

Abstract
OBJECTIVE—11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) regenerates cortisol from cortisone. 11β-HSD1 mRNA and activity are increased in vitro in subcutaneous adipose tissue from obese patients. Inhibition of 11β-HSD1 is a promising therapeutic approach in type 2 diabetes. However, release of cortisol by 11β-HSD1 from adipose tissue and its effect on portal vein cortisol concentrations have not been quantified in vivo.
RESEARCH DESIGN AND METHODS—Six healthy men underwent 9,11,12,12-[2H]4-cortisol infusions with simultaneous sampling of arterialized and superficial epigastric vein blood sampling. Four men with stable chronic liver disease and a transjugular intrahepatic porto-systemic shunt in situ underwent tracer infusion with simultaneous sampling from the portal vein, hepatic vein, and an arterialized peripheral vein.
RESULTS—Significant cortisol and 9,12,12-[2H]3-cortisol release were observed from subcutaneous adipose tissue (15.0 [95% CI 0.4–29.5] and 8.7 [0.2–17.2] pmol · min−1 · 100 g−1 adipose tissue, respectively). Splanchnic release of cortisol and 9,12,12-[2H]3-cortisol (13.5 [3.6–23.5] and 8.0 [2.6–13.5] nmol/min, respectively) was accounted for entirely by the liver; release of cortisol from visceral tissues into portal vein was not detected.
CONCLUSIONS—Cortisol is released from subcutaneous adipose tissue by 11β-HSD1 in humans, and increased enzyme expression in obesity is likely to increase local glucocorticoid signaling and contribute to whole-body cortisol regeneration. However, visceral adipose 11β-HSD1 activity is insufficient to increase portal vein cortisol concentrations and hence to influence intrahepatic glucocorticoid signaling.
Cortisol has potent effects in adipose tissue, influencing insulin sensitivity, fatty acid metabolism, adipocyte differentiation, adipokine expression, and body fat distribution (1). Adrenal secretion of cortisol is controlled by the hypothalamic-pituitary-adrenal axis; however, recent evidence suggests that cortisol is also generated from inert cortisone within adipose tissue by the enzyme 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) (2,3). Conversion of cortisone to cortisol occurs in vitro in human adipocytes cultured from visceral and subcutaneous adipose depots (4) and in vivo during infusion of [3H]2-cortisone into subcutaneous adipose tissue by microdialysis (5). In obesity, 11β-HSD1 mRNA and activity are increased in subcutaneous adipose tissue biopsies (6) and either increased or unchanged in visceral adipose tissue (rev. in 7). 11β-HSD1 inhibitors are being developed to lower intracellular cortisol concentrations in adipose tissue and liver in type 2 diabetes and obesity, with promising preclinical and early clinical results (8).
In addition to influencing intra-adipose cortisol concentrations, it has been suggested that cortisol release into the portal vein from visceral adipose tissue contributes to hepatic insulin resistance associated with central obesity (4). Transgenic overexpression of 11β-HSD1 in adipose tissue in mice results in a two- to threefold increase in portal vein glucocorticoid concentrations without altering systemic levels (9). However, the extent to which cortisol generated by 11β-HSD1 is released into the portal or systemic circulation from visceral or subcutaneous adipose tissue, respectively, in humans is unknown. In arteriovenous samples across subcutaneous adipose tissue, cortisol concentrations do not change, although there is net removal of cortisone (10,11). Similarly, sampling from portal or omental veins during intra-abdominal surgery has not revealed higher cortisol concentrations than in arterial blood (12,13).
Measuring cortisol concentrations in arterial and venous samples may not detect cortisol release by 11β-HSD1 if cortisol is also removed by other enzymes. This occurs, for example, in the liver, where cortisol concentrations are lower in hepatic vein than in arterial blood (14). A tracer technique is required to detect cortisol production in the liver in the face of additional cortisol clearance. We devised a stable isotope deuterated tracer—9,11,12,12-[2H]4-cortisol (d4-cortisol)—for this purpose (15). During d4-cortisol infusion, there is removal of the 11α-2H by 11β-HSD type 2 to form d3-cortisone, which is then regenerated to d3-cortisol by 11β-HSD1 (Fig. 1). The dilution of d4-cortisol by d3-cortisol therefore indicates 11β-HSD1 reductase activity and is independent of removal of both d4-cortisol and d3-cortisol by other enzymes. In tissues in which there is no source of cortisol production other than by 11β-HSD1, the dilution of d4-cortisol by cortisol also indicates 11β-HSD1 activity. Using this technique, we and others have quantified substantial cortisol release into the hepatic vein by 11β-HSD1 in the splanchnic circulation (visceral organs plus liver) (16,17). Moreover, by extrapolating from the rate of cortisol release into hepatic vein during first-pass liver metabolism of an oral dose of cortisone, we estimated that a substantial proportion of splanchnic cortisol production occurs in visceral tissues and liver (16). However, direct cannulation of veins draining adipose tissue depots during tracer cortisol infusion has not been reported, and portal vein sampling has only been performed in dogs in which cortisol release by visceral tissues was undetectable (18).
