

Abstract
OBJECTIVE—To determine the contribution of liver and viscera to splanchnic cortisol production in humans.
RESEARCH DESIGN AND METHODS—D4 cortisol was infused intravenously; arterial, portal venous, and hepatic venous blood was sampled; and liver and visceral fat were biopsied in subjects undergoing bariatric surgery.
RESULTS—Ratios of arterial and portal vein D4 cortisol/cortisoltotal (0.06 ± 0.01 vs. 0.06 ± 0.01) and D4 cortisol/D3 cortisol (1.80 ± 0.14 vs. 1.84 ± 0.14) did not differ, indicating that no visceral cortisol production or conversion of D4 cortisol to D3 cortisol via 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD-1) occurred. Conversely, ratios of both D4 cortisol to cortisoltotal (0.05 ± 0.01; P < 0.05) and D4 cortisol to D3 cortisol (1.33 ± 0.11; P < 0.001) were lower in the hepatic vein than in the portal vein, indicating production of both cortisol and D3 cortisol by the liver. The viscera did not produce either cortisol (−8.1 ± 2.6 μg/min) or D3 cortisol (−0.2 ± 0.1 μg/min). In contrast, the liver produced both cortisol (22.7 ± 3.90 μg/min) and D3 cortisol (1.9 ± 0.4 μg/min) and accounted for all splanchnic cortisol and D3 cortisol production. Additionally, 11β-HSD-1 mRNA was approximately ninefold higher (P < 0.01) in liver than in visceral fat. Although 11β-HSD-2 gene expression was very low in visceral fat, the viscera released cortisone (P < 0.001) and D3 cortisone (P < 0.01) into the portal vein.
CONCLUSIONS—The liver accounts for all splanchnic cortisol production in obese nondiabetic humans. In contrast, the viscera releases cortisone into the portal vein, thereby providing substrate for intrahepatic cortisol production.
Although it has been long known that glucocorticoids are potent regulators of glucose, fat, and protein metabolism, glucocorticoids have not been thought to cause insulin resistance in either obese or diabetic individuals because plasma concentrations do not differ from those present in lean nondiabetic subjects. However, extra-adrenal conversion of cortisone to cortisol via 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD-1) can result in high local concentrations of cortisol. This observation focused attention on the possibility that tissue-specific synthesis of glucocorticoids may contribute to the pathogenesis of insulin resistance and other components of the so called “metabolic syndrome” (1). The enzyme 11β-HSD-2 (which converts cortisol to cortisone) is present primarily in the kidney, whereas 11β-HSD-1 (which converts cortisone to cortisol) is present in both liver and adipose tissue with in vitro activity being greater in omental than subcutaneous fat deposits (2–5). Inhibition (6) or knockout (7–9) of 11β-HSD-1 in mice improves hepatic insulin action and protects against obesity and hyperglycemia. Conversely, selective overexpression of 11β-HSD-1 in adipose tissue in mice results in development of visceral obesity, hyperglycemia, hyperlipidemia, and hypertension (7–11).
Using a novel tracer infusion method, Andrew et al. (12) demonstrated that infusion of [9,11,12,12-2H4] cortisol (D4 cortisol) in fasting, nondiabetic humans resulted in the formation of measurable amounts of plasma [9,12,12-2H3] cortisol (D3 cortisol). Because conversion of D4 cortisol to D3 cortisone by 11β-HSD-2 results in the loss of the 11 α-deuterium and the generation of D3 cortisone that in turn forms D3 cortisol when D3 cortisone is converted back to cortisol, this observation provides strong experimental evidence that the conversion of cortisone to cortisol occurs in humans (12). More recently, we used the same method in combination with the hepatic venous and leg catheterization techniques to determine the site(s) of conversion of cortisone to cortisol. Those studies (13) led to the discovery that rates of splanchnic cortisol production in healthy nondiabetic individuals equaled or even exceeded those produced by extrasplanchnic tissues (e.g., the adrenals). However, because concomitant uptake of cortisol also occurred within the splanchnic bed, only a small net amount of cortisol was released into the systemic circulation.
