 | 55 | 3.6 | −2.0 | −1.8 | 7 (100) | 3 (43) |
| At codon 46, 82, or 84 | 3 | 11 (8–11) | 10 (9–11) | 200 | 50 | 4.6 | −1.7 | −1.8 | 2 (67) | 1 (33) |
| L90M | 6 | 6 (4–7) | 10 (8–19) | 345 | 5 | 4.6 | −1.5 | −1.2 | 2 (33) | 0 |
| Any two mutations | 10 | 7 (0–10) | 12 (4–16) | 140 | 40 | 5.0 | −0.9 | −0.5 | 2 (20) | 0 |
| Three or more mutations | 17 | 7 (2–10) | 14 (7–24) | 170 | 0 | 5.0 | −0.2 | 0.0 | 0 | 0 |
Response defined as <500 or <50 copies/mL by 12 weeks.
Based on “major” protease mutations, defined as those at codons 30, 46, 48, 54, 82, 84, and 90 (see Methods section).
The 11 reverse transcriptase mutations associated with resistance to nucleoside reverse transcriptase inhibitors were evaluated as predictors of virologic response. Mutations M41L, D67N/G, L210W, T215Y/F, and K219Q/E, which are associated with zidovudine resistance, and mutation T69D/N, which is associated with didanosine and zalcitabine resistance, were bivariate predictors of poor virologic response by week 12 (Figure 2).
Multivariate Prediction Models
Table 3 compares multivariate prediction models using baseline clinical and antiretroviral treatment characteristics with models using baseline HIV-1 genotype. The clinical and treatment history model was derived through stepwise regression of the bivariate predictors in Table 1; baseline plasma HIV-1 RNA level, duration of previous reverse transcriptase inhibitor therapy, number of previous protease inhibitors taken, and previous treatment with nelfinavir were independent predictors of virologic response by week 12. The clinical prediction model (model 1) composed of these four variables explains 45% of the variation in virologic outcomes.
Prediction models based on all seven major protease mutations in multivariate stepwise regression analysis did not identify a stable subset of independent predictors. Given the difficulty in choosing a subset of these mutations for a multivariate model and the likelihood that these mutations are not independent events, we used a single variable: the signed sum of the seven major mutations. The signed sum explained 59% of variation in outcomes by week 12 (model 2, Table 3). It is important to note that mutation D30N is positively associated with virologic response, whereas all other mutations are negatively associated with response.
To further explore protease mutations as predictors of response, we took the three additional protease mutations found to be bivariate predictors and created a signed-sum variable. We then added this variable to the signed sum of the major mutations (model 2). The resulting protease genotype model (model 3, Table 3) that includes these two variables explains 66% of the variation in outcomes, an improvement over the model based only on the signed sum of the major protease mutations (P < 0.01).
Through stepwise regression, mutations at codons 69 and 210 were selected as the strongest predictors of the 11 reverse transcriptase mutations associated with drug resistance. In model 4 (Table 3), the signed sum of these two reverse transcriptase mutations was combined with the signed sum of the protease mutations (model 3), producing a genotypic model that explains 67% of the variance in outcomes. This additional variable did not significantly improve the predictive capacity of model 3 (P > 0.2).
Finally, in comprehensive model 1 (model 5), the genotypic predictors were combined with the clinical and antiretroviral treatment history variables of model 1 (Table 3). The protease genotypic variables from model 3 remained strong predictors of response; however, of the clinical and treatment history variables from model 1, only number of previous protease inhibitors made a borderline contribution to the overall predictive capacity of the model. The resulting model explains 69% of the variance in virologic response to saquinavir–ritonavir by week 12. Moreover, the genotypic predictors from model 3 remained significant predictors in comprehensive model 2, in which all of the clinical predictors of model 1 were forced into the model (model 6) (P < 0.01). The explained variance of 71% for comprehensive model 2 is slightly improved over that shown for model 5, but none of the clinical variables are statistically significant in this prediction model (P = 0.16 compared with model 3). It is therefore likely to be an overfit model.