FIG. 1.

Quantifying cortisol production using deuterated cortisol. d4-Cortisol is converted mainly in the kidney to d3-cortisone, with the loss of the deuterium on C11. The d3-cortisone is then reduced by 11β-HSD1, predominantly in the liver and adipose tissue, with the addition of an unlabeled hydrogen to form d3-cortisol. Differences between d3-cortisol and d4-cortisol metabolism therefore reflect 11β-HSD1 reductase activity.
Here, we report results of deuterated cortisol infusions with selective venous cannulation to measure arteriovenous differences across subcutaneous adipose tissue and visceral tissues, to quantify cortisol release by 11β-HSD1 from adipose tissue for the first time in humans.
RESEARCH DESIGN AND METHODS
Participants were men, aged 20–70 years, with BMI 20–45 kg/m2 and normal full blood count and renal and thyroid function, and received no glucocorticoid therapy in the previous 6 months. Six men with normal liver function tests and alcohol intake <21 units/week were recruited for the subcutaneous adipose tissue study. Three subjects had no concurrent medical conditions, two had hypertension, and one had Parkinson's disease. Felodipine, bendroflumethiazide, enalapril, levodopa, entacapone, and pramipexole were each taken by one subject. Four men with alcoholic cirrhosis and transjugular intrahepatic portal-systemic shunts (TIPSS) in situ were recruited for the portal vein study. The TIPSS had been inserted at least 1 year previously for portal hypertension. These patients were currently abstinent from alcohol and attending an annual check of TIPSS patency. Three TIPSS patients had no additional medical conditions; one had type 2 diabetes. Three were taking proton pump inhibitors; insulin, metoprolol, spironolactone, furosemide, and aspirin were each taken by one subject. Local ethical approval and written informed consent were obtained.
Subjects were given oral dexamethasone (1 mg in TIPSS patients and 1.5 mg in healthy men) at 2300 h and fasted until attending the Clinical Research Facility at 0800 h. Measurements were taken of height, weight, total fat mass, and percentage body fat by bioimpedance. Blood was taken for fasting glucose and lipids. 21G cannulae were placed in a right antecubital fossa vein for infusion and a left dorsal hand vein for sampling; the left hand was placed in a box heated to 60°C to achieve arterialization. d4-Cortisol (Cambridge Isotopes, Andover, MA) was infused at 40% molar enrichment with 60% cortisol (Calbiochem, Nottingham, U.K.) at 1.74 mg/h for 210 min after a priming 3.5-mg bolus. The combination of dexamethasone and unlabeled cortisol administration aimed to maintain circulating cortisol and cortisone concentrations at stable nonstressed physiological levels during steady-state measurements. d4-Cortisol was infused at sufficiently high enrichment to ensure detectable levels of d3-cortisol and d3-cortisone, even in hepatic vein. Dexamethasone is not known to interfere with cortisol kinetics.
Cortisol release from subcutaneous adipose tissue in healthy men.
Subjects were served breakfast (30 g cornflakes and 300 ml skim milk) at 0800–0830 h, and 5% dextrose (50 ml/h) was infused intravenously throughout the study. Once the deuterated cortisol infusion was established, a 20G 15-cm catheter was sited in a superficial epigastric vein, as previously described (19). To ensure that blood was collected from subcutaneous adipose tissue and not from deeper structures, O2 saturation was confirmed to be >85%. After 2 h of d4-cortisol infusion, 1–2 MBq 133Xenon (gas) was injected subcutaneously beside the umbilicus, and radioactivity was measured continuously with a NaI detector to assess blood flow (20,21). Blood samples were taken from arterialized hand vein and superficial epigastric vein at intervals (Fig. 2).
FIG. 2.

Left panel: subcutaneous sampling; right panel: visceral sampling. Plasma measurements during deuterated cortisol infusion. Data are means ± SE for n = 6 (subcutaneous measurements) and n = 4 (visceral measurements) during deuterated cortisol infusion, with plasma samples from arterialized (▪), portal or subcutaneous (⋄), and hepatic (▴) cannulae. Plasma cortisol concentrations (A), plasma d4-cortisol enrichment (C), and d4-cortisol–to–d3-cortisol ratio (E) for subcutaneous study. Plasma cortisol concentrations (B), plasma d4-cortisol enrichment (D), d4-cortisol–to–d3-cortisol ratio (F) for visceral study. Statistical comparison of mean values in steady state (180–210 min) is shown in Table 2.
Cortisol release into portal and hepatic veins in TIPSS patients.