Because portal venous blood was not sampled in those studies, we could not determine the individual contributions of the viscera and the liver to splanchnic cortisol production. We therefore addressed this question in a chronically catheterized conscious dog model that permitted simultaneous selective sampling of blood from an artery, the portal vein, and the hepatic vein during intravenous infusion of D4 cortisol (14). Surprisingly, we showed that the liver accounted for all of the splanchnic cortisol production in the dog without discernable release by the viscera. However, the dogs were lean, and it is unknown if the pattern of splanchnic cortisol production in dogs reflects that in humans. Therefore, it remained possible that visceral fat releases cortisol into the portal vein in obese humans, thereby exposing the liver to high local glucocorticoid concentrations.
The present experiments addressed this question by selectively obtaining simultaneous samples of arterial, portal venous, and hepatic venous blood during a D4 cortisol infusion in severely obese subjects undergoing bariatric surgery. In addition, mRNA for the glucocorticoid receptor (NR3C1), 11β-HSD-1, and 11β-HSD-2 was measured in liver and visceral fat obtained during surgery. We report that the liver accounts for all of the splanchnic cortisol production in obese nondiabetic humans. In contrast, there was no detectible release of cortisol into the portal vein by the viscera. On the other hand, although the mRNA for 11β-HSD-2 in visceral fat was very low, the viscera released cortisone into the portal vein, thereby providing the liver with substrate for intrahepatic cortisol production.
RESEARCH DESIGN AND METHODS
After approval from the Mayo Institutional Review Board, 10 nondiabetic subjects (8 women and 2 men) gave informed written consent to participate in the study. All volunteers were undergoing elective bariatric surgery via a celiotomy and met the inclusion and exclusion criteria for the surgical procedure. Subjects had a mean age of 52 ± 3 years, a mean BMI of 46 ± 3 kg/m2, fasting plasma glucose of 97 ± 2 mg/dl, alkaline phosphatase of 98 ± 15 units/l (reference range 45–115 units/l), aspartate aminotransferase (AST) of 24 ± 1 unit/l (reference range 8–48 units/l), and A1C of 5.2 ± 0.5%. The liver was biopsied during surgery. Histopathology indicated that 7 of the 10 subjects had mild steatohepatitis and that the remaining three had normal livers. Subjects were on stable doses of thyroxine, estrogen replacement therapy, selective serotonin reuptake inhibitor antidepressants, low-dose thiazide, calcium channel blocker, losartan, or lipid-lowering agent at the time of surgery.
Subjects were admitted to the Mayo Clinic Center for Translational Science Activities Clinical Research Unit at 1700 h on the evening before the study. A standard 10-cal/kg meal (55% carbohydrate, 30% fat, and 15% protein) was eaten between 1730 and 1800 h.
At ∼0600 h on the morning of operation, an intravenous catheter was placed in a forearm vein for the infusions. A primed, continuous infusion of [9,11,12,12-2H4] cortisol (0.22 mg prime, 0.19 mg/h continuous; Cambridge Isotope Laboratories, Andover, MA) was started. Subjects then were taken to an interventional radiology suite where a hepatic venous catheter was placed selectively via the internal jugular vein under fluoroscopic control. An artery catheter was placed by the anesthesiologist as part of the operative procedure. Approximately 1 h before the start of the operation, an infusion of indocyanine green dye (Akorn, Buffalo Grove, IL) was begun via the forearm vein. During the intraoperative period, portal venous samples were obtained by catheterizing a small mesenteric vein of a segment of the proximal jejunum and threading a 3-French intravenous catheter into the portal vein (16). Two blood samples were taken simultaneously (5 min apart) from the hepatic vein, the portal vein, and the arterial catheter. The portal venous catheter then was removed, and that section of mesentery and jejunum was resected as per surgical technique (16). The hepatic venous catheter was also removed, and the infusions of D4 cortisol and indocyanine green were discontinued. Samples of liver and omental fat were also obtained during the operation.
Analytical techniques.
All samples were placed on ice, centrifuged at 4°C, and separated. Plasma concentrations of indocyanine green were measured spectrophotometrically at 805 nm on the day of study as described previously (14). All other samples were stored at −80°C until analysis.
Concentrations of hepatic venous, arterial, and portal venous cortisol; D4 cortisol; D3 cortisol; cortisone; and D3 cortisone were measured using liquid chromatography tandem mass spectrometry as described previously (13). In brief, methylprednisolone was added as an internal standard, and methylene chloride was used to extract the relevant steroids. The dried extract was then reconstituted and injected into a liquid chromatography tandem mass spectrometer. Cortisol, D4 cortisol, and D3 cortisol ions were generated with electrospray source in positive mode and were detected with multiple reactions monitoring the specific transitions for m/z 363 to 121, 367 to 121, and 366 to 121, respectively. This approach enabled simultaneous monitoring of both the pronated parent ion and fragmented daughter ion, thereby increasing specificity.
Liver and omental fat tissue were quick frozen with liquid nitrogen and stored in dry ice. Then 150-mg (liver) and 200-mg (fat) sections were weighed and lysed using an appropriate amount of Qiazol Lysis Reagent (Qiagen, Valencia, CA) and mechanical homogenization. Qiazol homogenates were used to isolate total RNA using automated Qiagen RNeasy technology on the Qiagen BioRobot 3000. Reverse transcription and PCR were performed at standard temperatures using the Invitrogen SuperScript III Platinum One-Step Quantitative RT-PCR System (Invitrogen, Carlsbad, CA). All samples were analyzed for RNA integrity using the Agilent 2100 Bioanalyzer. The samples were run in triplicate. Data analysis was performed by first normalizing all data to the Total Human Reference RNA standard curve for each gene to achieve a relative amount of target RNA (ng). Then, each sample was normalized to the average amount of RNA (ng) for the three control housekeeper genes (PPIA, RPLPO, and RPL19; Clontech, Mountain View, CA). This approach permitted assessment of relative expression between samples of interest for a specific target gene; however, comparison of absolute amounts of expression between genes was not possible.
Calculations.
Portal and splanchnic plasma flows were calculated by dividing the indocyanine green infusion rate by the arterial-to-portal venous and arterial-to-hepatic venous concentration gradient of the dye, respectively (13,14). Hepatic artery plasma flow was calculated by subtracting portal plasma flow from splanchnic plasma flow.
Fractional extraction of D4 cortisol across the liver was calculated as
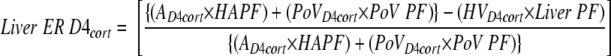 |
(1) |
where ER is the extraction ratio, A is arterial, HV is hepatic venous, PoV is portal venous, HAPF (ml/min) is hepatic artery plasma flow, PoV PF (ml/min) is portal venous plasma flow, and Liver PF (ml/min) is liver plasma flow.
Fractional extraction across the viscera was calculated as
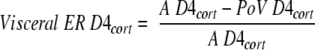 |
(2) |
Viscera net cortisol balance was calculated as
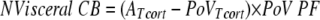 |
(3) |
where N is net and CB is cortisol balance and Tcort (referred to as cortisoltotal in the text) is the sum of unlabeled cortisol, D4 cortisol, and D3 cortisol.
Liver net cortisol balance was calculated as
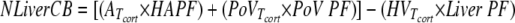 |
(4) |
Liver cortisol uptake was calculated as
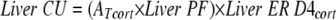 |
(5) |
where CU is cortisol uptake.
Visceral cortisol uptake was calculated as
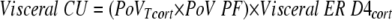 |
(6) |
Visceral and liver cortisol productions were calculated
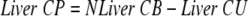 |
(7) |
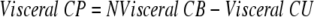 |
(8) |
For the sake of clarity of presentation, hepatic cortisol and D3 cortisol productions are expressed as positive numbers in the figures and the text.
D3 visceral and liver net balance, uptake, and production were calculated using Eqs. 3–8, except that D3 cortisol concentration was substituted for total cortisol concentration.
Statistical analyses.
Data in the text and figures are expressed as means ± SEM. Rates are expressed as micrograms per minute. Responses were determined by averaging the results of the two sets of samples taken 5 min apart. Student's paired t test was used to determine whether rates differed across the viscera and liver. ANOVA also was used to compare results among the artery, portal vein, and hepatic vein. A P value of <0.05 was considered as statistically significant.
RESULTS
Arterial, portal venous, and hepatic venous total cortisol, D4 cortisol, and D3 cortisol concentrations.
Portal venous cortisol concentrations were lower (P < 0.001) than arterial cortisol concentrations (17.6 ± 2.3 vs. 20.3 ± 2.3 μg/dl), indicating net uptake of cortisol by the viscera. Hepatic venous cortisol concentrations (16.7 ± 1.6 μg/dl) were lower (P < 0.001) than arterial but did not differ from portal venous cortisol concentrations (Fig. 1).
FIG. 1.

Concentrations of cortisoltotal, D4 cortisol, and D3 cortisol in the artery, portal vein, and hepatic vein. §P < 0.05 vs. portal vein; #P < 0.001 vs. portal vein; *P < 0.01 vs. artery; †P < 0.001 vs. artery.