The explained variances of the linear regression models shown in Table 3 were similar when a continuous virologic outcome of change in plasma HIV-1 RNA from baseline to either the first or second follow-up evaluation (whichever was larger) was used in place of the ordinal outcomes. For example, the R2 value for the comprehensive model 5 was 0.66 when the continuous outcome variable maximal change (log10) in HIV-1 RNA level by week 12 was used.
Prediction of Virologic Response at Week 26
The regression models in Table 3 were also used to predict the virologic response at the third evaluation. Compared to the results in Table 3, the explained variance decreased for each of the models tested. Using the virologic response at this time point, the explained variance decreased to 27% for model 1; 45% for model 2; and 49% for models 3, 4, and 5. None of the clinical variables (model 1) made a statistically significant contribution to a prediction model with genotypic variables, and forcing the clinical variables into a comprehensive model did not improve the explained variance above that of model 3. The protease genotypic variables remained the strongest predictors of response at week 26 and remained independent of the clinical variables (data not shown).
Discussion
This study demonstrates that HIV-1 genotype is a strong predictor of virologic response to saquinavir–ritonavir in persons in whom previous protease inhibitor–containing antiretroviral therapy had failed to achieve maximal suppression of HIV replication. In the multivariate analysis of virologic response, the baseline HIV-1 genotype adds significantly to the predictive capacity of baseline clinical features. A model of baseline clinical features in this study can, at best, explain 45% of the variation in virologic outcomes; in contrast, the explained variance of a model that includes genotypic predictors is almost 70%. This clinical cohort study, therefore, represents a proof of principle that HIV-1 genotypic resistance testing provides information that cannot otherwise be derived from standard clinical assessment of persons in whom antiretroviral therapy is failing virologically.
Our retrospective cohort study has several limitations. First, it does not prove that HIV-1 resistance testing will improve clinical outcomes in patients who are experiencing less-than-optimal virologic suppression with antiretroviral therapy, thus warranting a change in therapy according to current guidelines (48, 49). However, two recent pilot studies comparing HIV-1 genotype–guided choice with standard practice choice of new antiretroviral agents for patients in whom therapy is failing suggested improved virologic outcomes with genotypic guidance (50, 51). Second, the size of our study did not allow for detailed analysis of drug history in all its complexity, nor could we analyze all possible combinations of mutations that might affect outcome. Finally, our clinical cohort had uniformly good adherence to therapy; in groups with poorer adherence, the resistance predictors would probably not be as strongly associated with virologic response.
The sequencing technique used in this study cannot detect viral variants that make up less than 20% of the viral population present in the plasma (42). The relative lack of sensitivity to “minority” populations of viral quasi-species has raised concerns about the clinical utility of this technology, because these minority populations, if drug-resistant, may result in virologic rebound (40). An additional concern with the sequencing technique is that the results cannot indicate whether the drug-resistance mutations identified exist in a single quasi-species or are spread across multiple viral variants. Despite these limitations, HIV-1 genotype remained a strong predictor of virologic response to saquinavir– ritonavir therapy. Lack of standardized methods and interpretation of genotypic and phenotypic assays currently limit application of HIV-1 resistance testing, but progress is rapid (38, 39, 41, 42).
A clinical diagnosis of AIDS, baseline CD4 counts, and HIV-1 viral load were bivariate predictors of virologic response, but only the baseline plasma HIV-1 RNA level remained significant in multivariate models. Previous antiretroviral therapy was also a predictor of virologic response. Both the number and the duration of previous therapy with nucleoside reverse transcriptase and protease inhibitors were bivariate predictors. Although the drug history variables were not as strong as HIV-1 genotype in predicting response to saquinavir–ritonavir therapy, the number of previous protease inhibitors contributed to the predictive capacity of a model with HIV-1 genotype predictors.