These patients were not given breakfast or infused with dextrose. During the d4-cortisol infusion, dexamethasone was concurrently infused at 240 μg/h. Twenty minutes after beginning the tracer infusion, the right internal jugular vein was cannulated under local anesthesia (5 ml 2% lidocaine), and a 5F pigtail catheter (Cordis, Berkshire, U.K.) was passed into the TIPSS under X-ray guidance. After confirming patency of the TIPSS, the catheter was positioned in the portal vein for sampling. A 5F vertebral catheter (Merrit Medical, Lanarkshire, U.K.) was then placed in a separate tributary of the hepatic vein for sampling. From 2 h after beginning the tracer infusion, indocyanine green (ICG) (Pulsion Medical, Middlesex, U.K.) was infused into the antecubital vein at 30 mg/h. Blood samples were taken from the portal, hepatic, and arterialized veins at intervals (Fig. 2).
Laboratory analyses.
Plasma cortisol, d3-cortisol, d4-cortisol, cortisone, and d3-cortisone were measured by liquid chromatography–tandem mass spectrometry (LC-MS/MS). Epi-cortisol (500 ng) was added to 1.5 ml plasma and extracted using 15 ml chloroform. Samples were evaporated and then reconstituted in mobile phase (60% methanol and 40% 5 mmol/l ammonium acetate) before injection into a Thermo Finnigan LC-MS/MS, consisting of a TSQ Quantum Discovery Mass Spectrometer and a Surveyor Liquid Chromatogram using an Allure biphenyl column (50 mm × 4.6 mm × 5 μm; Thames Restek), with column temperature 25°C and mobile phase flow rate 0.5 ml/min. Ionization was achieved by positive electrospray. The precursor and product mass–to–charge ratios used were as follows: cortisol (363→121), d3-cortisol (366→121), d4-cortisol (367→121), cortisone (361→163), and d3-cortisone (364→164). Compounds were quantified by the ratio of area under peak of interest to area under peak of internal standard against a standard curve.
Serum ICG was measured by adapting a previous method (22) using diazepam as internal standard. Briefly, acetonitrile (ACN) was added to serum to sediment protein, the supernatant was mixed with ammonium sulfate and centrifuged, and the organic phase was added to water before analysis by high-performance liquid chromatography (HPLC) using a P680 HPLC pump and a PDA-100 photodiode array detector (Dionex, Sunnyvale, CA) with a Nova-pak C18 column (300 mm × 3.9 mm × 4 μm) at a temperature of 35°C. Analytes were eluted under linear gradient conditions at 1 ml/min mobile phase (initially, 80% water, 12% ACN, and 8% methanol; 0–17 min, 25% water, 65% ACN, and 8% methanol; and 17–29 min, 80% water, 12% ACN, and 8% methanol) and detected at λ230 (diazepam) and λ784 nm (ICG).
Plasma glucose, lipids, and liver function were measured using enzymatic colorimetric methods on an Olympus Diagnostics analyzer (County Clare, Ireland). Plasma A1C was analyzed by HPLC using a Variant II analyzer (Bio-Rad Laboratories, Hertfordshire, U.K.).
Kinetic analyses.
Calculations in each subject used the mean of measurements in steady state, between 180 and 210 min of d4-cortisol infusion (Fig. 2). Where possible, kinetic calculations relying on tracer-to-tracee ratios rather concentrations were favored to minimize variability. The equations are derived from Wolfe and Chinkes (23).
The concentration of ICG in the artery (A) and hepatic vein (HV) in steady state (ss) were used to calculate hepatic blood flow (HBF) (24) using
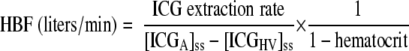 |
(1) |
Whole-body cortisol kinetics were calculated from measurements in arterialized samples (15,16) using Eqs. 2 and 3, where “cortisol” denotes unlabeled cortisol. Clearances were calculated by dividing the infusion rate (of cortisol or d4-cortisol) by the steady-state arterial concentration (of cortisol or d4-cortisol) (15).
 |
(2) |
 |
(3) |
Subcutaneous adipose tissue production of cortisol and d3-cortisol were calculated from measurements in arterialized (A) and superficial epigastric vein (V) samples using Eqs. 4 and 5. “Tissue delivery” is synonymous with “influx.”
 |
(4) |
where
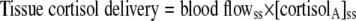 |
 |
(5) |
where
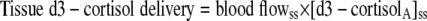 |
Splanchnic cortisol and d3-cortisol production (from visceral tissues and liver combined) were calculated from measurements in arterialized (A) and hepatic vein (HV) samples as previously described (16) using Eqs. 6 and 7, in which “tissue delivery” was calculated as above.
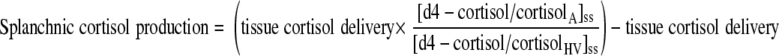 |
(6) |
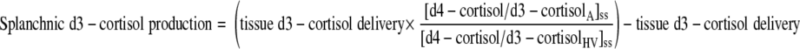 |
(7) |
Liver production of cortisol and d3-cortisol were calculated from measurements in portal vein (PV) and hepatic vein (HV) using Eqs. 8 and 9. HBF was measured by ICG extraction (Eq. 1) but portal blood flow (PBF) and hepatic arterial blood flow (HABF) were not measured. In health, the portal vein provides ∼80% and the hepatic artery ∼20% of total liver blood flow (25). In cirrhosis, PBF decreases and HABF increases (26,27), such that as little as 10% of HBF may originate from the portal vein (28). To account for this unknown, we have modeled PBF to range between 10 and 80% of HBF and concordantly for HABF to range between 90 and 20% of HBF.
 |
(8) |
where
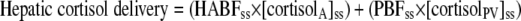 |
 |
(9) |
where
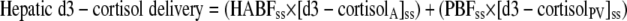 |
Visceral production of cortisol and d3-cortisol (i.e., release into the portal vein) was calculated using measurements from arterialized blood and portal vein with estimates of PBF as above, using Eqs. 10 and 11, in which tissue delivery was calculated as above.
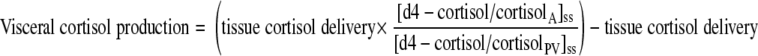 |
(10) |
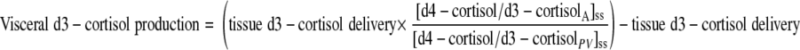 |
(11) |
Net production of cortisone and d3-cortisone across the viscera, liver, and the splanchnic tissues were calculated using Eqs. 12 and 13, in which the relevant measurements of blood flow and venous concentrations were substituted as appropriate.
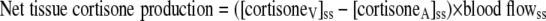 |
(12) |
 |
(13) |
Statistical analysis.
Using variance of steady-state kinetic parameters, a power calculation showed that including four patients in the portal vein sampling study gives >80% power to detect (to P < 0.05) release of 10 nmol/min cortisol into the portal vein. This provides ample power to detect the ∼30 nmol/min cortisol, which was estimated to be released into the portal vein from indirect modeling (16).
Using SPSS version 14, comparisons were by paired t tests or repeated measures ANOVA with post hoc Fisher's least significant differences test, as appropriate. Differences from zero were determined using the one-sample t test. P < 0.05 was considered significant. Data are presented as mean ± SE or, for calculated kinetic parameters, mean (95% CI).
RESULTS
Subject characteristics.
Participants’ anthropometric and biochemical characteristics are shown in Table 1.
TABLE 1.
Anthropometry and fasting plasma biochemistry
| Subcutaneous arteriovenous sampling | Visceral arteriovenous sampling | |
|---|---|---|
| n | 6 | 4 |
| Age (years) | 53.8 ± 5.4 | 50.9 ± 4.1 |
| BMI (kg/m2) | 30.8 ± 3.7 | 32.3 ± 1.1 |
| Fat mass (%) | 28.9 ± 4.1 | 30.6 ± 2.6 |
| Fat mass (kg) | 29.4 ± 7.1 | 32.5 ± 4.3 |
| Plasma glucose (mmol/l) | 5.5 ± 0.4 | 7.8 ± 2.0 |
| Plasma A1C (%) | 4.3 ± 0.3 | 6.3 ± 1.2 |
| Total plasma cholesterol (mmol/l) | 5.4 ± 0.3 | 4.3 ± 0.7 |
| Plasma triglycerides (mmol/l) | 1.7 ± 0.6 | 1.0 ± 0.3 |
| Serum alanine aminotransferase (units/l) | 30.5 ± 6.0 | 26.3 ± 2.6 |
| Serum bilirubin (μmol/l) | 8.7 ± 1.7 | 23.5 ± 6.3 |
Data are means ± SE.
Superficial epigastric vein cannulation study.
Adrenal cortisol production was suppressed by dexamethasone, with fasting morning plasma cortisol concentrations of 23 ± 9 nmol/l and cortisone concentrations of 9 ± 4 nmol/l. Steady state was achieved between 180 and 210 min of d4-cortisol infusion (Fig. 2).
Mean plasma measurements at steady state are in Table 2. Cortisol and d4-cortisol clearance was 0.56 ± 0.09 and 0.90 ± 0.09 l/min, respectively. Plasma cortisol concentrations were not different between artery and superficial epigastric vein; however, there was a trend for increased d3-cortisol levels in the vein (P < 0.06). d4-Cortisol, cortisone, and d3-cortisone concentrations were unchanged between artery and vein.
TABLE 2.
Steady-state concentrations and ratios during deuterated cortisol infusion
| Subcutaneous measurements
|
Visceral measurements
|
||||
|---|---|---|---|---|---|
| Artery | Subcutaneous vein | Artery | Portal vein | Hepatic vein | |
| Cortisol (nmol/l) | 97 ± 16 | 103 ± 15 | 186 ± 17 | 176 ± 22 | 180 ± 18 |
| d3-Cortisol (nmol/l) | 33 ± 4 | 36 ± 4 | 59 ± 4 | 56 ± 4 | 65 ± 6*† |
| d4-Cortisol (nmol/l) | 37 ± 3 | 37 ± 4 | 71 ± 9 | 67 ± 11 | 57 ± 6* |
| Cortisone (nmol/l) | 17 ± 2 | 14 ± 2 | 75 ± 6 | 91 ± 6 | 22 ± 16*† |
| d3-Cortisone (nmol/l) | 8 ± 1 | 6 ± 2 | 36 ± 4 | 45 ± 4* | 9 ± 8*† |
| d4-Cortisol–to–cortisol ratio | 0.40 ± 0.03 | 0.38 ± 0.03* | 0.38 ± 0.02 | 0.38 ± 0.02 | 0.32 ± 0.01*† |
| d4-Cortisol–to–d3-cortisol ratio | 1.13 ± 0.04 | 1.02 ± 0.04‡ | 1.19 ± 0.08 | 1.17 ± 0.10 | 0.87 ± 0.07*† |
Data are means ± SE for n = 6 (subcutaneous measurements) and n = 4 (visceral measurements) participants. The mean data from samples obtained between 180 and 210 min during the deuterated cortisol infusion was used to calculate steady-state concentrations. Comparisons were made by paired t test (subcutaneous study) or repeated measures ANOVA with post hoc testing with Fisher's least significant differences test (visceral study).
P < 0.05 and
P < 0.001 compared with artery;
P < 0.05 compared with portal vein.
The rates of appearance in arterial blood of cortisol (Eq. 2) and d3-cortisol (a specific measure of whole-body 11β-HSD1 activity; Eq. 3) were 33.8 (95% CI 16.0–51.5) and 28.3 (25.6–31.0) nmol/min, respectively. Adipose blood flow measured 2.5 ± 0.7 ml · min−1 · 100 g−1 adipose tissue. There was significant release across the subcutaneous adipose bed of both cortisol (15.0 [0.4–29.5] pmol · min−1 · 100 g−1 adipose tissue; Eq. 4) and d3-cortisol (8.7 [0.2–17.2] pmol · min−1 · 100 g−1 adipose tissue; Eq. 5) (both P < 0.05 vs. zero).
Hepatic and portal vein cannulation study.
Fasting morning plasma cortisol and cortisone concentrations were suppressed by dexamethasone, measuring 15 ± 5 and 11 ± 2 nmol/l, respectively. Steady state was achieved between 180 and 210 min of d4-cortisol infusion (Fig. 2).
Mean plasma measurements at steady state are in Table 2. Cortisol and d4-cortisol clearance was 0.27 ± 0.03 and 0.47 ± 0.06 l/min, respectively. In the hepatic vein, d3-cortisol concentrations were increased, and cortisone and d3-cortisone concentrations decreased, consistent with substantial intrahepatic steroid extraction and 11β-HSD1 reductase activity. Conversely, d4-cortisol, which cannot be regenerated by 11β-HSD1, was lower in the hepatic vein, consistent with substantial intrahepatic cortisol metabolism. In the portal vein, cortisol, d3-cortisol and d4-cortisol concentrations were unaltered compared with arterial blood. However, d3-cortisone concentrations were significantly increased, and there was a trend for increased cortisone (P = 0.051) in portal vein, consistent with visceral 11β-dehydrogenase activity.
The whole-body rates of appearance of cortisol (Eq. 2) and d3-cortisol (Eq. 3) in the artery were 36.3 (95% CI 24.4–48.2) and 26.9 (21.0–32.7) nmol/min, respectively. HBF by ICG extraction was 0.40 ± 0.08 l/min. Splanchnic production of cortisol (13.5 [3.6–23.5] nmol/min; Eq. 6) and d3-cortisol (8.0 [2.6–13.5] nmol/min; Eq. 7) was substantial (both P < 0.05 vs. zero). This could be accounted for entirely by the liver, because hepatic cortisol (Eq. 8) and d3-cortisol (Eq. 9) production rates were 13.3 (1.3–25.4) and 7.7 (1.3–14.2) nmol/min, respectively (both P < 0.05 vs. zero and not different from splanchnic production rates) when PBF was estimated as 40% of total HBF (28). No visceral cortisol (Eq. 10) or d3-cortisol (Eq. 11) release into the portal vein was detected (0.0 [−1.7 to 1.7] and 0.1 [−0.7 to 1.1] nmol/min, respectively). Modeling for portal venous flow from 10–80% of HBF did not significantly alter these results (not shown).
Net cortisone and d3-cortisone production across the splanchnic tissues (Eqs. 12 and 13) measured −18.2 (95% CI −33.3 to −3.2) and −9.2 (−15.4 to −3.0) nmol/min, respectively (both P < 0.05 vs. zero). This was accounted for by cortisone and d3-cortisone extraction across the liver (−20.8 [−39.4 to −2.2] and −10.6 [−18.3 to −2.8] nmol/min, estimating portal flow as 40% of HBF), consistent with substantial hepatic cortisone metabolism and 11β-HSD1 reductase activity. However, across the viscera, net cortisone production rate did not differ from zero (2.6 [−1.2 to 6.3] nmol/min; P = 0.12), and there was a trend for net generation of d3-cortisone (1.4 [−0.4 to 3.1] nmol/min; P = 0.09 vs. zero).
DISCUSSION
These data quantify for the first time the contributions of subcutaneous adipose tissue, visceral tissues, and liver to whole-body cortisol production by 11β-HSD1 in humans. We confirmed that splanchnic cortisol production is substantial, and we attribute this entirely to 11β-HSD1 activity in the liver. However, although we could not detect release of cortisol by 11β-HSD1 into the portal vein, which drains a number of visceral organs, we found significant cortisol release into veins draining exclusively subcutaneous adipose tissue. Similar results were obtained using the equations derived by Basu et al. (18). These results allow us to put in context the variations in 11β-HSD1 activity described in biopsied tissue, for example, in obesity.
The absolute rates of appearance of cortisol and d3-cortisol in steady state are sensitive to the prevailing concentrations of cortisone and d3-cortisone, the substrates for 11β-HSD1, which are determined by the rates of exogenous cortisol and d4-cortisol infusion. Moreover, the implications for intra-adipose cortisol concentrations are impossible to estimate, because venous changes in concentration at a given rate of appearance are highly dependent on tissue blood flow. However, with these and other caveats in mind, we can attempt to extrapolate from these data what some of the consequences might be for endogenous cortisol metabolism. If the release of cortisol by adipose tissue in the anterior abdominal wall were mirrored in all adipose depots, then the observed cortisol production rate of 15 pmol · min−1 · 100 g−1 adipose tissue would equate with a whole-body production rate of 4.0 ± 1.5 nmol/min, based on a total adipose mass of 29.4 ± 7.1 kg measured in our participants using bioimpedance. This represents ∼12% of whole-body cortisol regeneration by 11β-HSD1. Significant 11β-HSD1 activity in subcutaneous adipose tissue is supported by the trend for a fall in cortisone concentrations from artery to vein (Table 2), which, although not statistically significant, was of similar magnitude to previous larger studies (10,11). However, release of cortisol from subcutaneous adipose tissue may be offset by intra-adipose cortisol clearance, e.g., by 5α-reductase type 1 (29), so that arterial and venous cortisol concentrations are unaltered (Table 2), as previously described (10,11). Nevertheless, these data are consistent with the hypothesis that variations in 11β-HSD1 have a significant impact on intra-adipose cortisol concentrations (2,3,7).
Cortisol release from subcutaneous adipose tissue into the systemic circulation is unlikely to have effects in other organs, because the feedback control by the hypothalamic-pituitary-adrenal axis will adjust adrenal cortisol secretion to maintain circulating cortisol concentrations. Therefore, the most likely impact of this source of cortisol will be intracrine or paracrine in the local adipose environment. However, release from visceral adipose tissue into the portal vein could deliver cortisol directly to the liver, contributing to the association of central obesity with hepatic insulin resistance and dyslipidemia (4,16). We did not, however, detect release of cortisol from 11β-HSD1 in visceral tissues into portal vein, in agreement with a study in dogs (18). Assuming portal venous blood accounts for 40% of total HBF (28), the mean estimate for visceral cortisol release from these subjects was 0.0 (95% CI −1.7 to 1.7) nmol/min. Although the number of subjects included was small (n = 4), this provides 97.5% confidence that visceral cortisol production is <1.7 nmol/min. Because hepatic cortisol delivery was 72.7 nmol/min, we can conclude that any cortisol released by visceral 11β-HSD1 would not significantly impact on hepatic cortisol delivery.
To access portal vein samples, we studied patients with alcoholic liver disease and TIPSS. Although sufficiently unwell with portal hypertension to require a TIPSS, these patients were stable and were all overweight or obese. Cortisol metabolism is abnormal in cirrhosis (30–32), although specific measurements of 11β-HSD1 activity have not been reported. Compared with healthy volunteers undergoing subcutaneous adipose tissue measurements, our cirrhotic patients had lower whole-body clearance of cortisol and d4-cortisol and hence higher endogenous and deuterated cortisol and cortisone concentrations in steady state (Table 2). Splanchnic cortisol release was less than one-third of that reported previously under similar conditions (16,17,33), even though whole-body cortisol regeneration by 11β-HSD1 was not unusually low. This paradox may reflect either upregulation of 11β-HSD1 in nonsplanchnic tissues or an underestimation of splanchnic cortisol release because of misleading blood flow measurement. Although it is plausible that chronic liver disease may reduce hepatic 11β-HSD1 activity, it seems less likely that it would affect extrahepatic 11β-HSD1. As previously described in TIPSS patients (34), HBF estimated by ICG extraction was substantially lower than in healthy volunteers. Although none of our patients had reversed flow in the portal vein (hepatofugal flow) (35), a proportion of blood in the portal circulation is shunted away from the portal vein in patients with liver disease via anastomoses with the systemic circulation (36). However, none of these alterations predict that cortisol release into the portal vein should be artifactually low.
The portal vein drains blood from other organs, including gut, pancreas, and spleen. Although our results suggest that none of these organs releases cortisol by 11β-HSD1 reductase activity, it remains possible that venous drainage from the other visceral tissues dilutes any observable change in d3-cortisol–to–d4-cortisol ratios in blood from visceral adipose tissue. HBF was measured at 400 ml/min, of which portal vein flow may be up to 320 ml/min. This compares with blood flow of just 2.5 ml · min−1 · 100 g−1 in subcutaneous adipose tissue. If adipose blood flow were the same in the visceral as in the subcutaneous adipose depot and the visceral depot weighed 3 kg (37), then the contribution of visceral fat to PBF may be 75 ml/min, as little as a quarter of total flow. This dilution effect could only be overcome by cannulating an omental vein during d4-cortisol infusion, which is unlikely to be achievable during steady-state tracer cortisol infusion in unstressed subjects.
Steady-state plasma d3-cortisone concentrations were significantly higher in portal vein than in artery, and there was a similar trend for cortisone concentrations (Table 2). Cortisol, d3-cortisol, and d4-cortisol concentrations showed opposite trends. Although not confirmed by statistically significant differences in visceral d3-cortisone production, these results suggest 11β-dehydrogenase activity, converting cortisol to cortisone in the viscera. This is likely due to 11β-HSD type 2 activity in the gut, although it is conceivable that 11β-HSD1 may be functioning in the dehydrogenase direction in visceral adipose tissue (38).
In a previous study, we measured splanchnic cortisol production in steady state and modeled the relative contribution of liver and visceral tissues by measuring first pass conversion of oral cortisone into cortisol in the hepatic vein (16). We estimated that up to two-thirds of splanchnic cortisol production occurs in visceral tissues and that portal vein cortisol concentrations were likely to be ∼30 nmol/l higher than arterial concentrations. The current data do not support these estimates, at least in patients with cirrhosis. This discrepancy is most likely due to portal vein cortisone concentrations being higher than we predicted; our model was based in part on removal of cortisone by 11β-HSD1 in visceral adipose tissue, as occurs in subcutaneous adipose tissue (see above). Revisiting our model in light of the new finding of higher cortisone concentrations in the portal vein, we have confirmed that it predicts much higher steady-state cortisol regeneration in the liver and hence a much lower contribution from visceral adipose tissue.
What implications do these observations have for patients with obesity? The mean BMI in our participants was in the obese range (31 kg/m2 in the subcutaneous adipose tissue study and 32 kg/m2 in the portal vein study), although the numbers (n = 4–6) were too small to allow meaningful correlations with indexes of 11β-HSD1 activity. Previous measurements in biopsies suggest that 11β-HSD1 activity is ∼2.5-fold higher per gram of adipose tissue in obese people (BMI ∼31 kg/m2) than in lean controls with BMI ∼9 kg/m2 lower (6,39). Given the Km of human 11β-HSD1 for cortisone of ∼1 μmol/l (40), it is reasonable to assume a linear relationship between 11β-HSD1 protein concentrations and cortisol generation rates within the physiological range of cortisone concentrations of ∼10–100 nmol/l. Therefore, we anticipate that a 10-kg/m2 increase in BMI might elevate the cortisol production rate in subcutaneous adipose tissue by up to 2.5-fold (i.e., 37.5 pmol · min−1 · 100 g−1) and that, accounting for an associated ∼15 kg increase in fat mass, this equates with an increase in whole-body adipose cortisol production of ∼12.7 nmol/min. In obesity, hepatic 11β-HSD1 activity is decreased by ∼50% (6,39,41). The predicted increase in cortisol release from adipose tissue may cancel out the decrease in cortisol release from the liver in obesity, potentially explaining the lack of change in whole-body d3-cortisol production rate (5,33).
These data support the concept that 11β-HSD1 is a key determinant of intra-adipose cortisol concentrations but appear to refute the concept that 11β-HSD1 substantially elevates cortisol concentrations in the portal vein.
Acknowledgments
This work was funded by the British Heart Foundation, the Translational Medicine Research Institute, the Northern Sweden County Council, the Sweden Research Council, and the Sweden Heart and Lung Foundation and Heart Centre.
Within the last two years, T.O. has received consultant fees from Wyeth Pharmaceuticals and lecture fees from GlaxoSmithKline. Within the past two years, B.R.W. has consulted for AstraZeneca, Dainippon Sumitomo, Merck, Johnson & Johnson, Incyte, Ipsen, Roche, Vitae, Wyeth, and Zydus Research Centre, received lecture fees from Abbott and Bristol Myers Squibb, and received research funding from Wyeth. B.R.W. is also an inventor with relevant patents held by the University of Edinburgh. No other potential conflicts of interest relevant to this article were reported.
We thank Jill Harrison, Scott Denham, Natalie Homer, Kim MacBeth, Inger Arnesjo, Britt-Inger Norberg, Alistair Millar, Jean Antonelli, Lesley Breen, and the staff of the Welcome Trust Clinical Research Facility and its Mass Spectrometry Core Laboratory for their assistance and technical advice.
This research has been presented in part at the British Endocrine Society, Harrogate, U.K., 7–10 April 2008.
Published ahead of print at http://diabetes.diabetesjournals.org on 13 October 2008.
R.H.S. and J.A. contributed equally to this work.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C Section 1734 solely to indicate this fact.
See accompanying commentary, p. 14
REFERENCES
- 1.Macfarlane DP, Forbes S, Walker BR: Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J Endocrinol 197: 189–204, 2008 [DOI] [PubMed] [Google Scholar]
- 2.Seckl JR, Walker BR: 11β-Hydroxysteroid dehydrogenase type 1: a tissue-specific amplifier of glucocorticoid action. Endocrinology 142: 1371–1376, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, Hewison M, Stewart PM: 11Beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 25: 831–866, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bujalska IJ, Kumar S, Stewart PM: Does central obesity reflect ‘Cushing's disease of the omentum’? Lancet 349: 1210–1213, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Sandeep TC, Andrew R, Homer NZ, Andrews RC, Smith K, Walker BR: Increased in vivo regeneration of cortisol in adipose tissue in human obesity and effects of the 11β-hydroxysteroid dehydrogenase type 1 inhibitor carbenoxolone. Diabetes 54: 872–879, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DEW, Johnson O, Walker BR: Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab 86: 1418–1421, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Walker BR: Glucocorticoids and cardiovascular disease. Eur J Endocrinol 157: 545–559, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Hughes KA, Webster SP, Walker BR: 11-Beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) inhibitors in type 2 diabetes mellitus and obesity. Expert Opin Investig Drugs 17: 481–496, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS: A transgenic model of visceral obesity and the metabolic syndrome. Science 294: 2166–2170, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Katz JR, Mohamed-Ali V, Wood PJ, Yudkin JS, Coppack SW: An in vivo study of the cortisol-cortisone shuttle in subcutaneous abdominal adipose tissue. Clin Endocrinol 50: 63–68, 1999 [DOI] [PubMed] [Google Scholar]
- 11.Wake DJ, Stimson RH, Tan GD, Homer NZ, Andrew R, Karpe F, Walker BR: Effects of peroxisome proliferator-activated receptor (PPAR) {alpha} and {gamma} agonists on 11{beta}-hydroxysteroid dehydrogenase type 1 in subcutaneous adipose tissue in men. J Clin Endocrinol Metab 92: 1848–1856, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Aldhahi W, Mun E, Goldfine AB: Portal and peripheral cortisol levels in obese humans. Diabetologia 47: 833–836, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Tarantino G, Lobello R, Scopacasa F, Contaldo F, Pasanisi F, Cirillo M, De Caterina M, Conca P, Terracciano D, Gennarelli N, Ariello M, Mazzarella C, Grimaldi E, Macchia V: The contribution of omental adipose tissue to adipokine concentrations in patients with the metabolic syndrome. Clin Invest Med 30: E192–E199, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Walker BR, Campbell JC, Fraser R, Stewart PM, Edwards CRW: Mineralocorticoid excess and inhibition of 11β-hydroxysteroid dehydrogenase in patients with ectopic ACTH syndrome. Clin Endocrinol (Oxf) 27: 483–492, 1992 [DOI] [PubMed] [Google Scholar]
- 15.Andrew R, Smith K, Jones GC, Walker BR: Distinguishing the activities of 11beta-hydroxysteroid dehydrogenases in vivo using isotopically labelled cortisol. J Clin Endocrinol Metab 87: 277–285, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Andrew R, Westerbacka J, Wahren J, Yki-Jarvinen H, Walker BR: The contribution of visceral adipose tissue to splanchnic cortisol production in healthy humans. Diabetes 54: 1364–1370, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Basu R, Singh RJ, Basu A, Chittilapilly EG, Johnson CM, Toffolo G, Cobelli C, Rizza RA: Splanchnic cortisol production occurs in humans: evidence for conversion of cortisone to cortisol via the 11-beta hydroxysteroid dehydrogenase (11beta-hsd) type 1 pathway. Diabetes 53: 2051–2059, 2004 [DOI] [PubMed] [Google Scholar]
- 18.Basu R, Edgerton DS, Singh RJ, Cherrington A, Rizza RA: Splanchnic cortisol production in dogs occurs primarily in the liver: evidence for substantial hepatic specific 11{beta} hydroxysteroid dehydrogenase type 1 activity. Diabetes 55: 3013–3019, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Coppack SW, Fisher RM, Gibbons GF, Humphreys SM, McDonough MJ, Potts JL, Frayn KN: Postprandial substrate deposition in human forearm and adipose tissues in vivo. Clin Sci (Lond) 79: 339–348, 1990 [DOI] [PubMed] [Google Scholar]
- 20.Larsen OA, Lassen NA, Quaade F: Blood flow through human adipose tissue determined with radioactive xenon. