

Abstract
It has been suggested that disturbances in endocannabinoid signaling contribute to the development of depressive illness; however, at present there is insufficient evidence to allow for a full understanding of this role. To further this understanding, we performed an analysis of the endocannabinoid system in an animal model of depression. Male rats exposed to chronic, unpredictable stress (CUS) for 21 days exhibited a reduction in sexual motivation, consistent with the hypothesis that CUS in rats induces depression-like symptoms. We determined the effects of CUS, with or without concurrent treatment with the antidepressant imipramine (10 mg/kg), on CP55940 binding to the cannabinoid CB1 receptor; whole tissue endocannabinoid content; and fatty acid amide hydrolase (FAAH) activity in the prefrontal cortex, hippocampus, hypothalamus, amygdala, midbrain and ventral striatum. Exposure to CUS resulted in a significant increase in CB1 receptor binding site density in the prefrontal cortex and a decrease in CB1 receptor binding site density in the hippocampus, hypothalamus and ventral striatum. Except in the hippocampus, these CUS-induced alterations in CB1 receptor binding site density were attenuated by concurrent antidepressant treatment. CUS alone produced a significant reduction in N-arachidonylethanolamine (anandamide) content in every brain region examined, which was not reversed by antidepressant treatment. These data suggest that the endocannabinoid system in cortical and subcortical structures is differentially altered in an animal model of depression and that the effects of CUS on CB1 receptor binding site density are attenuated by antidepressant treatment while those on endocannabinoid content are not.
Keywords: 2-AG, cannabinoid, anhedonia, depression, HPA axis, glucocorticoid
Introduction
Depressive illness is widespread and exhibits a lifetime prevalence rate of 16% (Kessler et al., 2003). Depressive illnesses are characterized by an array of disturbances in emotional behavior, memory, neurovegetative functions and hedonic processing. The neurobiological mechanisms subserving the development, manifestation and treatment of depression are complex, and there is ample evidence that disturbances in monoaminergic signaling, glucocorticoid activity and neurotrophic/neuroplastic processes are involved (Ressler and Nemeroff, 2000; Holsboer, 2000; Duman and Monteggia, 2006; McEwen, 2005).
Our understanding of the biochemical processes in depression has been shaped by preclinical work employing animal models of depression. While an unequivocal animal model of depression is not available, several models have demonstrated a high degree of biological and behavioral concordance with major depression in humans (Nestler et al., 2002; O'Neil and Moore, 2003). In particular, the chronic unpredictable stress model (CUS; also referred to as chronic mild stress [CMS]) has been found to have reasonable face validity as a model of depression, largely because this model incorporates etiological factors that are similar to human depression (protracted exposure to mild, but unpredictable stressors) and elicits both biochemical (eg. alterations in monoaminergic receptors and glucocorticoid signaling) and behavioral (eg. anhedonia) responses that parallel those seen in human major depression (Lopez et al., 1998; Willner, 2005; however see Reid et al., 1997; Forbes et al., 1996 for a critique of the model). Accordingly, the CUS model represents an useful tool to examine potential systems involved in the pathophysiology of depression.
There is an increasing evidence to support a role for the endocannabinoid system (ECS) in the neurobiology of depression (Hill and Gorzalka, 2005a; Witkin et al., 2005; Vinod and Hungund, 2006; Mangieri and Piomelli, 2007). The central ECS is composed of a G-protein coupled receptor (CB1 receptor; Howlett, 2002) and two endogenous ligands, N-arachidonylethanolamide (anandamide or AEA; Devane et al., 1992) and 2-arachidonoylglycerol (2-AG; Sugiura et al., 1995). There is predominant expression of the CB1 receptor on axon terminals where it inhibits transmitter release via activation of Gi/o proteins and subsequent attenuation of axonal calcium influx (Freund et al., 2003). Signaling at the CB1 receptor is elicited by both endocannabinoids, which are likely released from postsynaptic cells by changes in neuronal activity (Chevaleyre et al., 2006; Marsicano and Lutz, 2006). The functional lifespan of these molecules is regulated by hydrolytic enzymes; AEA is exclusively metabolized in the brain by fatty acid amide hydrolase (FAAH), while 2-AG is metabolized primarily by monoacylglycerol lipase, although it is likely that other hydrolytic enzymes, including FAAH, can also inactivate 2-AG in the brain (Ligresti et al., 2005; Hillard, 2000a).
The ECS is present in moderate to high levels in limbic brain regions, such as the prefrontal cortex, hippocampus and amygdala, where neuronal activity is known to be altered in depression (Herkenham et al., 1991; Egertova et al., 2003; Bisogno et al., 1999). Genetic deletion of the CB1 receptor in mice results in a phenotype that is reminiscent of the symptom profile of melancholic depression (reviewed in Hill and Gorzalka, 2005a), such as increased depressive-like and anxiety-like behaviors (Steiner et al., 2008b; Uriguen et al., 2004); perseverance of emotionally aversive memories (Marsicano et al., 2002); enhanced activation and impaired inhibition of the hypothalamic-pituitary-adrenal (HPA) axis (Cota et al., 2007); diminished feeding behavior (Ravinet Trillou et al., 2004) and impaired responsiveness to rewarding stimuli (Sanchis-Segura et al., 2004). Consistent with this hypothesis, at the clinical level, we have recently reported that serum 2-AG concentrations are reduced in women with major depression compared to a matched control group (Hill et al., 2008b) and post-mortem analysis has revealed that the expression of CB1 receptors on glial cells in the anterior cingulate cortex is significantly reduced in depressed individuals (Koethe et al., 2007).
Pharmacological facilitation of the ECS has been shown to produce an antidepressant response alone and to enhance the effects of conventional antidepressants in several rodent models of antidepressant efficacy (Bortolato et al., 2007; Filip et al., 2006; Gobbi et al., 2005; Hill and Gorzalka, 2005b; Hill et al., 2007b). Further, several modalities known to produce antidepressant effects in humans have been shown to increase endocannabinoid/CB1 receptor signaling in the brain, including chronic treatment with the antidepressant desipramine (Hill et al., 2006), repeated electroconvulsive shock treatment (Hill et al., 2007a) and sleep deprivation (Chen and Bazan, 2005). Collectively, these studies support the hypotheses that hypofunctional endocannabinoid signaling contributes to the etiology or symptom spectrum of depressive illness and that increasing endocannabinoid signaling is associated with anti-depressant efficacy.
On the other hand, there is also evidence that hyperfunctional ECS is associated with depression. There is a reliable association between long-term cannabis use and the development of suicidal ideations and depressive illness (Bovasso, 2001; Degenhardt et al., 2003; Lynskey et al., 2004), suggesting that overactive CB1 receptor signaling promotes the development of depression. CB1 receptor protein expression, binding site density and signal transduction are up-regulated in the prefrontal cortex of depressed individuals who have committed suicide (Hungund et al., 2004). Similarly, chronic mild stress exposure to rats (Bortolato et al., 2007) and CUS exposure to mice (Hillard et al., 2006) increases CB1 receptor mRNA in the prefrontal cortex. Both acute and chronic treatments with a CB1 receptor antagonist elicit antidepressant responses in the forced swim test and chronic mild stress paradigm (Griebel et al., 2005; Shearman et al., 2005; Steiner et al., 2008a; Tzavara et al., 2003). Finally, some interventions that reduce depressive symptoms also down-regulate the ECS; including electroconvulsive shock (Hill et al., 2007a) and chronic treatment with the antidepressant fluoxetine (Oliva et al., 2005). Taken together, these results suggest reducing ECS signaling can alleviate depressive symptoms.
In an attempt to reconcile these apparently discrepant findings, we have previously suggested that the endocannabinoid system could differentially contribute to the symptomatology of depression in a region-specific manner (Hill et al., 2007a). Thus, the aim of the current study is to carry out a regional analysis of the endocannabinoid system in rats exposed to CUS model and to explore the sensitivity of these changes to the antidepressant, imipramine. Given the equivocal nature of the CUS model for evoking “depression”-like symptoms (refer to Willner, 2005; Reid et al., 1997; Forbes et al., 1996) we have included a study of the effects of CUS on sexual motivation, a parameter which represents a measure of natural reward and motivational drive and is often impaired in depression (Williams and Reynolds, 2006). The current data confirm the hypothesis that endocannabinoid signaling is regionally modulated in an animal model of depression, and that imipramine normalizes several, but not all, of these changes.
Methods
Subjects
Male, Long-Evans rats (300 g; 70 days of age) housed in groups of three in triple mesh wire caging maintained at 21 °C, and on a 12 h light/dark cycle, with lights on at 0900 h. All rats were given ad libitum access to Purina Rat Chow and tap water, except during deprivation periods of the stressing protocol (outlined below). In addition to the experimental animals, female, Long-Evans rats were used as stimuli for sexual behavior testing. All females were bilaterally ovariectomized at 3 months of age using standard surgical procedures while anesthetized with 75 mg/kg ketamine hydrochloride and 7 mg/kg xylazine (intraperitoneal). All animal protocols were approved by the Canadian Council for Animal Care and the standards of the Animal Ethics Committee of the University of British Columbia.
Chronic Unpredictable Stress Protocol
For behavioral testing, one cohort of animals were either subjected to 21 days of CUS or acted as cage controls and were handled weekly. At the conclusion of this period, all animals were tested for copulatory behavior.
For biochemical assays, a different cohort of animals was employed with four treatment conditions generated to incorporate concurrent antidepressant treatment: 1) vehicle / no stress; 2) imipramine / no stress; 3) vehicle / CUS; 4) imipramine / CUS. Imipramine was dissolved in 0.9 % saline and was administered via intraperitoneal injection at a dose of 10 mg/kg, and at a volume of 1 ml/kg. Injections were given daily between 1200h–1400h using 26 gauge 1/2" needles. The CUS paradigm employed has been repeatedly utilized by our laboratory for both behavioral and biochemical analysis (Brotto et al., 2001; Hill et al., 2005; Hill and Gorzalka, 2004), and is adapted from the original CMS paradigm (Willner et al., 1987). Despite the fact that the original CMS paradigm was designed as an 8–10 week regimen, a large body of research has demonstrated that exposure of rodents to 14 to 21 days of CUS is sufficient to induce neurobiological alterations that parallel those documented in individuals suffering from major depression (Iredale et al., 1996; Lopez et al., 1998; Ossowska et al., 2001, 2002; Xu et al., 2007). Additionally, concurrent administration of antidepressant agents throughout the duration of a 2–3 week CUS paradigm has also been demonstrated to reliably reverse neurochemical changes evoked by this regimen (Lopez et al., 1998; Ossowska et al., 2001, 2002). The CUS paradigm consisted of 2–3 stressors a day from the following list: 30 min tube restraint; 30 min exposure to social crowding with white noise/stroboscopic illumination; 5 min forced swim; 18 h food and/or water deprivation; 3 h cage rotation; and 18 h social isolation in damp bedding. All stressors were separated by a period of at least 2 h and were applied daily over the 21 day period. For the biochemical assays, all animals were rapidly decapitated in the morning after the 21st day of stress exposure following 18 h of overnight social isolation. Brains were removed and the prefrontal cortex (consisting of medial prefrontal cortex and anterior cingulate cortex), hippocampus, amygdala (consisting of central, basolateral and medial nuclei), hypothalamus, ventral striatum (consisting of the nucleus accumbens) and midbrain (including the cell bodies of monoaminergic nuclei in the raphe, ventral tegmental area and locus coeruleus) were sectioned out as previously described (Hill et al., 2006). Brains were frozen in liquid nitrogen within 5–7 min of decapitation and stored at −80 °C until analysis. Two independent cohorts of tissue were collected. One cohort of tissue (n = 7–8) was used for lipid extraction to determine endocannabinoid ligand content. The other cohort of tissue (n=4–5) was used as the source of membranes for CB1 receptor binding and FAAH activity assays. Trunk blood was collected upon decapitation for measurement of serum endocannabinoid content.
Sexual Behavior Testing
Females were induced into estrus using 10 µg estradiol benzoate 48 h prior to testing and 500 µg progesterone 4 h prior to testing. Both hormones were received from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in peanut oil and injected subcutaneously at a volume of 0.1 ml.
Prior to testing, males were exposed to an estrus-induced female on five occasions and then screened for sexual activity in two independent sessions. To achieve the criterion for sexual proficiency, male subjects had to ejaculate at least once during both of two 30 min screening sessions with a receptive female. Males that achieved this criterion were randomly assigned to be exposed to CUS or functioned as cage controls. All behavioral testing occurred during the middle third of the light cycle and was performed by trained observers who were blinded to the treatment groups. Testing occurred in cubical Plexiglas (30×30×30 cm) and cylindrical glass (30 cm diameter×45 cm height) chambers lined with contact bedding. Males were habituated to the chambers for 5 min prior to the beginning of testing. Test sessions began with the presentation of an estrus-induced female to a male in an individual testing chamber. The sexual behavior parameters scored were: frequency of mounts with pelvic thrusting prior to ejaculation, frequency of penile intromissions prior to ejaculation, frequency of ejaculations, latency to initiate mounting behavior, latency to initiate intromitting behavior, ejaculation latency (i.e., the period between the first intromission and the first ejaculation) and the postejaculatory interval (i.e., the period between ejaculation and the first intromission of the next copulatory bout). Stimulus females were rotated between males every 10 min to maintain sexual interest.
Membrane Preparation
Dissected brain sections were homogenized in 10 volumes of 0.32 M sucrose containing 3 mM HEPES (pH 7.5) and 1 mM EDTA. The homogenates were centrifuged at 18,000 × g for 20 min after which the supernatant was rapidly decanted. The remaining pellet, which is the membrane fraction, was resuspended in 1–2 ml TME buffer (50 mM Tris HCl, pH 7.4; 1 mM EDTA and 3 mM MgCl2) containing 1 mM sodium orthovanadate. Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA, USA).
CB1 Receptor Binding Assay
CB1 receptor binding assays were performed using a Multiscreen Filtration System with Durapore 1.2-µM filters (Millipore, Bedford, MA) as described previously (Hillard et al., 1995a). Incubations (total volume = 0.2 mL) were carried out using TME buffer containing 1 mg/mL bovine serum albumin (TME/BSA). Membranes (10 µg protein per incubate) were added to the wells containing 0.25, 0.5, 1.0, or 2.5 nM [3H]CP 55,940. Ten µM Δ9-tetrahydrocannabinol was used to determine non-specific binding. KD and Bmax values were determined by nonlinear curve fitting to the single site binding equation using GraphPad Prism (San Diego, CA, USA).
Fatty Acid Amide Hydrolase Activity Assay
FAAH activity was measured as the conversion of AEA to arachidonic acid and ethanolamine by membrane preparations (Hillard et al., 1995b). AEA labeled with [3H] in the ethanolamine portion of the molecule ([3H]AEA; Omeir et al., 1995) was the radiolabeled substrate. Membranes were incubated in a final volume of 0.5 ml of TME buffer (50 mM Tris-HCl, 3.0 mM MgCl2, and 1.0 mM EDTA, pH 7.4) containing 1.0 mg/ml fatty acid-free bovine serum albumin and 0.2 nM [3H]AEA. Isotherms were constructed using eight concentrations of AEA at concentrations between 10 nM and 10 µM. Incubations were carried out at 37°C and were stopped with the addition of 2 ml of chloroform/methanol (1:2). After standing at ambient temperature for 30 min, 0.67 ml of chloroform and 0.6 ml of water were added. Aqueous and organic phases were separated by centrifugation at 1,000 rpm for 10 min. The amount of [3H] in 1 ml each of the aqueous and organic phases was determined by liquid scintillation counting and the conversion of [3H]AEA to [3H]ethanolamine was calculated. The KI and Vmax values for this conversion were determined by fitting the data to a single site competition equation using Prism. The r2 value for the goodness of fit of the data to the single site, hyperbolic equation was always greater than 0.9 and typically closer to 0.98.
Endocannabinoid Extraction and Analysis
For analysis of endocannabinoid content, brain regions were subjected to a lipid extraction process as described previously (Patel et al., 2003). Briefly, tissue samples were weighed and placed into borosilicate glass culture tubes containing 2 ml of acetonitrile with 84 pmol of [2H8]anandamide and 186 pmol of [2H8]2-AG for extraction. Tissue was homogenized with a glass rod and sonicated for 30 min. Samples were incubated overnight at −10°C to precipitate proteins, and subsequently centrifuged at 1,500 × g. The supernatants were removed to a new glass tube and evaporated to dryness under N2 gas. The samples were resuspended in 300 µl of methanol to recapture any lipids adhering to the glass tube, and dried again under N2 gas. Finally, lipid extracts were suspended in 20 µl of methanol, and stored at −80°C until analysis.
Trunk blood collected upon decapitation was allowed to settle for 1 h after which it was centrifuged at 1200 × g for 15 min, following which 1 ml of plasma was extracted from each sample and frozen at −80°C until extraction. Plasma extractions were performed similar to that previously described for human serum (Hill et al., 2008b). All plasma extractions were performed using Bond Elut C18 solid-phase extraction columns (1 ml; Varian Inc, Lake Forest, CA). Plasma samples were thawed and made up to 15% ethanol, to which the internal standards [2H8]-AEA (16.9 pmol) and [2H8]-2-AG (46.5 pmol) (Cayman Chemicals, Ann Arbor, MI) were added. Samples were then vortexed and centrifuged at 1000 × g for 4 min. The supernatant was loaded on C18 columns, which had been conditioned with 1 ml redistilled ethanol and 3 ml of double distilled water (ddH2O). The remaining pellet was washed with 100 µl of 15% ethanol and centrifuged again for 3 min. The resulting supernatant was also loaded onto the C18 column. Columns were washed with 5 ml ddH2O and eluted with 1 ml of ethyl acetate. The ethyl acetate layer in the resulting elute was removed and dried under N2. Lipids in the residual ddH2O phase were extracted by mixing with an additional 1 ml of ethyl acetate, which was added to the original ethyl acetate solution. Once dried, samples were resuspended in 20 µl of methanol and stored at −80°C until analysis.
The contents of the two primary endocannabinoids AEA and 2-AG within lipid extracts in methanol from brain tissue and plasma were determined using isotope-dilution liquid chromatography–mass spectrometry as described previously (Patel et al., 2005a).
Statistics
Comparison of the effects of CUS on parameters of sexual behavior was performed using independent t-tests. Comparison of the effects of CUS exposure and/or imipramine administration on parameters of CB1 receptor binding, FAAH activity and endocannabinoid ligand content in different brain regions were analyzed using a univariate analysis of variance (ANOVA), with stress exposure and drug treatment acting as fixed factors. When applicable, post-hoc analysis of each of these variables in each region was performed using a Tukey’s test. Significance was established against an alpha level of 0.05.
Results
The Effects of CUS on Sexual Behavior
Exposure of male rats to CUS resulted in a significant increase in the latency of rats to initiate both mounting [t (12) = 3.41, p < 0.01; Table 1] and intromitting [t (12) = 3.67, p < 0.01; Table 1] behaviors with a receptive female rat. There was a near significant trend in the ability of CUS to increase the latency required for male rats to achieve ejaculation during copulation [t (12) = 1.87, p = 0.08; Table 1] as well as to reduce the total number of ejaculations [t (12) = 1.70, p = 0.1; Table 1]. There was no effect of CUS exposure on the frequency of mounts [t (12) = 1.21, p > 0.05; Table 1]; intromissions [t (12) = 0.34, p > 0.05; Table 1];or the latency required to re-initiate sexual activity following the first ejaculation [t (12) = 0.01, p > 0.05; Table 1]. Thus, exposure of male rats to CUS resulted in a significant diminution of motivation for sexual activity, but did not significantly impair sexual performance once it was initiated.
Table 1. The effects of exposure to chronic, unpredictable stress (CUS) on parameters of male sexual behavior.
Exposure of male rats to 21 days of CUS resulted in a significant impairment in sexual motivation as demonstrated by the significant increase in the latency to engage in sexual activity with a receptive female rat. The specificity of these effects are demonstrated by the fact that sexual performance itself was not otherwise compromised by CUS exposure. Significant differences from control animals (p < 0.05) denoted by *. Both treatment conditions are n = 7. Data are presented as mean values +/− SEM.
| Control | CUS | |
|---|---|---|
| Mounts: | 5.86 +/− 1.18 | 8.29 +/− 1.63 |
| Intromissions: | 11.57 +/− 0.95 | 12.29 +/− 1.26 |
| Ejaculations: | 2.00 +/− 0.31 | 1.29 +/− 0.29 |
| Mount Latency (s): | 19.43 +/− 6.26 | 68.57 +/− 12.97* |
| Intromission Latency (s): | 26.57 +/− 7.39 | 80.57 +/− 12.72* |
| Ejaculation Latency (s): | 303.43 +/− 18.52 | 358.00 +/− 22.51 |
| Post Ejaculatory Interval (s): | 311.71 +/− 12.29 | 311.43 +/− 16.27 |
Effects of CUS and/or Imipramine Treatment on CB1 Receptor Binding Parameters
In the prefrontal cortex (PFC), the agonist binding site density (Bmax) of the CB1 receptor was significantly increased in rats exposed to CUS compared to non-stressed controls [F (1, 16) = 5.09, p < 0.04; Fig. 1a]; there was no effect of imipramine treatment [F (1, 16) = 2.77, p > 0.05]; and there was no significant interaction between CUS and imipramine treatment [F (1, 16) = 2.62, p > 0.05]. The equilibrium dissociation constant (Kd) for [3H]CP55940 binding to the CB1 receptor was not affected by CUS exposure [F (1, 16) = 2.31, p > 0.05; Table 2] but was significantly reduced by imipramine compared to saline treated rats [F (1, 16) = 5.09, p < 0.04]. There was no significant interaction between CUS and imipramine treatment [F (1, 16) = 0.09, p > 0.05]. Therefore, CUS increased CB1 receptor binding site density in the PFC and imipramine treatment increased agonist affinity for the CB1 receptor.
Figure 1.
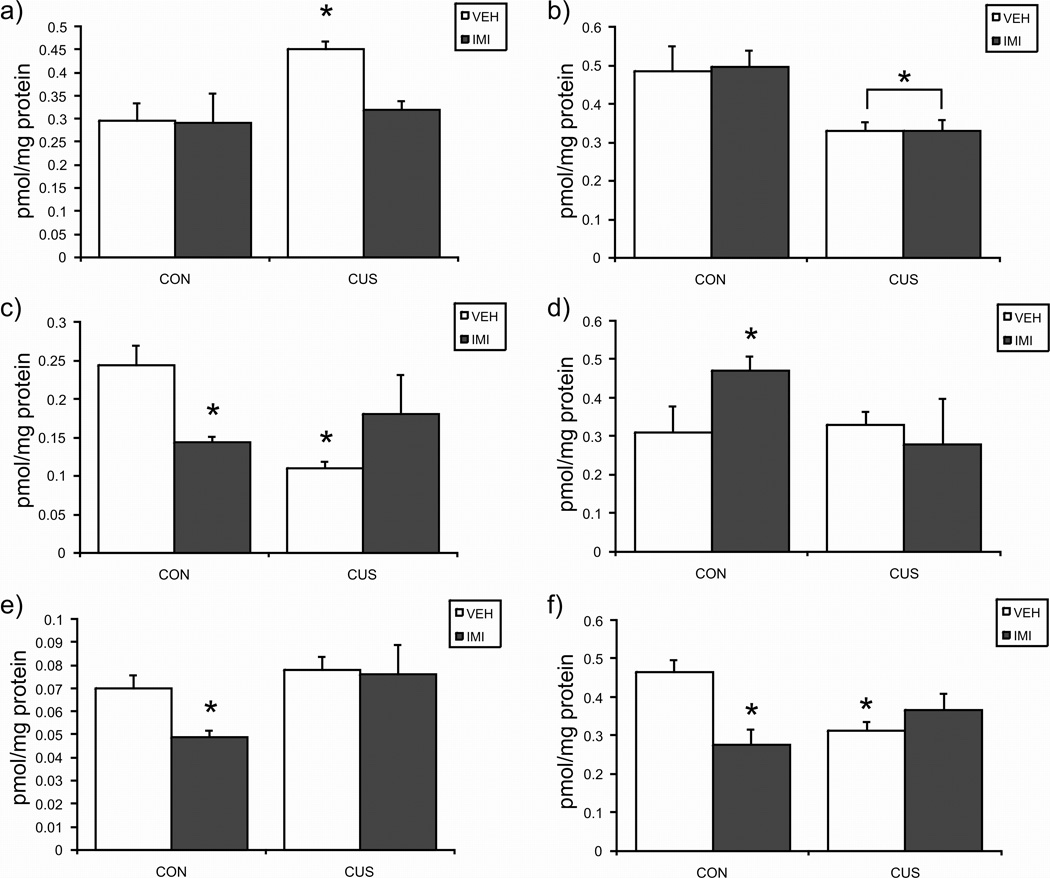
The effects of exposure to 21 days of chronic unpredictable stress (CUS), imipramine (10 mg/kg; IMI) administration, or both regimens combined, on the maximal binding (Bmax) of [3H]CP55940 to the cannabinoid CB1 receptor in the a) prefrontal cortex; b) hippocampus; c) hypothalamus; d) amygdala; e) midbrain; f) ventral striatum. Values are denoted as means ± SEM. Significant differences from non-stressed (CON), vehicle (VEH) treated animals (p < 0.05) denoted by *. For all treatment conditions, n = 4–5.
Table 2. The effects of exposure to chronic, unpredictable stress (CUS) and/or concurrent treatment with the antidepressant imipramine (IMI; 10 mg/kg) on the dissociation constant (Kd) of 3H-CP55940 from the cannabinoid CB1 receptor.
Exposure of male rats to CUS for 21 days resulted in a significant reduction in the binding affinity (Kd) of the cannabinoid CB1 receptor in the hypothalamus, and this effect was prevented by concurrent IMI treatment. IMI treatment for 21 days reduced the Kd of the CB1 receptor in the prefrontal cortex and the ventral striatum. Significant differences from non-stressed (CON), vehicle (VEH) treated animals (p < 0.05) denoted by *. For all treatment conditions, n = 4–5. Data are presented as mean nM of 3H-CP55940 +/− SEM.
| CON/VEH | CUS/VEH | CON/IMI | CUS/IMI | |
|---|---|---|---|---|
| Prefrontal Cortex | 0.61 +/− 0.14 | 0.83 +/− 0.10 | 0.38 +/− 0.15* | 0.52 +/− 0.08 |
| Hippocampus | 0.60 +/− 0.11 | 0.59 +/− 0.11 | 0.79 +/− 0.15 | 0.55 +/− 0.06 |
| Hypothalamus | 1.54 +/− 0.15 | 0.57 +/− 0.07* | 1.02 +/− 0.06 | 1.22 +/− 0.45 |
| Ventral Striatum | 0.65 +/− 0.08 | 0.55 +/− 0.12 | 0.34 +/− 0.05* | 0.37 +/− 0.02* |
| Amygdala | 0.56 +/− 0.06 | 0.56 +/− 0.03 | 0.95 +/− 0.19 | 0.49 +/− 0.12 |
| Midbrain | 0.40 +/− 0.08 | 0.25 +/− 0.05 | 0.25 +/− 0.04 | 0.27 +/− 0.04 |
In the hippocampus, CB1 receptor agonist binding site density was significantly decreased by CUS [F (1, 15) = 11.51, p < 0.005; Fig. 1b] and was unaffected by imipramine [F (1, 15) = 0.36, p > 0.05]. There was no significant interaction between the effects of CUS and imipramine treatment on the Bmax of the CB1 receptor [F (1, 15) = 0.40, p > 0.05]. There were no significant effects of CUS [F (1, 15) = 1.14, p > 0.05; Table 2] or imipramine [F (1, 15) = 0.46, p > 0.05], or a significant interaction between these two factors [F (1, 15) = 1.00, p > 0.05], on the Kd of [3H]CP55490 for binding to the CB1 receptor in the hippocampus. Therefore, the only effect of CUS in the hippocampus was a large reduction in CB1 receptor binding site density; this change was unaffected by imipramine treatment.
In the hypothalamus, analysis of variance revealed that there was a significant interaction between CUS and imipramine treatment on CB1 receptor binding site density [F (1, 15) = 10.44, p < 0.01; Fig. 1c]. Post-hoc analysis of these data indicate that the Bmax for [3H]CP55940 binding to the CB1 receptor binding site density was decreased both by CUS (p < 0.01) and imipramine treatment (p < 0.05); however, when CUS and imipramine were administered together, there was no significant difference in this group relative to control animals (p > 0.05). A similar pattern was seen in the effects of CUS and imipramine on the Kd of [3H]CP55940 for the CB1 receptor in that there was a significant interaction between CUS and imipramine treatment [F (1, 15) = 5.43, p < 0.05; Table 2]. Post hoc analyses reveal that CUS produced a significant decrease in the Kd of [3H]CP55940 the CB1 receptor (p < 0.05) while there was no effect of imipramine treatment alone (p > 0.05). When imipramine and CUS were administered concurrently, there was no difference in the Kd of [3H]CP55940 for binding to the CB1 receptor relative to control animals (p > 0.05). Therefore, CUS both reduces CB1 receptor binding site density and increases agonist affinity in the hypothalamus. Both of these effects of CUS are reversed by co-treatment with imipramine. Imipramine alone did not affect CB1 receptor binding parameters in the hypothalamus.
In the ventral striatum, there was a significant interaction between CUS and imipramine treatment on the Bmax of the CB1 receptor [F (1, 15) = 11.03, p < 0.01; Fig. 1f]. Post hoc analyses revealed that both CUS (p < 0.05) and imipramine treatment (p < 0.01) induced significant reductions in Bmax that were not seen when both treatments were administered concurrently (p > 0.05). The Kd for [3H]CP55940 binding to the CB1 receptor was not affected by CUS [F (1, 15) = 0.20, p > 0.05; Table 2] but was significantly reduced by imipramine [F (1, 15) = 12.17, p < 0.01]. There was no interaction between CUS and imipramine treatment [F (1, 15) = 0.87, p > 0.05]. In sum, the primary effect of CUS on CB1 agonist binding in ventral striatum is to reduce the Bmax; an effect that is both mimicked and reversed by co-administration of imipramine.
In the amygdala, there was a significant interaction between CUS and imipramine treatments on the Bmax of the CB1 receptor [F (1, 14) = 5.10, p < 0.05; Fig. 1d]; however, post hoc analysis demonstrated that this effect was due to an increase in Bmax following imipramine treatment (p < 0.05) that did not occur in rats exposed to CUS and imipramine treatment combined (p > 0.05). CUS alone did not affect the Bmax for the CB1 receptor (p > 0.05). The Kd of [3H]CP55940 binding in the amygdala was not affected by CUS [F (1, 14) = 3.76, p > 0.05; Table 2]; imipramine [F (1, 14) = 1.18, p > 0.05]; nor was there an interaction between the treatments [F (1, 14) = 3.76, p > 0.05]. Therefore, there were no effects of CUS on CB1 agonist binding in the amygdala; however, imipramine alone increased CB1 receptor binding site density in control but not CUS treated rats.
Within the midbrain, there was no effect of CUS exposure on CB1 receptor binding site density [F (1, 16) = 2.04, p > 0.05; Fig. 1e], while imipramine produced a significant reduction in Bmax [F (1, 16) = 4.54, p < 0.05]. There was no interaction between CUS exposure and imipramine treatment on the Bmax of the CB1 receptor [F (1, 16) = 1.43, p > 0.05; Fig. 1f]. Neither CUS [F (1, 16) = 1.49, p > 0.05; Table 2] nor imipramine [F (1, 16) = 1.92, p > 0.05], affected the Kd of [3H]CP55940 for the CB1 receptor in the midbrain, nor was there a significant interaction between these two treatment regimens on this parameter [F (1, 16) = 2.75, p > 0.05]. Therefore, midbrain CB1 receptor binding parameters are not affected by CUS; however, imipramine alone decreased CB1 receptor binding site density.