The baseline HIV-1 genotype proved to be a robust prognostic tool. In particular, the seven major protease mutations defined a priori were strong predictors of virologic response in this cohort. Although several reverse transcriptase mutations (at codons 69 and 210) had some predictive power, the protease mutations were much stronger predictors of response. This probably reflects the extensive previous exposure to nucleoside reverse transcriptase inhibitors in this cohort; as a result, the virologic response was primarily due to the dual protease inhibitor therapy. All eight patients with the D30N mutation (seven who had it as a single major mutation and one who also had the L90M mutation) were complete responders to saquinavir–ritonavir therapy. The “positive” association of this mutation with virologic outcome is more accurately viewed as the lack of negative association; negative associations were seen with the other major mutations. The virologic response in this group supports clinical trial results in patients switched to saquinavir–ritonavir therapy after nelfinavir-containing regimens failed (52).
Our study has several notable features. First, the clinical cohort, with its complex and varied antiretroviral treatment histories, reflects the diversity of drug treatment in HIV-infected persons in clinical practice. Inclusion of patients with a wide variety of treatment histories allowed evaluation of treatment history as a predictor of virologic outcome. Second, HIV-1 genotype was not used as a criterion for selection of patients into the study. Knowing the genotype before enrollment could have introduced bias into subject selection. Third, the signed-sum method used in the multivariate analysis of the genotypic patterns helped to simplify complex data and avoid the potential problem of colinearity between mutation predictors. Finally, recent retrospective studies support our findings, although not all of these studies have shown that the predictive capacity of resistance testing is independent of standard clinical predictors (37, 53–56).
Our results indicate that HIV-1 genotypic resistance testing provides prognostic information in patients who are experiencing less-than-optimal virologic response to antiretroviral therapy that contains protease inhibitors and that this information is not available through standard clinical evaluation. In randomized, controlled pilot trials (50, 51), this prognostic capacity may translate into improved treatment outcomes of HIV-1 infected persons, but further study is required.
Acknowledgments
The authors thank Mitch Katz, MD, of the Department of Public Health, San Francisco, for critical review of the manuscript, and John Sninsky, PhD, of Roche Molecular Systems, Alameda, California, for contributing PCR kits and reagents used in the HIV-1 sequencing reactions.
References
- 1.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337:734–9. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 2.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337:725–33. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 3.Montaner JS, Reiss P, Cooper D, Vella S, Harris M, Conway B, et al. A randomized, double-blind trial comparing combinations of nevirapine, didanosine, and zidovudine for HIV-infected patients: the INCAS trial. The Netherlands, Canada and Australia Study. JAMA. 1998;279:930–7. doi: 10.1001/jama.279.12.930. [DOI] [PubMed] [Google Scholar]
- 4.Cameron DW, Heath-Chiozzi M, Danner S, Cohen C, Kravcik S, Maurath C, et al. Randomized placebo-controlled trial of ritonavir in advanced HIV-1 disease. The Advanced HIV Disease Ritonavir Study Group. Lancet. 1998;351:543–9. doi: 10.1016/s0140-6736(97)04161-5. [DOI] [PubMed] [Google Scholar]