Portal venous D4 cortisol concentrations (0.84 ± 0.10 μg/dl) were lower (P < 0.01) than arterial D4 cortisol concentrations (0.97 ± 0.11 μg/dl) but greater (P < 0.001) than hepatic venous D4 cortisol concentrations (0.73 ± 0.10 μg/dl), indicating 13 ± 2 and 15 ± 2% extraction of cortisol by the viscera and the liver, respectively.
Portal venous D3 cortisol concentrations were lower (P < 0.01) than arterial D3 cortisol concentrations (0.49 ± 0.07 vs. 0.57 ± 0.09 μg/dl), indicating net uptake of D3 cortisol by the viscera. Hepatic venous D3 cortisol concentrations (0.60 ± 0.10 μg/dl) were greater (P < 0.05) than portal venous concentrations but did not differ from arterial cortisol concentrations.
Arterial, portal venous, and hepatic venous total cortisone and D3 cortisone concentrations.
Portal venous cortisone concentrations were higher (P < 0.001) than arterial cortisone concentrations (3.55 ± 0.44 vs. 1.45 ± 0.12 μg/dl), indicating net release of cortisone by the viscera. Hepatic venous cortisone concentrations (0.10 ± 0.03 μg/dl) were lower (P < 0.001) than both arterial and portal venous cortisone concentrations, indicating net extraction of cortisone by the liver (Fig. 2).
FIG. 2.

Concentrations of cortisonetotal and D3 cortisone in the artery, portal vein, and hepatic vein. §P < 0.05 vs. portal vein; #P < 0.001 vs. portal vein; †P < 0.001 vs. artery.
A similar pattern was observed for D3 cortisone. Portal venous D3 cortisone concentrations were higher (P < 0.05) than arterial D3 cortisone concentrations (0.16 ± 0.05 vs. 0.06 ± 0.02 μg/dl), indicating net release by the viscera. Hepatic venous D3 cortisone concentrations (0.00 ± 0.00 μg/dl) were lower (P < 0.05) than portal venous concentrations, indicating net extraction of D3 cortisone by the liver.
Plasma D4 cortisol–to–cortisoltotal and D4 cortisol–to–D3 cortisol ratios.
The plasma ratio of D4 cortisol to cortisoltotal only is influenced by a change in the rate of cortisol production because both D4 cortisol and cortisol are cleared in parallel. The plasma ratio of D4 cortisol to cortisoltotal was identical in the artery and portal vein (0.06 ± 0.01 vs. 0.06 ± 0.01), indicating that the viscera did not produce cortisol. In contrast, the ratio of D4 cortisol to cortisoltotal in the hepatic vein (0.05 ± 0.01) was lower (P < 0.05) than that in the portal vein and artery, indicating cortisol production by the liver (Fig. 3).
FIG. 3.

Ratios of D4 cortisol to cortisoltotal and D4 cortisol to D3 cortisol in the artery, portal vein, and hepatic vein. §P < 0.05 vs. portal vein; #P < 0.001 vs. portal vein.
Similarly, the ratio of D4 cortisol to D3 cortisol did not differ in the artery and the portal vein (1.80 ± 0.14 vs. 1.84 ± 0.14) but was lower (P < 0.001) in the hepatic vein (1.33 ± 0.11) than in the portal vein, indicating generation of D3 cortisol from D4 cortisol via the 11 β-HSD-1 pathway in the liver but not in the viscera.
Visceral and liver net cortisol balance, cortisol uptake, and cortisol production.
Arterial and portal venous plasma flow averaged 77 ± 11 and 983 ± 94 ml/min, respectively. Net visceral cortisol balance (26.94 ± 4.81 μg/min) was positive, indicating net cortisol uptake across the viscera. In contrast, net cortisol balance across the liver did not differ from zero (11.87 ± 8.08 μg/min). Visceral cortisol uptake tended to be lower (P = 0.07) than liver cortisol uptake (18.84 ± 2.42 vs. 34.52 ± 7.32 μg/min). Cortisol production by the liver averaged 22.65 ± 3.85 μg/min, accounting for all cortisol produced within the splanchnic bed. No cortisol was produced by the viscera (−8.10 ± 2.62 μg/min) (Figs. 4 and 5).
FIG. 4.

Viscera and liver net cortisol balance, cortisol uptake, and cortisol production. *P < 0.001 vs. viscera.
FIG. 5.

Schematic representation of visceral and liver cortisol and D3 cortisol uptake and production. LCP, liver cortisol production; LCU, liver cortisol uptake; VCP, viscera cortisol production; VCU, viscera cortisol uptake.
Viscera and liver net D3 cortisol balance, D3 cortisol uptake, and D3 cortisol production.
D4 cortisol is converted to D3 cortisone by 11β-HSD-2, which in turn is converted to D3 cortisol by 11β-HSD-1. Therefore, measurement of D3 cortisol production provides an index of flux through the 11β-HSD-1 pathway. Net balance of D3 cortisol across the viscera averaged 0.8 ± 0.2 μg/min, indicating net uptake. Net D3 cortisol balance across the liver averaged −1.0 ± 0.3 μg/min, indicating there was net release of D3 cortisol. Uptake of D3 cortisol by the visceral and the liver averaged 0.6 ± 0.1 and 0.9 ± 0.2 μg/min, respectively. Whereas significant amounts (P < 0.001 vs. zero production) of D3 cortisol were produced by the liver (1.9 ± 0.4 μg/min), no D3 cortisol was produced by the viscera (−0.2 ± 0.1 μg/min) (Figs. 5 and 6).
FIG. 6.

Viscera and liver D3 cortisol net balance, D3 cortisol uptake, and D3 cortisol production. †P < 0.01 and *P < 0.001 vs. viscera.
Gene expression of glucocorticoid receptor, 11β-HSD-1, and 11β-HSD-2 in visceral fat and the liver.
Gene expression (mRNA) for the glucocorticoid receptor (NR3C1) did not differ in the liver and visceral fat. In contrast, 11β-HSD-1 mRNA expression was approximately ninefold higher (P < 0.01) in the liver than visceral fat. 11β-HSD-2 mRNA expression was low but detectible (<0.5 relative ng of human reference RNA standard curve) in both liver and visceral fat (Fig. 7).
FIG. 7.

Relative mRNA expression for glucocorticoid receptor (NR3C1), 11β-HSD-1, and 11β-HSD-2. †P < 0.01 vs. viscera.
DISCUSSION
These data indicate that the liver accounts for virtually all splanchnic cortisol production in obese humans. Although 11β-HSD-1 gene expression is present in visceral fat, albeit at levels far lower than in the liver, the viscera does not appear to release cortisol into the portal vein nor does it convert D4 cortisol to D3 cortisol. In contrast, despite very low expression of mRNA for 11β-HSD-2 in visceral fat, the viscera releases cortisone and D3 cortisone (derived from D4 cortisol) into the portal vein, thereby providing substrate for intrahepatic cortisol production.
The conclusion that the liver accounts for virtually all splanchnic cortisol production in obese humans is consistent with what we previously observed in chronically catheterized lean dogs (17). These observations differ from the report by Walker and colleagues (18) that visceral fat accounts for approximately two-thirds and the liver approximately one-third of splanchnic cortisol production. However, as appropriately pointed out by those authors (18), this conclusion was based on estimates rather than direct measurement of visceral and liver cortisol production. The lack of detectible cortisol release into the portal vein by the viscera in the present experiments does not exclude the possibility that cortisol is generated within visceral adipocytes and that the resultant high intracellular concentrations do not equilibrate with the extracellular space; however, because cortisol diffuses readily across plasma membranes, we believe this to be unlikely. The subjects in the present experiments were undergoing bariatric surgery. Therefore, it is possible that visceral 11β-HSD-1 activity was inhibited by anesthesia. However, the fact that substantial cortisone-to-cortisol conversion occurred within the liver and the rates of splanchnic cortisol production observed in the present experiments are comparable with those observed in our previous experiments in the absence of anesthesia argue against this possibility (13).
Calculation of rates of cortisol production requires measurement of blood flow. Therefore, variability in blood flow could have confounded detection of the release of small amounts of cortisol by the viscera. However, the virtually identical tracer-to-tracee ratios (i.e., D4 cortisol to cortisoltotal and D4 cortisol to D3 cortisol) across the viscera provide blood flow–and model-independent evidence that there was no release of cortisol by the viscera into the portal vein. Cortisol could have been released by visceral fat into the portal vein that was not detected because of dilution by blood flow from other tissues within the viscera that did not release cortisol. Because the subjects were severely obese and therefore had massive amounts of visceral fat, if such release did occur, it was not sufficient to increase portal cortisol delivery to the liver in these subjects and therefore is extremely unlikely to do so in less obese humans. Perhaps more importantly, cortisol production was measured across the entire viscera. Different patterns of conversion could occur in different tissues. For example, visceral fat could convert cortisone to cortisol via 11β-HSD-1 reductase and gut epithelium could convert cortisol to cortisone via 11β-HSD-2. However, conversion of cortisol to cortisone in one tissue (e.g., gut) does not influence calculation of visceral cortisol or D3 cortisol production in another, because this would be reflected by an increase in fractional extraction of D4 cortisol, which in turn is used to calculate cortisol production (see Eqs. 6 and 8).
It should be noted that although none of the subjects were known to have diabetes, only fasting glucose concentrations were measured. Therefore, it is possible (in fact likely) that at least some of the subjects would have been diagnosed as having diabetes if two oral glucose tolerance tests had been performed. However, because visceral cortisol production was negligible in all subjects, we do not believe that oral glucose tolerance test status would change our conclusions.
Consistent with previous reports, 11β-HSD-2 mRNA was barely detectible in visceral fat (15). We therefore were surprised to find that there was net release of cortisone by the viscera into the portal vein. There also was net release of D3 cortisone derived from the D4 cortisol entering the viscera. The increment in cortisone between the artery and portal vein averaged ∼21 μg/l. Because portal blood flow averaged ∼1 l/min, this resulted in ∼21 μg/min cortisone being delivered to the liver, which essentially equaled the ∼23 μg/min cortisol produced by the liver. Although rates of D3 cortisone release by the viscera (∼1.0 μg/min) and hepatic D3 cortisol production by the liver (∼1.9 μg/min) were not matched as closely, the potential contribution of viscerally derived D3 cortisone to hepatic D3 cortisol production still was substantial. If this observation is confirmed in subsequent studies, then it would suggest that whereas the viscera do not increase hepatic cortisol exposure by releasing cortisol into the portal vein, cortisone release could increase cortisol concentrations within the liver by providing the substrate for hepatic 11β-HSD-1. If so, this would be particularly intriguing, because the reduction in portal cortisol by conversion of cortisol to cortisone by the viscera would decrease local cortisol concentrations in areas of the liver where 11β-HSD-1 is not expressed but increase concentrations in areas where 11β-HSD-1 is expressed (e.g., the periportal region) (19). The observation that substantial conversion of cortisol to cortisone occurred in the viscera despite the fact that 11β-HSD-2 message was barely detectible in visceral fat is intriguing. However, because 11β-HSD-1 is a bidirectional enzyme and because substantial amounts of mRNA for this enzyme were present in omental fat, it could account for most if not all of visceral cortisol to cortisone conversion (1,2).
In conclusion, the liver accounts for virtually all splanchnic cortisol production in obese, nondiabetic humans. There is no evidence for visceral cortisol production whether measured as dilution of tracee-to-tracer ratios across the viscera or by calculation of rates of production of visceral cortisol or D3 cortisol. On the other hand, the viscera release cortisone into the portal vein, thereby providing the liver additional substrate for intrahepatic cortisol production. The extent to which hepatic cortisol production is regulated by the rate of cortisone delivery and/or other factors remains to be determined.
Acknowledgments
R.A.R. is the Earl and Annette R. McDonough Professor of Medicine. This study was supported by U.S. Public Health Service grants DK-29953, RR-00585, and U-54RR-24150-1 and by the Mayo Clinic.
No potential conflicts of interest relevant to this article were reported.
We thank Barbara Norby, Betty Dicke, P. Reich, and G. DeFoster for technical assistance; Cynthia Nordyke and Monica Le for assistance with graphics; and the staff of the Mayo Clinical Research Unit and Center for Clinical and Translation Science Activities for assistance with the studies.
Published ahead of print at http://diabetes.diabetesjournals.org on 13 October 2008.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C Section 1734 solely to indicate this fact.