Effects of CUS and/or Imipramine Treatment on Regional, Tissue Endocannabinoid Contents
CUS produced a significant reduction in AEA content in all brain regions examined (Fig. 2). Imipramine treatment alone had no effect on AEA content in any brain region and there were no significant interactions between CUS and imipramine. Therefore, CUS exerts a global and imipramine-insensitive reduction in AEA throughout the limbic system. ANOVA results for the effects of CUS, imipramine treatment, or both on the tissue content of AEA are as follows: Prefrontal cortex [CUS × IMI: F (1, 27) = 0.20, p > 0.05; CUS: F (1, 27) = 25.14, p < 0.001; IMI: F (1, 27) = 0.56, p > 0.05]; hippocampus [CUS × IMI: F (1, 26) = 0.08, p > 0.05; CUS: F (1, 26) = 20.86, p < 0.001; IMI: F (1, 26) = 0.13, p > 0.05]; hypothalamus [CUS × IMI: F (1, 25) = 0.89, p > 0.05; CUS: F (1, 25) = 8.41, p < 0.01; IMI: F (1, 25) = 0.62, p > 0.05]; ventral striatum [CUS × IMI: F (1, 27) = 2.06, p > 0.05; CUS: F (1, 27) = 25.28, p < 0.001; F (1, 27) = 0.02, p > 0.05]; amygdale [CUS × IMI: F (1, 25) = 0.24, p > 0.05; CUS: F (1, 25) = 12.14, p < 0.005; IMI: F (1, 25) = 0.12, p > 0.05]; and midbrain [CUS × IMI: F (1, 27) = 0.24, p > 0.05; CUS: F (1, 27) = 16.18, p < 0.001; IMI: F (1, 27) = 2.02, p > 0.05].
Figure 2.
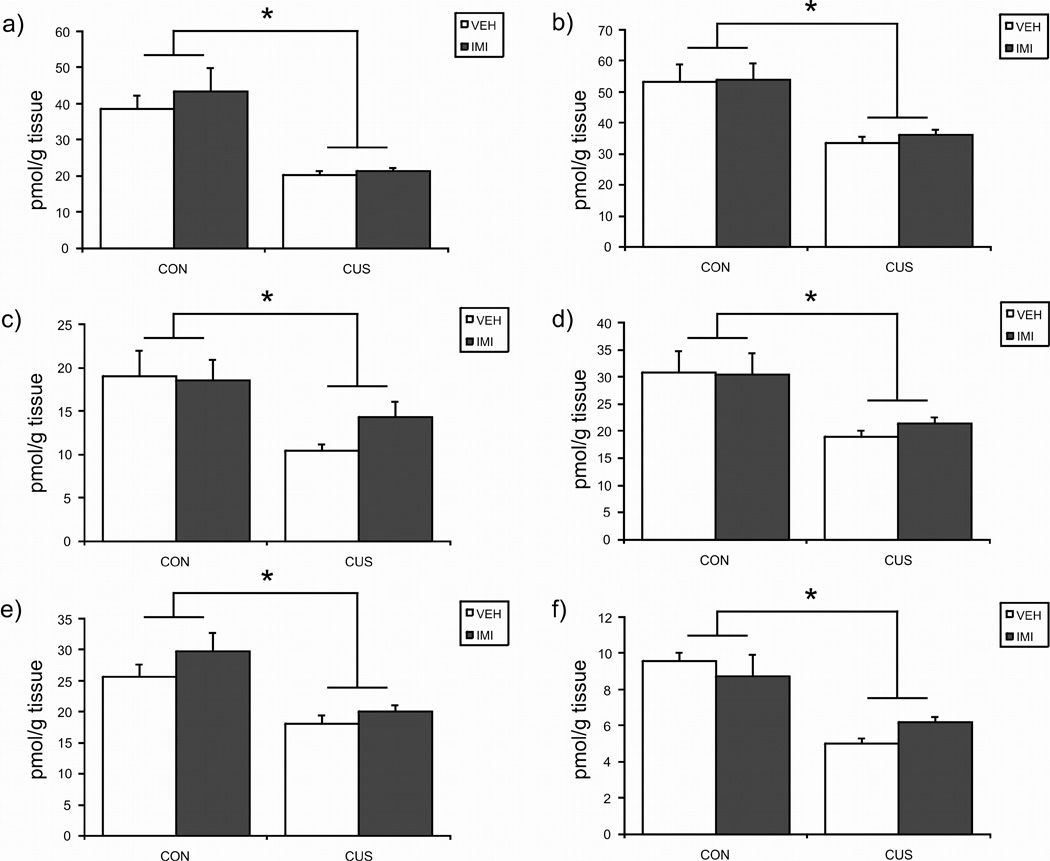
The effects of exposure to 21 days of chronic unpredictable stress (CUS), imipramine (10 mg/kg; IMI) administration, or both regimens combined, on the tissue content of the endocannabinoid ligand anandamide (AEA) in the a) prefrontal cortex; b) hippocampus; c) hypothalamus; d) amygdala; e) midbrain; f) ventral striatum. Values are denoted as means ± SEM. Significant differences\between groups (p < 0.05) denoted by *. For all treatment conditions are, = 7–8.
In contrast, tissue 2-AG contents were increased in rats exposed to CUS only in the hypothalamus and midbrain. These regional increases were unaffected by imipramine treatment (see Table 3 for both data and ANOVA results).
Table 3. The effects of exposure to chronic, unpredictable stress (CUS) and/or concurrent treatment with the antidepressant imipramine (IMI; 10 mg/kg) on the tissue content (nmol/g tissue) of the endocannabinoid 2-arachidonylglycerol (2-AG).
Exposure of male rats to 21 days of CUS resulted in a significant increase in 2-AG content within the hypothalamus and the midbrain, neither of which was reversed by concurrent IMI treatment. Significant differences from non-stressed (CON), vehicle (VEH) treated animals (p < 0.05) denoted by *. For all treatment conditions, n = 7–8. Data are presented as mean nmol/g tissue +/− SEM.
| CON/VEH | CUS/IMI | CON/VEH | CUS/IMI | |
|---|---|---|---|---|
| Prefrontal Cortex | 4.35 +/− 0.29 | 5.16 +/− 0.14 | 5.60 +/− 0.45 | 5.63 +/− 0.52 |
| CUS × IMI F (1, 27) = 0.82, p > 0.05; CUS F (1, 27) = 0.93, p > 0.05; IMI F (1, 27) = 3.93, p > 0.05 | ||||
| Hippocampus | 6.37 +/− 0.64 | 6.11 +/− 0.45 | 6.82 +/− 0.69 | 7.04 +/− 0.59 |
| CUS × IMI F (1, 26) = 0.08, p > 0.05; CUS F (1, 26) = 0.00, p > 0.05; IMI F (1, 26) = 1.28, p > 0.05 | ||||
| Hypothalamus | 8.25 +/− 0.96 | 12.92 +/− 0.60* | 8.38 +/− 0.76 | 13.56 +/− 0.98* |
| CUS × IMI F (1, 25) = 0.10, p > 0.05; CUS F (1, 25) = 35.76, p < 0.001; IMI F (1, 25) = 0.22, p > 0.05 | ||||
| Ventral Striatum | 5.81 +/− 0.77 | 6.26 +/− 0.45 | 4.71 +/− 0.27 | 5.68 +/− 0.26 |
| CUS × IMI F (1, 27) = 0.32, p > 0.05; CUS F (1, 27) = 2.41, p > 0.05; IMI F (1, 27) = 3.33, p > 0.05 | ||||
| Amygdala | 9.57 +/− 0.68 | 8.84 +/− 1.04 | 7.91 +/− 0.94 | 10.60 +/− 1.08 |
| CUS × IMI F (1, 25) = 3.00, p > 0.05; CUS F (1, 25) = 0.98, p > 0.05; IMI F (1, 25) = 0.00, p >0.05 | ||||
| Midbrain | 4.00 +/− 0.31 | 5.71 +/− 0.44* | 5.02 +/− 0.40 | 5.73 +/− 0.19* |
| CUS × IMI F (1, 27) = 2.03, p > 0.05; CUS F (1, 27) = 11.89, p < 0.005; IMI F (1, 27) = 2.20, p > 0.05 | ||||
Effects of CUS and/or Imipramine Treatment on Plasma Endocannabinoid Concentrations
CUS produced a significant increase in plasma AEA content [F (1, 25) = 61.24, p < 0.001; Fig. 3]; there was neither an effect of imipramine [F (1, 25) = 0.36, p > 0.05] nor an interaction between CUS and imipramine treatment [F (1, 25) = 0.14, p > 0.05] on plasma AEA. Plasma 2-AG content was unaffected by CUS exposure [F (1, 25) = 1.27, p > 0.05; Fig. 3] and imipramine treatment [F (1, 25) = 0.22, p > 0.05]. There was also no interaction between CUS and imipramine [F (1, 25) = 0.14, p > 0.05] on plasma 2-AG content.
Figure 3.
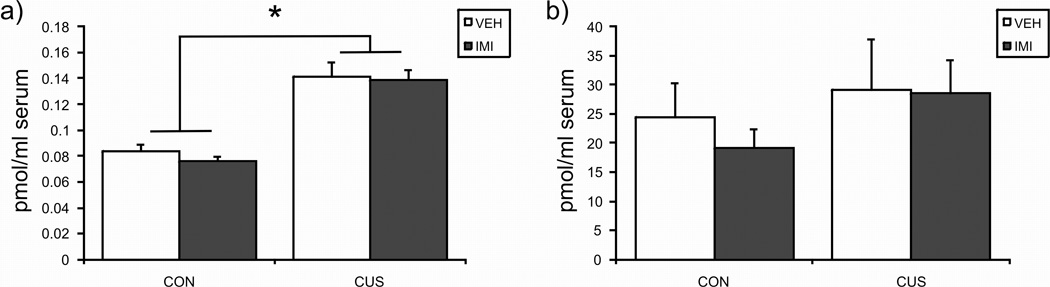
The effects of exposure to 21 days of chronic unpredictable stress (CUS), imipramine (10 mg/kg; IMI) administration, or both regimens combined, on the plasma content of the endocannabinoids anandamide (AEA; left panel) and 2-arachidonylglycerol (2-AG; right panel). Values are denoted as means ± SEM. Significant differences from non-stressed (CON), vehicle (VEH) treated animals (p < 0.05) denoted by *. For all treatment conditions, n = 6–8.
Effects of CUS and/or Imipramine Treatment on FAAH Activity
The Vmax (see Fig. 4) values for the hydrolysis of AEA by membranes were determined in a region-specific manner. There were no effects of either CUS or imipramine alone on the maximal hydrolytic activity of FAAH in any brain region examined; however, within the ventral striatum and midbrain there was a significant interaction between CUS and imipramine treatment such that the combination of these treatments increased the Vmax of FAAH in these structures. ANOVA results for the effects of CUS, imipramine treatment, or both on the maximal hydrolytic activity (Vmax) of FAAH are as follows: prefrontal cortex [CUS × IMI: F (1, 15) = 0.48, p > 0.05; CUS: F (1, 15) = 0.66, p > 0.05; IMI: F (1, 15) = 0.00, p > 0.05]; hippocampus [CUS × IMI: F (1, 16) = 0.52, p > 0.05; CUS: F (1, 16) = 0.41, p > 0.05; IMI: F (1, 16) = 0.08, p > 0.05]; hypothalamus [CUS × IMI: F (1, 14) = 2.13, p > 0.05; CUS: F (1, 14) = 0.59, p > 0.05; IMI: F (1, 14) = 0.09, p > 0.05]; amygdala [CUS × IMI: F (1, 12) = 0.22, p > 0.05; CUS: F (1, 12) = 1.00, p > 0.05; IMI: F (1, 12) = 0.06, p > 0.05]; ventral striatum [CUS × IMI: F (1, 14) = 9.57, p < 0.01; post hoc analysis revealed that CUS/IMI is significantly different (p < 0.05) relative to no stress/vehicle, CUS/vehicle and no stress/IMI); midbrain [CUS × IMI: F (1, 16) = 4.76, p < 0.05; post hoc analysis revealed that CUS/IMI is significantly different (p < 0.05) relative to no stress/vehicle, CUS/vehicle and no stress/IMI).
Figure 4.
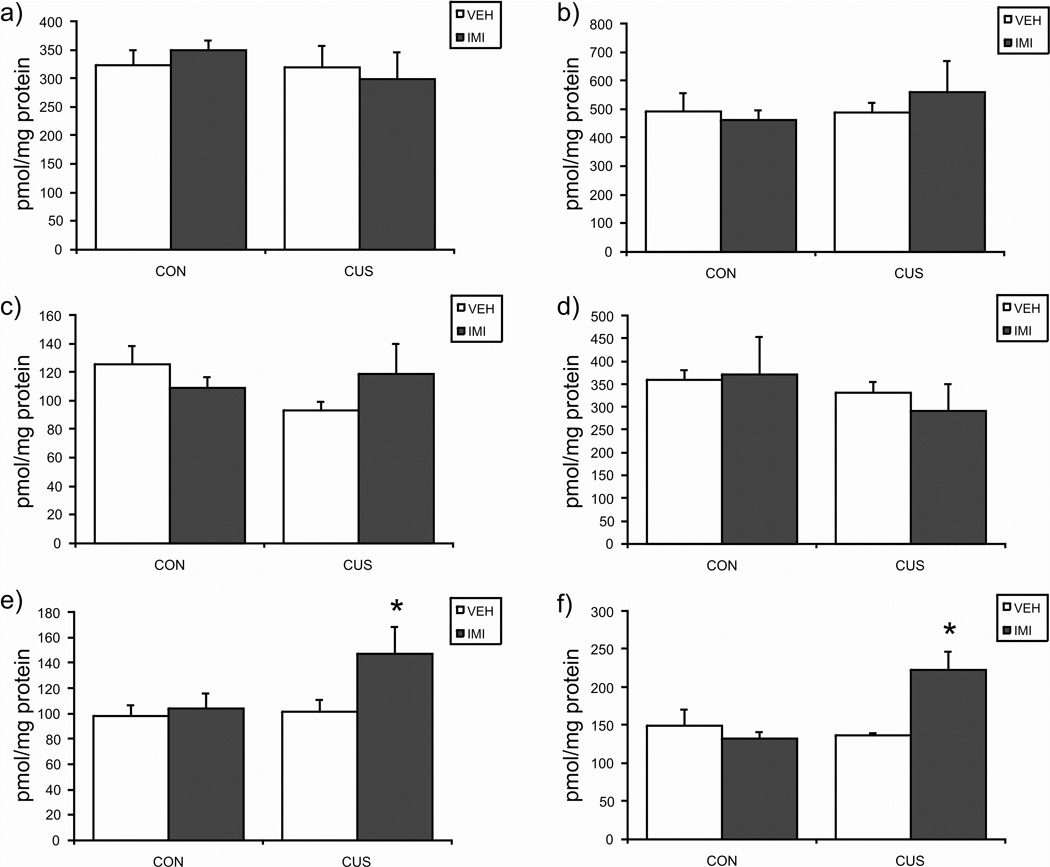
The effects of exposure to 21 days of chronic unpredictable stress (CUS), imipramine (10 mg/kg; IMI) administration, or both regimens combined, on the the maximal hydrolysis of anandamide by fatty acid amide hydrolase (Vmax) in the a) prefrontal cortex; b) hippocampus; c) hypothalamus; d) amygdala; e) midbrain; f) ventral striatum. Values are denoted as means ± SEM. Significant differences from non-stressed (CON), vehicle (VEH) treated animals (p < 0.05) denoted by *. For all treatment conditions, n = 4–5.
The binding affinity of AEA for FAAH (Km) was also determined in a region specific manner. The Km of FAAH was not effected by CUS alone in any brain region. Similarly, the Km of FAAH was not effected by imipramine alone, except in the midbrain. The combination of CUS and imipramine increased the Km of FAAH in the ventral striatum exclusively (data and ANOVA results in Table 4).
Table 4. The effects of exposure to chronic, unpredictable stress (CUS) and/or concurrent treatment with the antidepressant imipramine (IMI; 10 mg/kg) on the binding affinity (Km) of fatty acid amide hydrolase for anandamide.
Exposure of male rats to 21 days of CUS did not modify the binding affinity of fatty acid amide hydrolase (FAAH) for anandamide. Administration of IMI alone increased the Km of FAAH, exclusively in the midbrain. However, the joint administration of CUS and IMI resulted in an increase in the Km of FAAH in the ventral striatum and the midbrain. Significant differences from non-stressed (CON), vehicle (VEH) treated animals (p < 0.05) denoted by *. For all treatment conditions, n = 4–5. Data are presented as mean nM of 3H-arachidonoylethanolamide +/− SEM.
| CON/VEH | CUS/VEH | CON/IMI | CUS/IMI | |
|---|---|---|---|---|
| Prefrontal Cortex | 0.61 +/− 0.08 | 0.65 +/− 0.09 | 0.65 +/− 0.10 | 0.46 +/− 0.05 |
| CUS × IMI: F (1, 15) = 1.92, p > 0.05; CUS F (1, 15) = 0.68, p > 0.05; IMI F (1, 15) = 0.80, p > 0.05 | ||||
| Hippocampus | 0.64 +/− 0.12 | 0.69 +/− 0.08 | 0.80 +/− 0.28 | 0.71 +/− 0.10 |
| CUS × IMI F (1, 16) = 0.17, p > 0.05; CUS F (1, 16) = 0.02, p > 0.05; IMI F (1, 16) = 0.28, p > 0.05 | ||||
| Hypothalamus | 0.62 +/− 0.10 | 0.42 +/− 0.05 | 0.44 +/− 0.03 | 0.55 +/− 0.12 |
| CUS × IMI F (1, 14) = 2.81, p > 0.05; CUS F (1, 14) = 0.20, p > 0.05; IMI F (1, 14) = 0.07, p > 0.05 | ||||
| Ventral Striatum | 0.37 +/− 0.05 | 0.37 +/− 0.06 | 0.37 +/− 0.04 | 0.69 +/− 0.08* |
| CUS × IMI F (1, 14) = 7.32, p < 0.02; CUS F (1, 14) = 2.11, p > 0.05; IMI F (1, 14) = 1.90 | ||||
| Amygdala | 0.81 +/− 0.14 | 0.83 +/− 0.17 | 0.95 +/− 0.14 | 0.53 +/− 0.10 |
| CUS × IMI F (1, 12) = 2.42, p > 0.05; CUS F (1, 12) = 2.11, p > 0.05; IMI F (1, 12) = 0.24, p > 0.05 | ||||
| Midbrain | 0.30 +/− 0.03 | 0.45 +/− 0.05 | 0.60 +/− 0.07* | 0.61 +/− 0.11* |
| CUS × IMI F (1, 16) = 1.11, p > 0.05; CUS F (1, 16) = 1.36, p > 0.05; IMI F (1, 16) = 10.35, p < 0.01 | ||||
Discussion
There has been considerable debate over the validity of the CUS/CMS model to induce behavioral responses akin to the anhedonia seen in human depression (Forbes et al., 1996; Reid et al., 1997). In animal models, anhedonia is typically defined and measured as a deficit in motivational drive for a normally rewarding behavior (Willner et al., 1992). One of the prominent symptoms of depression is a lack of motivation for rewarding activities, which is frequently manifested as a loss of sex drive (Williams and Reynolds, 2006). In the present study, we found that exposure of male rats to three weeks of CUS resulted in a significant impairment in motivation for sexual activity without affecting performance after sexual activity was initiated. These data are consistent with the hypothesis that CUS results in a deficit in motivation for a natural reward and is a useful model for the studies of the neurochemical changes that accompany anhedonia.
In this study, we found that CUS produced changes in both CB1 receptor agonist binding site density and in the tissue content of the endocannabinoid ligands. Exposure of rats for 21 days to CUS produced a reduction in the tissue content of AEA in all of the brain regions examined (PFC, hippocampus, hypothalamus, ventral striatum, amygdala and midbrain) compared to group housed control rats. This finding stands in contrast to two earlier studies in which CUS did not affect brain AEA contents (Hill et al., 2005; Bortolato et al., 2007). While the reasons for this discrepancy is not clear, it is likely that methodological differences are responsible. For example, in the present study, rats were sacrificed at the end of the dark cycle while both of the other studies collected brain tissue towards the end of light cycle. AEA exhibits a diurnal rhythm in that AEA levels are higher in the dark phase than in the light phase (Valenti et al., 2004). It is possible that a CUS-induced decrease in tissue AEA is only detectable when overall content is high. Regardless of the mechanism, these results demonstrate that tissue eCB changes in response to interventions such as CUS must be interpreted with caution, particularly when only one time point is examined.
In the present study, the CUS-induced reduction in AEA content was not due to accelerated metabolism, as membranes from CUS exposed rats did not exhibit changes in the hydrolysis of AEA. The widespread nature of this reduction is surprising in light of the generally accepted view that endocannabinoid synthesis/release is driven by local changes in neuronal activity. These data support an alternative hypothesis that AEA content in the brain is regulated more broadly and suggest that one of the functions of AEA is to provide a common set point for CB1 receptor activation throughout the brain. Interestingly, this effect of CUS was unaltered by concurrent treatment with the tricyclic antidepressant, imipramine. These data argue against the hypothesis that the global change in AEA is secondary to the effects of CUS on monoaminergic signaling.
In addition to the effects of CUS on the ECS, it was also found that imipramine treatment alone exerted significant modulation of specific parameters of the ECS. In particular, imipramine treatment was found to increase CB1 receptor binding in the amygdala, while reducing CB1 receptor binding in the midbrain; neither of these effects were seen in animals that were concurrently subjected to CUS. The increase in amygdalar CB1 receptor binding following imipramine treatment is consistent with prior studies from our group which collectively demonstrate that several treatments which are beneficial to depression, such as electroconvulsive shock and tricyclic antidepressant treatment, increase CB1 receptor activity in subcortical limbic structures, such as the hippocampus, amygdala and hypothalamus (Hill et al., 2006), 2007a. The functional relevance of these changes to the behavioral effects of antidepressant treatment has yet to be elucidated; however, preclinical studies have demonstrated the CB1 receptor is required for the behavioral effects of noradrenergic based antidepressants but is dispensable for the behavioral effect of serotonergic based antidepressants (Mato et al., 2007; Steiner et al., 2008a).
Rats exposed to CUS exhibited a significant increase in CB1 receptor binding site density in the PFC, a finding that is consistent with other reports that CUS/CMS results in a significant increase in CB1 receptor mRNA in the PFC (Bortolato et al., 2007; Hillard et al., 2006). Therefore, it is likely that the increase in PFC CB1 receptor agonist binding site density reflects an up-regulation of CB1 receptor expression within specific neuronal populations in the PFC. However, the identity of the cells which exhibit increased CB1 receptor expression has yet to be determined and will greatly influence the interpretation of the current data. Additionally, the tissue chunk of PFC which was used for membrane preparations in this study (as well as past studies from our group; Hill et al., 2006, 2007a) was composed of both the anterior cingulate cortex, as well as the ventromedial PFC. Neuroimaging studies in human patients suffering from depression have demonstrated that distinct differences exist in the neural alterations seen in the cingulate cortex and more ventral regions of the PFC (Mayberg, 1997; Brody et al., 2001). Thus, future work examining the effects of CUS on CB1 receptor expression patterns within the anterior cingulate cortex and the ventromedial PFC is required to determine how these changes may relate to alterations in frontocortical neural activity in depressive illness.
Regardless of these caveats, these data are consistent with clinical studies demonstrating that CB1 receptor protein expression, binding site density and signal transduction are also increased in the PFC of depressed humans who died by suicide (Hungund et al., 2004; Pazos et al., 2006; Vinod et al., 2005). Recent work has demonstrated that increased endocannabinoid/CB1 receptor signaling in the PFC can increase voluntary alcohol intake (Hansson et al., 2007), suggesting that increased prefrontal cortical CB1 receptor binding in depression could be related to the high prevalence of alcoholism associated with this disease, as has previously been suggested (Vinod et al., 2005). Moreover, increases in CB1 receptor signaling within the PFC enhances the association of aversive cues (Laviolette and Grace, 2005), which suggests that increased CB1 receptor binding in the PFC in depression could contribute to the enhancement in aversive emotional memory that often characterizes this disease. On the other hand, CB1 receptors in the ventromedial PFC mediate the antidepressant actions of CB1 receptor agonists through trans-synaptic activation of dorsal raphe serotonergic neurons (Bambico et al., 2007), suggesting that increased CB1 receptor density in the PFC could be a compensatory response that dampens the effects of CUS and/or depression.
In contrast to the PFC, CUS exposure resulted in a significant reduction in CB1 receptor agonist binding site density in the hippocampus. This finding is in agreement with other reports that CUS reduces hippocampal CB1 receptor mRNA and protein expression, binding site density and activation of GDP/GTP exchange (Hill et al., 2005; Perez-Rial et al., 2004; Hillard et al., 2006; Reich et al., 2007) but not with a recent report that CB1 receptor mRNA was not altered in the hippocampus of rats exposed to 10 weeks of CMS (Bortolato et al., 2007). However, in the latter study, the control rats were also exposed to isolation, and food and water deprivation which are stressors themselves, thus could have caused stress-induced changes in CB1 receptor expression in the controls that would mask changes in the CUS exposed rats.
The effect of CUS to reduce hippocampal CB1 receptor expression is likely mediated by increased glucocorticoid secretion since CUS elicits glucocorticoid hypersecretion (Herman et al., 1995; Hill and Gorzalka, 2004; Hill et al., 2005) and 21 days of corticosterone administration, in the absence of stress, elicits a comparable reduction in CB1 receptor binding and protein expression in the hippocampus (Hill et al., 2008a). Imipramine treatment did not affect hippocampal CB1 receptor density in either control or CUS rats. These findings are in disagreement with earlier findings that a similar tricyclic antidepressant, desipramine, results in an up-regulation of CB1 receptor binding in the hippocampus of unstressed rats (Hill et al., 2006); however, this discrepancy may be due to the neurochemical targets of the agents of use as desipramine is known to exert a higher preferential affinity for norepinephrine transport over imipramine, which has greater effects on 5-HT reuptake (Langer and Schoemaker, 1988). While this result does not support a role for hippocampal CB1 receptor density effects in the mechanism of action of imipramine, it does not rule out a role for these changes in the spectrum of CUS-induced behavioral and neurochemical effects.
Similar to what was seen in the hippocampus, CUS reduced the CB1 receptor agonist binding site density in the hypothalamus. Several studies have established a critical role of the hypothalamic endocannabinoid system in the regulation of the hypothalamic-pituitary-adrenal (HPA) axis; in particular, endocannabinoid/CB1 receptor signaling inhibits activation of the HPA axis while loss of CB1 receptors and CB1 receptor antagonists enhance HPA axis reactivity (Patel et al., 2004, 2005b; Cota et al., 2003, 2007; Steiner et al., 2008a). Disturbances in the HPA axis, such as overactivity and/or resistance to feedback inhibition, are common in melancholic depression (Wong et al., 2000) and are a consequence of CUS (Herman et al., 1995; Hill et al., 2005; Hill and Gorzalka, 2004). Therefore, the reduction in the CB1 receptor density in the hypothalamus is consistent with the increased activation of the HPA axis seen following CUS. Normalization of HPA axis dysfunction is correlated with remission of depression (Ribeiro et al., 1993; Greden et al., 1983) and antidepressant therapies can diminish HPA axis responsivity in depressed humans and rats exposed to CUS (Duncan et al., 1996; Lopez et al., 1998; Deuschle et al., 2003; Hill et al., 2006). We reported previously that chronic treatment with the antidepressant, desipramine, increases hypothalamic CB1 receptor agonist binding site density in unstressed rats, which contributes desipramine’s ability to dampen HPA axis activation (Hill et al., 2006). Similarly, the present data demonstrate that concurrent imipramine attenuates the effects of CUS to decrease CB1 receptor binding site density in the hypothalamus. Taken together, these and earlier findings are consistent with the hypotheses that reduced CB1 receptor expression contributes to HPA axis hyper-reactivity following CUS and that anti-depressant-induced reversal of this change contributes to the mechanism by which this class of drugs normalizes HPA function in stressed rodents and, possibly, depressed humans.
The tissue content of 2-AG in the hypothalamus was increased following CUS. We have shown previously that acute stress reduces hypothalamic 2-AG content in mice which correlates with increased HPA axis activation (Patel et al., 2004). Furthermore, we found that repeated exposure to a homotypic stressor resulted in both a significant increase in hypothalamic 2-AG and habituation of HPA axis activation response to the stressor. These data support the hypothesis that elevated 2-AG content in the hypothalamus dampens HPA axis activation; if this is correct, then increased hypothalamic 2-AG following CUS would function to oppose HPA hyperactivity. As discussed above, however, CUS also reduced hypothalamic CB1 receptor density which would produce the opposite effect on HPA activity. It is possible that the change in 2-AG compensates for the loss of CB1 receptors; or, alternatively, the increase in 2-AG results in CB1 receptor down-regulation.
The effects of CUS and imipramine on the CB1 receptor in the ventral striatum were similar to those in the hypothalamus; CB1 receptor binding densities were reduced by CUS and partially reversed by concurrent imipramine treatment. Activation of CB1 receptors in the nucleus accumbens within the ventral striatum increases dopamine release and enhances the rewarding properties of both natural and artificial rewards (Caille et al., 2007; Mahler et al., 2007), suggesting, in turn, that decreased CB1 receptor signaling in this structure could impair hedonic processing. Anhedonia is a core symptom of major depression, and a behavioral phenomenon typically seen in rodents following CUS/CMS (Willner et al., 1992; Willner, 2005). It has recently been shown that the administration of a FAAH inhibitor during CMS (Bortolato et al., 2007) and to mice exposed to repeated restraint (Rademacher and Hillard, 2007) attenuates the development of anhedonia. Furthermore, the induction of anhedonia in response to repeated restraint stress is associated with a reduction in ECS signaling in the ventral striatum (Rademacher and Hillard, 2007). The current data present the first evidence that CUS-induced anhedonia could be due to local impairments in CB1 receptor activity within the nucleus accumbens, and that the reversal of this effect by antidepressants (or FAAH inhibition) may, in part, be related to normalization of CB1 receptor signaling in this region; however, this remains to be experimentally demonstrated.
Surprisingly, an interaction between CUS and imipramine occurred in the ventral striatum and midbrain, such that co-treatment of imipramine and CUS resulted in a robust activation of FAAH activity although neither alone had an effect on activity. These data are interesting and suggest that FAAH activity is sensitive to imipramine only in the context of CUS and only in select brain regions; and, alternatively, that FAAH activity is sensitive to CUS only in the context of imipramine treatment. If these data are interpreted in relationship to the model above that a decrease in ventral striatal AEA content is associated with anhedonia, then they suggest that this effect of imipramine to increase FAAH activity in the presence of CUS is an undesirable side effect of the antidepressant that is countering the clinical efficacy of these drugs. In support of this suggestion, preclinical data demonstrate that administration of a FAAH inhibitor can augment the behavioral effects of imipramine treatment (Filip et al., 2006).
We also examined the effects of CUS on circulating endocannabinoid concentrations. Plasma AEA content was significantly elevated following CUS, while plasma 2-AG was not affected. This profile mirrors the plasma endocannabinoid contents in women diagnosed with minor depression (Hill et al., 2008b). While the functional significance of circulating endocannabinoids is not known, peripheral endocannabinoid signaling is known to be important for immune and cardiovascular parameters (Hillard, 2000b; Kunos et al., 2000; Klein et al., 2003), both of which are often disturbed in depressive illness (Carney et al., 2002; Kim et al., 2007).
Accumulating evidence implicates the endocannabinoid system in the pathophysiology of depression (Gorzalka et al., in press; Hill and Gorzalka, 2005a; Witkin et al., 2005; Vinod and Hungund, 2006; Mangieri and Piomelli, 2007). The results reported herein support that role and demonstrate significant and region-specific changes in the CB1 receptor and/or endocannabinoids. CUS in rats, which produces some of the important symptoms of human depression (Willner, 2005), significantly reduced CB1 receptor binding site density in the hippocampus, hypothalamus and ventral striatum and increased density in the PFC. The increase in PFC CB1 receptor binding site densities following CUS could represent either a compensatory response to maintain serotonergic firing activity (Bambico et al., 2007) or a driving force in the development of depression (Hungund et al., 2004). Alternately, the reduction of CB1 receptor signaling in subcortical structures could contribute to the changes in HPA axis drive, hedonic valuation and emotional behavior seen in depression. The fact that AEA is so profoundly reduced, that this effect is insensitive to antidepressant treatment and that antidepressants even appear to activate FAAH in the face of concurrent stress in some brain regions, supports the contention that inhibition of FAAH should be investigated as a pharmacological target for the development of novel antidepressants or adjuncts to conventional antidepressants (Gobbi et al., 2005; Bortolato et al., 2007; Hill et al., 2007b).
Acknowledgements
This research was supported by operating grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institute of Health Research (CIHR) to BBG; NIH grant DA16967 to CJH; a; a MSFHR senior graduate trainee award and a NSERC Canadian Graduate Scholarship to MNH; and a MSFHR junior graduate trainee award to RJM.
Abbreviations
- CUS
(chronic unpredictable stress)
- CMS
(chronic mild stress)
- AEA
(N-Arachidonylethanolamide)
- 2-AG
(2-arachidonoylglycerol)
- FAAH
(fatty acid amide hydrolase)
- PFC
(prefrontal cortex); (hypothalamic-pituitary-adrenal)
References
- Bambico FR, Katz N, Debonnel G, Gobbi G. Cannabinoids elicit antidepressant-like behavior and activate serotonergic neurons through the medial prefrontal cortex. J. Neurosci. 2007;27:11700–11711. doi: 10.1523/JNEUROSCI.1636-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Berrendero F, Ambrosino G, Cebeira M, Ramos JA, Fernandez-Ruiz JJ, Di Marzo V. Brain regional distribution of endocannabinoids: implications for their biosynthesis and biological function. Biochem. Biophys. Res. Commun. 1999;256:377–380. doi: 10.1006/bbrc.1999.0254. [DOI] [PubMed] [Google Scholar]
- Bortolato M, Mangieri RA, Fu J, et al. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol. Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Bovasso GB. Cannabis abuse is a risk factor for depressive symptoms. Am. J. Psychiatry. 2001;158:2033–2037. doi: 10.1176/appi.ajp.158.12.2033. [DOI] [PubMed] [Google Scholar]
- Brody AL, Barsom MW, Bota RG, Saxena S. Prefrontal-subcortical and limbic circuit mediation of major depressive disorder. Semin. Clin. Neuropsychiatry. 2001;6:102–112. doi: 10.1053/scnp.2001.21837. [DOI] [PubMed] [Google Scholar]
- Brotto LA, Gorzalka BB, LaMarre AK. Melatonin protects against the effects of chronic stress on sexual behaviour in male rats. Neuroreport. 2001;12:3465–3469. doi: 10.1097/00001756-200111160-00018. [DOI] [PubMed] [Google Scholar]
- Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J. Neurosci. 2007;27:3695–3702. doi: 10.1523/JNEUROSCI.4403-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney RM, Freedland KE, Miller GE, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J. Psychosomatic Res. 2002;53:897–902. doi: 10.1016/s0022-3999(02)00311-2. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chen C, Bazan NG. Lipid signaling: sleep, synaptic plasticity, and neuroprotection. Prostaglandins Other Lipid Mediat. 2005;77:65–76. doi: 10.1016/j.prostaglandins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Tschop M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Invest. 2003;112:423–431. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Steiner MA, Marsicano G, et al. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148:1574–1581. doi: 10.1210/en.2005-1649. [DOI] [PubMed] [Google Scholar]
- Degenhardt L, Hall W, Lynskey M. Exploring the association between cannabis use and depression. Addiction. 2003;98:1493–1504. doi: 10.1046/j.1360-0443.2003.00437.x. [DOI] [PubMed] [Google Scholar]
- Deuschle M, Hamann B, Meichel C, Krumm B, Lederbogen F, Kniest A, Colla M, Heuser I. Antidepressive treatment with amitriptyline and paroxetine: effects on saliva cortisol concentrations. J. Clin. Psychiatry. 2003;23:201–205. doi: 10.1097/00004714-200304000-00014. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Knapp DJ, Johnson KB, Breese GR. Functional classification of antidepressants based on antagonism of swim stress-induced fos-like immunoreactivity. J. Pharmacol. Exp. Ther. 1996;277:1076–1089. [PubMed] [Google Scholar]
- Egertova M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- Filip M, Frankowska M, Wydra K, Nowak E, McCreary AC. Effects of combined administration of a selective inhibitor of FAAH (URB597) and imipramine or citalopram in the forced swimming test in rats. Eur. Neuropsychopharmacol. 2006;16:S341. [Google Scholar]
- Forbes NF, Stewart CA, Matthews K, Reid IC. Chronic mild stress and sucrose consumption: validity as a model of depression. Physiol. Behav. 1996;60:1481–1484. doi: 10.1016/s0031-9384(96)00305-8. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc. Natl. Acad. Sci. USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorzalka BB, Hill MN, Hillard CJ. Regulation of endocannabinoid signaling by stress: Implications for affective disorders. Neurosci. Biobehav. Rev. doi: 10.1016/j.neubiorev.2008.03.004. (in press) [DOI] [PubMed] [Google Scholar]
- Greden JF, Gardner R, King D, Grunhaus L, Carroll BJ, Kronfol Z. Dexamethasone suppression tests in antidepressant treatment of melancholia. The process of normalization and test-retest reproducibility. Arch. Gen. Psychiatry. 1983;40:493–500. doi: 10.1001/archpsyc.1983.01790050019002. [DOI] [PubMed] [Google Scholar]
- Griebel G, Stemmelin J, Scatton B. Effects of the cannabinoid CB1 receptor antagonist rimonabant in models of emotional reactivity in rodents. Biol. Psychiatry. 2005;57:261–267. doi: 10.1016/j.biopsych.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Bermudez-Silva FJ, Malinen H, et al. Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology. 2007;32:117–126. doi: 10.1038/sj.npp.1301034. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Adams D, Prewitt C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology. 1995;61:180–190. doi: 10.1159/000126839. [DOI] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. Enhancement of anxiety-like responsiveness to the cannabinoid CB(1) receptor agonist HU-210 following chronic stress. Eur. J. Pharmacol. 2004;499:291–295. doi: 10.1016/j.ejphar.2004.06.069. [DOI] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. Is there a role for the endocannabinoid system in the etiology and treatment of melancholic depression? Behav. Pharmacol. 2005a;16:333–352. doi: 10.1097/00008877-200509000-00006. [DOI] [PubMed] [Google Scholar]
- Hill MN, Gorzalka BB. Pharmacological enhancement of cannabinoid CB1 receptor activity elicits an antidepressant-like response in the rat forced swim test. Eur. Neuropsychopharmacol. 2005b;15:593–599. doi: 10.1016/j.euroneuro.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, Gorzalka BB. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30:508–515. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- Hill MN, Ho WS, Sinopoli KJ, Viau V, Hillard CJ, Gorzalka BB. Involvement of the endocannabinoid system in the ability of long-term tricyclic antidepressant treatment to suppress stress-induced activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2006;31:2591–2599. doi: 10.1038/sj.npp.1301092. [DOI] [PubMed] [Google Scholar]
- Hill MN, Barr AM, Ho WS, Carrier EJ, Gorzalka BB, Hillard CJ. Electroconvulsive shock treatment differentially modulates cortical and subcortical endocannabinoid activity. J. Neurochem. 2007a;103:47–56. doi: 10.1111/j.1471-4159.2007.04688.x. [DOI] [PubMed] [Google Scholar]
- Hill MN, Karacabeyli ES, Gorzalka BB. Estrogen recruits the endocannabinoid system to modulate emotionality. Psychoneuroendocrinology. 2007b;32:350–357. doi: 10.1016/j.psyneuen.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Hill MN, Ho WS, Carrier EJ, Shi L, Patel S, Gorzalka BB, Hillard CJ. Prolonged glucocorticoid treatment decreases cannabinoid CB1 receptor density in the hippocampus. Hippocampus. 2008a;18:221–226. doi: 10.1002/hipo.20386. [DOI] [PubMed] [Google Scholar]
- Hill MN, Miller GE, Ho WS, Gorzalka BB, Hillard CJ. Serum endocannabinoid content is altered in females with depressive disorders: A preliminary study. Pharmacopsychiatry. 2008b;41:48–53. doi: 10.1055/s-2007-993211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000a;61:3–18. doi: 10.1016/s0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Hillard CJ. Endocannabinoids and vascular function. J. Pharmacol. Exp. Ther. 2000b;294:27–32. [PubMed] [Google Scholar]
- Hillard CJ, Edgemond WS, Campbell WB. Characterization of ligand binding to the cannabinoid receptor of rat brain membranes using a novel method: application to anandamide. J. Neurochem. 1995a;64:677–683. doi: 10.1046/j.1471-4159.1995.64020677.x. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Wilkison DM, Edgemond WS, Campbell WB. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim. Biophys. Acta. 1995b;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Hill MN, Carrier EJ, Shi L, Cullinan WE, Gorzalka BB. Regulation of cannabinoid receptor expression by chronic unpredictable stress in rats and mice. Soc. Neurosci. Abstr. 2006 746.19. [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002;68– 69:619–631. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Vinod KY, Kassir SA, Basavarajappa BS, Yalamanchili R, Cooper TB, Mann JJ, Arango V. Upregulation of CB1 receptors and agonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Mol. Psychiatry. 2004;9:184–190. doi: 10.1038/sj.mp.4001376. [DOI] [PubMed] [Google Scholar]
- Iredale PA, Terwilliger R, Widnell KA, Nestler EJ, Duman RS. Differential regulation of corticotropin-releasing factor1 receptor expression by stress and agonist treatments in brain and cultured cells. Mol Pharmacol. 1996;50:1103–1110. [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Kim YK, Na KS, Shin KH, Jung HY, Choi SH, Kim JB. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2007;31:1044–1053. doi: 10.1016/j.pnpbp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton C, Larsen K, et al. The cannabinoid system and immune modulation. J. Leukoc. Biol. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- Koethe D, Llenos IC, Dulay JR, Hoyer C, Torrey EF, Leweke FM, et al. Expression of CB1 cannabinoid receptor in the anterior cingulate cortex in schizophrenia, bipolar disorder and major depression. J Neural Transm. 2007;114:1055–1063. doi: 10.1007/s00702-007-0660-5. [DOI] [PubMed] [Google Scholar]
- Kunos G, Jarai Z, Batkai S, Goparaju SK, Ishac EJ, Liu J, Wang L, Wagner JA. Endocannabinoids as cardiovascular modulators. Chem. Phys. Lipids. 2000;108:159–168. doi: 10.1016/s0009-3084(00)00194-8. [DOI] [PubMed] [Google Scholar]
- Langer SZ, Schoemaker H. Effects of antidepressants on monoamine transporters. Prog. Neuropsychopharmacol. Biol. Psychiatry. 1988;12:193–216. doi: 10.1016/0278-5846(88)90037-1. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA. Cannabinoids potentiate emotional learning plasticity in neurons of the medial prefrontal cortex through basolateral amygdala inputs. J. Neurosci. 2005;26:6458–6468. doi: 10.1523/JNEUROSCI.0707-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligresti A, Cascio MG, Di Marzo V. Endocannabinoid metabolic pathways and enzymes. Curr. Drug Targets CNS Neurol. Disord. 2005;4:615–623. doi: 10.2174/156800705774933104. [DOI] [PubMed] [Google Scholar]
- Lopez JF, Chalmers DT, Little KY, Watson SJ. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol. Psychiatry. 1998;43:547–573. doi: 10.1016/s0006-3223(97)00484-8. [DOI] [PubMed] [Google Scholar]
- Lynskey MT, Glowinski AL, Todorov AA, Bucholz KK, Madden PA, Nelson EC, Statham DJ, Matin NG, Heath AC. Major depressive disorder, suicidal ideation, and suicide attempt in twins discordant for cannabis dependence and early-onset cannabis use. Arch. Gen. Psychiatry. 2004;61:1026–1032. doi: 10.1001/archpsyc.61.10.1026. [DOI] [PubMed] [Google Scholar]
- Mahler SV, Smith KS, Berridge KC. Endocannabinoid hedonic hotspot for sensory pleasure: Anandamide in nucleus accumbens shell enhances 'liking' of a sweet reward. Neuropsychopharmacology. 2007;32:2267–2278. doi: 10.1038/sj.npp.1301376. [DOI] [PubMed] [Google Scholar]
- Mangieri RA, Piomelli D. Enhancement of endocannabinoid signaling and the pharmacotherapy of depression. Pharmacol. Res. 2007;56:360–366. doi: 10.1016/j.phrs.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Neuromodulatory functions of the endocannabinoid system. J. Endocrinol. Invest. 2006;29:27–46. [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- Mato S, Aso E, Castro E, Martin M, Valverde O, Maldonado R, Pazos A. CB1 knockout mice display impaired functionality of 5-HT1A and 5-HT2A/C receptors. J. Neurochem. 2007;103:2111–2120. doi: 10.1111/j.1471-4159.2007.04961.x. [DOI] [PubMed] [Google Scholar]
- Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J. Neuropsychiatry Clin. Neurosci. 1997;9:471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54:20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Gould E, Manji H, et al. Preclinical models: status of basic research in depression. Biol. Psychiatry. 2002;52:503–528. doi: 10.1016/s0006-3223(02)01405-1. [DOI] [PubMed] [Google Scholar]
- O'Neil MF, Moore NA. Animal models of depression: are there any? Hum. Psychopharmacol. 2003;18:239–254. doi: 10.1002/hup.496. [DOI] [PubMed] [Google Scholar]
- Oliva JM, Uriguen L, Perez-Rial S, Manzanares J. Time course of opioid and cannabinoid gene transcription alterations induced by repeated administration with fluoxetine in the rat brain. Neuropharmacology. 2005;49:618–626. doi: 10.1016/j.neuropharm.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Omeir RL, Chin S, Hong Y, Deutsch DG. Arachidonoyl ethanolamide-[1,2-14C] as a substrate for anandamide amidase. Life Sci. 1995;56:2033–2040. doi: 10.1016/0024-3205(95)00181-5. [DOI] [PubMed] [Google Scholar]
- Ossowska G, Nowak G, Kata R, Klenk-Majewska B, Danilczuk Z, Zebrowska-Lupina I. Brain monoamine receptors in a chronic unpredictable stress model in rats. J Neural Transm. 2001;108:311–319. doi: 10.1007/s007020170077. [DOI] [PubMed] [Google Scholar]
- Ossowska G, Nowak G, Klenk-Majewska B, Danilczuk Z, Zebrowska-Lupina I. Effect of imipramine on brain D-1 and 5-HT-2A receptors in a chronic unpredictable stress model in rats. Pol J Pharmacol. 2002;54:89–93. [PubMed] [Google Scholar]
- Patel S, Rademacher DJ, Hillard CJ. Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J. Pharmacol. Exp. Ther. 2003;306:880–888. doi: 10.1124/jpet.103.054270. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- Patel S, Carrier EJ, Ho WS, Rademacher DJ, Cunningham S, Reddy DS, Falck JR, Cravatt BF, Hillard CJ. The postmortal accumulation of brain N- 40 arachidonylethanolamine (anandamide) is dependent upon fatty acid amide hydrolase activity. J. Lipid Res. 2005a;46:342–349. doi: 10.1194/jlr.M400377-JLR200. [DOI] [PubMed] [Google Scholar]
- Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur. J. Neurosci. 2005b;21:1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- Pazos A, Mostany R, Mato S, et al. Constitutive activity of cannabinoid CB1 receptors in major depression. Soc. Neurosci. Abstr. 2006 191.18. [Google Scholar]
- Perez-Rial S, Uriguen L, Palomo T, Manzanares J. Acute and chronic stress alter cannabinoid CB1 receptor function and POMC gene expression in the rat brain. Eur. Neuropsychopharmacol. 2004;14:S318. [Google Scholar]
- Rademacher DJ, Hillard CJ. Interactions between endocannabinoids and stress-induced decreased sensitivity to natural reward. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2007;31:633–641. doi: 10.1016/j.pnpbp.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrie P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int. J. Obes. Relat. Metab. Disord. 2004;28:640–648. doi: 10.1038/sj.ijo.0802583. [DOI] [PubMed] [Google Scholar]
- Reich CG, Taylor ME, McCarthy MM. Stress-induced modulation of the endocannabinoid system in the HPA axis: Effects of gonadal status. Soc. Neurosci. Abstr. 2007 197.7. [Google Scholar]
- Reid I, Forbes N, Stewart C, Matthews K. Chronic mild stress and depressive disorder: a useful new model? Psychopharmacology. 1997;134:365–367. doi: 10.1007/s002130050471. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress. Anxiety. 2000;12:2–19. doi: 10.1002/1520-6394(2000)12:1+<2::AID-DA2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Ribeiro SC, Tandon R, Grunhaus L, Greden JF. The DST as a predictor of outcome in depression: a meta-analysis. Am. J. Psychiatry. 1993;150:1618–1629. doi: 10.1176/ajp.150.11.1618. [DOI] [PubMed] [Google Scholar]
- Sanchis-Segura C, Cline BH, Marsicano G, Lutz B, Spanagel R. Reduced sensitivity to reward in CB1 knockout mice. Psychopharmacology. 2004;176:223–232. doi: 10.1007/s00213-004-1877-8. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Rosko KM, Fleischer R, Wang J, Xu S, Tong XS, Rocha BA. Antidepressant-like and anorectic effects of the cannabinoid CB1 receptor inverse agonist AM251 in mice. Behav. Pharmacol. 2003;14:573–582. doi: 10.1097/00008877-200312000-00001. [DOI] [PubMed] [Google Scholar]
- Steiner MA, Marsicano G, Nestler EJ, Holsboer F, Lutz B, Wotjak CT. Antidepressant-like behavioral effects of impaired cannabinoid receptor type 1 signaling coincide with exaggerated corticosterone secretion in mice. Psychoneuroendocrinology. 2008a;33:54–67. doi: 10.1016/j.psyneuen.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner MA, Wanisch K, Monroy K, Marsicano G, Borroni E, Bachli H, Holsboer F, Lutz B, Wotjak CT. Impaired cannabinoid receptor type 1 signaling interferes with stress-coping behavior in mice. Pharmacogenomics J. 2008b;8:196–208. doi: 10.1038/sj.tpj.6500466. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Davis RJ, Perry KW, Li X, Salhoff C, Bymaster FP, Witkin JM, Nomikos GG. The CB1 receptor antagonist SR141716A selectively 42 increases monoaminergic neurotransmission in the medial prefrontal cortex: implications for therapeutic actions. Br. J. Pharmacol. 2003;138:544–553. doi: 10.1038/sj.bjp.0705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriguen L, Perez-Rial S, Ledent C, Palomo T, Manzanares J. Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmcology. 2004;46:966–973. doi: 10.1016/j.neuropharm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Valenti M, Vigano D, Casico MG, Rubino T, Steardo L, Parolaro D, Di Marzo V. Differential diurnal variations of anandamide and 2-arachidonoyl-glycerol levels in rat brain. Cell Mol. Life Sci. 2004;61:945–950. doi: 10.1007/s00018-003-3453-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinod KY, Hungund BL. Role of the endocannabinoid system in depression and suicide. Trends Pharmacol. Sci. 2006;27:539–545. doi: 10.1016/j.tips.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Vinod KY, Arango V, Xie S, Kassir SA, Mann JJ, Cooper TB, Hungund BL. Elevated levels of endocannabinoids and CB1 receptor-mediated G-protein signaling in the prefrontal cortex of alcoholic suicide victims. Biol. Psychiatry. 2005;57:480–486. doi: 10.1016/j.biopsych.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Williams K, Reynolds MF. Sexual dysfunction in major depression. CNS Spectr. 2006;11:19–23. doi: 10.1017/s1092852900026729. [DOI] [PubMed] [Google Scholar]
- Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52:90–110. doi: 10.1159/000087097. [DOI] [PubMed] [Google Scholar]
- Willner P, Towell A, Sampson D, Sophokleous S, Muscat R. Reduction of sucrose preference by chronic unpredictable mild stress, and its restoration by a tricyclic antidepressant. Psychopharmacology. 1987;93:358–364. doi: 10.1007/BF00187257. [DOI] [PubMed] [Google Scholar]
- Willner P, Muscat R, Papp M. Chronic mild stress-induced anhedonia: a realistic animal model of depression. Neurosci. Biobehav. Rev. 1992;16:525–534. doi: 10.1016/s0149-7634(05)80194-0. [DOI] [PubMed] [Google Scholar]
- Witkin JM, Tzavara ET, Davis RJ, Li X, Nomikos GG. A therapeutic role for cannabinoid CB1 receptor antagonists in major depressive disorders. Trends Pharnacol. Sci. 2005;26:609–617. doi: 10.1016/j.tips.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Wong ML, Kling MA, Munson PJ, et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc. Natl. Acad. Sci. USA. 2000;97:325–330. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ku B, Cui L, Li X, Barish PA, Foster TC, et al. Curcumin reverses impaired hippocampal neurogenesis and increases serotonin receptor 1A mRNA and brainderived neurotrophic factor expression in chronically stressed rats. Brain Res. 2007;1162:9–18. doi: 10.1016/j.brainres.2007.05.071. [DOI] [PubMed] [Google Scholar]