- 5.Powderly WG, Landay A, Lederman MM. Recovery of the immune system with antiretroviral therapy: the end of opportunism? JAMA. 1998;280:72–7. doi: 10.1001/jama.280.1.72. [DOI] [PubMed] [Google Scholar]
- 6.Autran B, Carcelain G, Li TS, Blanc C, Mathez D, Tubiana R, et al. Positive effects of combined antiretroviral therapy on CD4 + T cell homeostasis and function in advanced HIV disease. Science. 1997;277:112–6. doi: 10.1126/science.277.5322.112. [DOI] [PubMed] [Google Scholar]
- 7.Angel JB, Kumar A, Parato K, Filion LG, Diaz-Mitoma F, Daftarian P, et al. Improvement in cell-mediated immune function during potent anti-human immunodeficiency virus therapy with ritonavir plus saquinavir. J Infect Dis. 1998;177:898–904. doi: 10.1086/515244. [DOI] [PubMed] [Google Scholar]
- 8.Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–60. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 9.Hogg RS, O'Shaughnessy MV, Gataric N, Yip B, Craib K, Schechter MT, et al. Decline in deaths from AIDS due to new antiretrovirals [Letter] Lancet. 1997;349:1294. doi: 10.1016/S0140-6736(05)62505-6. [DOI] [PubMed] [Google Scholar]
- 10.Fatkenheuer G, Theisen A, Rockstroh J, Grabow T, Wicke C, Becker K, et al. Virologic treatment failure of protease inhibitor therapy in an unselected cohort of HIV-infected patients. AIDS. 1997;11:F113–6. doi: 10.1097/00002030-199714000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Deeks S, Hecht F, Swanson M, Elbeikt T, Loftus R, Cohen PT, et al. HIV RNA and CD4 cell count response to protease inhibitor therapy in an urban AIDS clinic: response to both initial and salvage therapy. AIDS. 1999;13:F35–F43. doi: 10.1097/00002030-199904160-00001. [DOI] [PubMed] [Google Scholar]
- 12.Flexner C. HIV-protease inhibitors. N Engl J Med. 1998;338:1281–92. doi: 10.1056/NEJM199804303381808. [DOI] [PubMed] [Google Scholar]
- 13.Vanhove GF, Schapiro JM, Winters MA, Merigan TC, Blaschke TF. Patient compliance and drug failure in protease monotherapy [Letter] JAMA. 1996;276:1955–6. [PubMed] [Google Scholar]
- 14.Schapiro JM, Winters MA, Stewart F, Efron B, Norris J, Kozal MJ, et al. The effect of high-dose saquinavir on viral load and CD4 + T-cell counts in HIV-infected patients. Ann Intern Med. 1996;124:1039–50. doi: 10.7326/0003-4819-124-12-199606150-00003. [DOI] [PubMed] [Google Scholar]
- 15.Mayers DL, Gallahan DL, Martin GJ, Emmons WW, Chung RC, Spooner KM, et al. Drug resistance genomes from plasma virus of HIV-infected patients failing combination drug therapy [Abstract]. Programme and Abstracts, First International Workshop on HIV Resistance, Treatment Strategies and Eradication; St. Petersburg, Florida. 25-28 June 1997; London: International Medical Pr; p. 52. [Google Scholar]
- 16.Lorenzi P, Yerly S, Abderrakim K, Fathi M, Rutschmann OT, von Overbeck J, et al. Toxicity, efficacy, plasma drug concentrations and protease mutations in patients with advanced HIV infection treated with ritonavir plus saquinavir. Swiss HIV Cohort Study. AIDS. 1997;11:F95–9. doi: 10.1097/00002030-199712000-00002. [DOI] [PubMed] [Google Scholar]
- 17.Hazuda D, Kuo L. Failure of AZT: a molecular perspective. Nat Med. 1997;3:836–7. doi: 10.1038/nm0897-836. [DOI] [PubMed] [Google Scholar]