See accompanying commentary, p. 14
REFERENCES
- 1.Walker BR: Is “Cushing's disease of the omentum” an affliction of mouse and men? Diabetologia 47: 767–769, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Moore JS, Monson JP, Kaltsas G, Putignano P, Wood PJ, Sheppard MC, Besser GM, Taylor NF, Stewart PM: Modulation of 11β-hydroxysteroid dehydrogenase isozymes by growth hormone and insulin-like growth factor: in vivo and in vitro studies. J Clin Endocrinol Metab 84: 4172–4177, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Jamieson PM, Chapman KE, Edwards CRW, Seckl JR: 11ß-hydroxysteroid dehydrogenase is an exclusive 11ß-reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology 136: 4754–4761, 1995 [DOI] [PubMed] [Google Scholar]
- 4.Paulmyer-Lacroix O, Boullu S, Oliver C, Alessi M-C, Grino M: Expression of the mRNA coding for 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: an in situ hybridization study. J Clin Endocrinol Metab 87: 2701–2705, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Bujalska I, Kumar S, Stewart PM: Does central obesity reflect “Cushing's disease of the omentum”? Lancet 349: 1210–1213, 1997 [DOI] [PubMed] [Google Scholar]
- 6.Barf T, Vallgarda J, Edmond R, Haggstrom C, Kurz G, Nygren A, Larwood V, Mosialou E, Axelsson K, Olsson R, Engblom L, Edling N, Rönquist-Nii Y, Öhman B, Alberts P, Abrahmsen L: Arylsulfonamiodothiazoles as a new class of potential antidiabetic drugs: discovery of potent and selective inhibitors of the 11β-hydroxysteroid dehydrogenase type 1. J Med Chem 45: 3813–3815, 2002 [DOI] [PubMed] [Google Scholar]
- 7.Kotelevtsev Y, Holmes MC, Burchell A, Houston PM, Schmoll D, Jamieson P, Best R, Brown R, Edwards CRW, Seckl JR, Mullins JJ: 11β-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci U S A 94: 14924–14929, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes MC, Kotelevtsev Y, Mullins JJ, Seckl JR: Phenotypic analysis of mice bearing targeted deletions of 11β-hydroxysteroid dehydrogenases 1 and 2 genes. Mol Cell Endocrinol 171: 15–20, 2001 [DOI] [PubMed] [Google Scholar]
- 9.Alberts P, Engblom L, Edling N, Forsgren M, Klingström G, Larsson C, Rönquist-Nii Y, Öhman B, Abrahmsén L: Selective inhibition of 11β-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia 45: 1528–1532, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS: A transgenic model of visceral obesity and the metabolic syndrome. Science 294: 2166–2170, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MGF, Fleming S, Mullins JJ, Seckl JR, Flier JS: Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest 112: 83–90, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrew R, Smith K, Jones GC, Walker BR: Distinguishing the activities of 11β-hydrogenases in vivo using isotopically labeled cortisol. J Clin Endocrinol Metab 87: 277–285, 2002 [DOI] [PubMed] [Google Scholar]
- 13.Basu R, Singh RJ, Basu A, Chittilapilly EG, Johnson CM, Toffolo G, Cobelli C, Rizza RA: Splanchnic cortisol production occurs in humans: evidence for conversion of cortisone to cortisol via the 11-β hydroxysteroid dehydrogenase (11-β HSD) type 1 pathway. Diabetes 53: 2051–2059, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Basu A, Basu R, Shah P, Vella A, Johnson CM, Nair KS, Jensen MD, Schwenk WF, Rizza RA: Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: evidence for a defect in hepatic glucokinase activity. Diabetes 49: 272–283, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Tomlinson JW, Stewart PM: Cortisol metabolism and the role of 11beta-hydroxysteroid dehydrogenase. Best Pract Res Clin Endocrinol Metab 15: 61–78, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Ikramuddiin S, Kendrick ML, Kellogg TA, Sarr MG: Open and laparoscopic Roux-en-Y gastric bypass: our techniques. J Gastrointest Surg 11: 217–228, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Basu R, Edgerton DS, Singh RJ, Cherrington A, Rizza RA: Splanchnic cortisol production in dogs occurs primarily in the liver: evidence for substantial hepatic specific 11β hydroxysteroid dehydrogenase type 1 activity. Diabetes 55: 3013–3019, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Andrew R, Westerbacka J, Wahren J, Yki-jarvinen H, Walker BR: The contribution of visceral adipose tissue to splanchnic cortisol production in healthy humans. Diabetes 54: 1364–1370, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, Stewart PM: Immunohistological localization of type 1 11β-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab 83: 1325–1335, 1998 [DOI] [PubMed] [Google Scholar]