- 18.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–9. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 19.Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper PJ, Mo HM, et al. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat Med. 1996;2:760–6. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- 20.Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero JC, et al. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature. 1995;374:569–71. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 21.Jacobsen H, Hanggi M, Ott M, Duncan IB, Owen S, Andreoni M, et al. In vivo resistance to a human immunodeficiency virus type 1 proteinase inhibitor: mutations, kinetics, and frequencies. J Infect Dis. 1996;173:1379–87. doi: 10.1093/infdis/173.6.1379. [DOI] [PubMed] [Google Scholar]
- 22.Shafer RW, Winters MA, Palmer S, Merigan TC. Multiple concurrent reverse transcriptase and protease mutations and multidrug resistance of HIV-1 isolates from heavily treated patients. Ann Intern Med. 1998;128:906–11. doi: 10.7326/0003-4819-128-11-199806010-00008. [DOI] [PubMed] [Google Scholar]
- 23.Condra JH, Holder DJ, Schleif WA, Blahy OM, Danovich RM, Gabryelski LJ, et al. Genetic correlates of in vivo viral resistance to indinavir, a human immunodeficiency virus type 1 protease inhibitor. J Virology. 1996;70:8270–6. doi: 10.1128/jvi.70.12.8270-8276.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patick AK, Mo H, Markowitz M, Appelt K, Wu B, Musick L, et al. Antiviral and resistance studies of AG1343, an orally bioavailable inhibitor of human immunodeficiency virus protease. Antimicrob Agents Chemother. 1996;40:292–7. doi: 10.1128/aac.40.2.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schinazi RF, Larder BA, Mellors JW. Mutations in retroviral genes associated with drug resistance. International Antiviral News. 1997;5:2–14. [Google Scholar]
- 26.D'Aquila RT, Johnson VA, Welles SL, Japour AJ, Kuritzkes DR, DeGruttola V, et al. Zidovudine resistance and HIV-1 disease progression during antiretroviral therapy. AIDS Clinical Trials Group Protocol 116B/117 Team and the Virology Committee Resistance Working Group. Ann Intern Med. 1995;122:401–8. doi: 10.7326/0003-4819-122-6-199503150-00001. [DOI] [PubMed] [Google Scholar]
- 27.Kozal MJ, Kroodsma K, Winters MA, Shafer RW, Efron B, Katzenstein DA, et al. Didanosine resistance in HIV-infected patients switched from zidovudine to didanosine monotherapy. Ann Intern Med. 1994;121:263–8. doi: 10.7326/0003-4819-121-4-199408150-00005. [DOI] [PubMed] [Google Scholar]
- 28.Shafer RW, Iversen AK, Winters MA, Aguiniga E, Katzenstein DA, Merigan TC. Drug resistance and heterogeneous long-term virologic responses of human immunodeficiency virus type 1-infected patients to zidovudine and didanosine combination therapy. The AIDS Clinical Trials Group 143 Virology Team. J Infect Dis. 1995;172:70–8. doi: 10.1093/infdis/172.1.70. [DOI] [PubMed] [Google Scholar]
- 29.Miller V, Phillips A, Rottmann C, Staszewski S, Pauwels R, Hertogs K, et al. Dual resistance to zidovudine and lamivudine in patients treated with zidovudine-lamivudine combination therapy: association with therapy failure. J Infect Dis. 1998;177:1521–32. doi: 10.1086/515304. [DOI] [PubMed] [Google Scholar]
- 30.Rey D, Hughes M, Pi JT, Winters MA, Merigan TC, Katzenstein DA. HIV-1 reverse transcriptase codon 215 mutation in plasma RNA: immunologic and virologic responses to zidovudine. The AIDS Clinical Trials Group Study 175 Virology Team. J Acquir Immune Defic Syndr. 1998;17:203–8. doi: 10.1097/00042560-199803010-00003. [DOI] [PubMed] [Google Scholar]
- 31.Larder BA, Kohli A, Kellem P, Kemp SD, Kronick M, Henfrey RD. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature. 1993;365:671–3. doi: 10.1038/365671a0. [DOI] [PubMed] [Google Scholar]
- 32.Stuyver L, Wyseur A, Rombout A, Louwagie J, Scarcez T, Verhofstede C, et al. Line probe assay for rapid detection of drug-selected mutations in the human immunodeficiency virus type 1 reverse transcriptase gene. Antimicrob Agents Chemother. 1997;41:284–91. doi: 10.1128/aac.41.2.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozal MJ, Shah N, Shen N, Yang R, Fucini R, Merigan TC, et al. Extensive polymorphism observed in HIV-1 clade B protease gene using high-density oligonucleotide arrays. Nat Med. 1996;2:753–9. doi: 10.1038/nm0796-753. [DOI] [PubMed] [Google Scholar]
- 34.Eastman PS, Boyer E, Mole L, Kolberg J, Urdea M, Holodniy M. Nonisotopic hybridization assay for determination of relative amounts of genotypic human immunodeficiency virus type 1 zidovudine resistance. J Clin Microbiol. 1995;33:2777–80. doi: 10.1128/jcm.33.10.2777-2780.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Japour AJ, Mayers DL, Johnson VA, Kuritzkes DR, Beckett LA, Arduino JM, et al. Standardized peripheral blood mononuclear cell culture assay for determination of drug susceptibilities of clinical human immunodeficiency virus type 1 isolates. The RV-43 Study Group, the AIDS Clinical Trials Group Virology Committee Resistance Working Group. Antimicrob Agents Chemother. 1993;37:1095–101. doi: 10.1128/aac.37.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hertogs K, de Bethune MP, Miller V, Ivens T, Schel P, Van Cauwenberge A, et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob Agents Chemother. 1998;42:269–76. doi: 10.1128/aac.42.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deeks SG, Hellman NS, Grant RM, Parkin N, Petropoulos CJ, Becker M, et al. Novel four-drug salvage treatment regimens after failure of a human immunodeficiency virus type 1 protease inhibitor-containing regimen: antiviral activity and correlation of baseline phenotype drug susceptibility with virologic outcome. J Infect Dis. 1999;179:1375–81. doi: 10.1086/314775. [DOI] [PubMed] [Google Scholar]
- 38.Condra JH. Resisting resistance: maximizing the durability of antiretroviral therapy [Editorial] Ann Intern Med. 1998;128:951–4. doi: 10.7326/0003-4819-128-11-199806010-00017. [DOI] [PubMed] [Google Scholar]
- 39.Vella S. Advances in the virology of HIV infection and implications for clinical management. AIDS Clin Care. 1998;10:17–9. [PubMed] [Google Scholar]
- 40.Condra JH, Holder DJ, Graham DJ, Shivaprakash M, Laird DT, Schleif WA, et al. Genotypic or phenotypic susceptibility testing may not predict clinical responses to indinavir [Abstract]. Programme and Abstracts, First International Workshop on HIV Resistance, Treatment Strategies and Eradication; St. Petersburg, Florida. 25-28 June 1997; London: International Medical Pr; pp. 31–2. [Google Scholar]
- 41.Deeks SG, Abrams DI. Genotypic-resistance assays and antiretroviral therapy [Editorial] Lancet. 1997;349:1489–90. doi: 10.1016/S0140-6736(05)62092-2. [DOI] [PubMed] [Google Scholar]
- 42.Hirsch MS, Conway B, D'Aquila RT, Johnson VA, Brun-Vezinet F, Clotet B, et al. Antiretroviral drug resistance testing in adults with HIV infection. International AIDS Society—USA Panel. JAMA. 1998;279:1984–91. doi: 10.1001/jama.279.24.1984. [DOI] [PubMed] [Google Scholar]
- 43.Sun R, Ku J, Jayakar H, Kuo JC, Brambilla D, Herman S, et al. Ultrasensitive reverse transcription-PCR assay for quantitation of human immunodeficiency virus type-1 RNA in plasma. J Clin Microbiol. 1998;36:2964–9. doi: 10.1128/jcm.36.10.2964-2969.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winters MA, Schapiro JM, Lawrence J, Merigan TC. Human immunodeficiency virus type 1 protease genotypes and in vitro protease inhibitor susceptibilities of isolates from individuals who were switched to other protease inhibitors after long-term saquinavir treatment. J Virol. 1998;72:5303–6. doi: 10.1128/jvi.72.6.5303-5306.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Human Retroviruses and AIDS: A Compilation and Analysis of Nucleic and Amino Acid Sequences. Los Alamos, NM: Theoretical Biology and Biophysics Group, T-10, Los Alamos National Laboratory; 1997. [Google Scholar]
- 46.Chatterjee S, Hadi AS. Sensitivity in Regression Analysis. New York: Wiley & Sons; 1988. p. 117. [Google Scholar]
- 47.Efron B. Estimating the error rate of a prediction rule: some improvements on cross-validation. Journal of the American Statistical Association. 1983;78:316–31. [Google Scholar]
- 48.Guidelines for the use of antiretroviral agents in HIV-infected adults and adolescents. Department of Health and Human Services and the Henry J. Kaiser Family Foundation. Ann Intern Med. 1998;128(12 Pt 2):1079–100. doi: 10.7326/0003-4819-128-12_part_2-199806151-00003. [DOI] [PubMed] [Google Scholar]
- 49.Carpenter CC, Fischl MA, Hammer SM, Hirsch MS, Jacobsen DM, Katzenstein DA, et al. Antiretroviral therapy for HIV infection in 1998: updated recommendations of the International AIDS Society—USA Panel. JAMA. 1998;280:78–86. doi: 10.1001/jama.280.1.78. [DOI] [PubMed] [Google Scholar]
- 50.Durant J, Clevenbergh P, Halfon P, Delgiudice P, Porsin S, Simonet P, et al. Drug-resistance genotyping in HIV-1 therapy: the VIRADAPT randomized controlled trial. Lancet. 1999;353:2195–9. doi: 10.1016/s0140-6736(98)12291-2. [DOI] [PubMed] [Google Scholar]
- 51.Baxter JD, Mayers DL, Wentworth DN, Neoton JD, Merigan TC. A pilot study of the short-term effects of antiretroviral management based on plasma genotypic antiretroviral resistance testing (GART) in patients failing antiretroviral therapy [Abstract]. CPCRA 046 Study Team; Proceedings of Sixth Conference on Retroviruses and Opportunistic Infectious; Chicago. 31 January-4 February 1999. [Google Scholar]
- 52.Tebas P, Patick AK, Kane EM, Klebert MK, Simpson JH, Erice A, et al. Virologic responses to a ritonavir-saquinavir-containing regimen in patients who have previously failed nelfinavir. AIDS. 1999;13:F23–8. doi: 10.1097/00002030-199902040-00002. [DOI] [PubMed] [Google Scholar]
- 53.Patick AK, Zhang M, Hertogs K, Griffiths L, Mazabel E, Pauwels R, et al. Correlation of virological response with genotype and phenotype of plasma HIV-1 variants in patients treated with nelfinavir in the US expanded access program [Abstract]. Programme and Abstracts, Second International Workshop on HIV Drug Resistance Treatment Strategies; 24-27 June 1998; Lake Maggiore, Italy. London: International Medical Pr; 1998. p. 39. [Google Scholar]
- 54.Harrigan PR, Montaner JS, Hogg RS, Yip B, Hertogs K, Pauwels R, et al. Baseline resistance profile predicts response to ritonavir/saquinavir therapy in a community setting. AIDS. doi: 10.1097/00002030-199910010-00008. In press. [DOI] [PubMed] [Google Scholar]
- 55.Lorenzi P, Opravil M, Hirschel B, Chave JP, Furrer HJ, Sax H, et al. Impact of drug resistance mutations on virologic response to salvage therapy. Swiss HIV Cohort Study. AIDS. 1999;13:F17–21. doi: 10.1097/00002030-199902040-00001. [DOI] [PubMed] [Google Scholar]
- 56.Lanier ER, Scott J, Steel H, Hetherington S, Ait-Khaled M, Pearce G, et al. Multivariate analysis of predictors of response to abacavir: comparison of prior antiretroviral therapy, baseline HIV RNA, CD4 count and viral resistance [Abstract] Antiviral Therapy. 1999;4(Suppl 1):56. [Google Scholar]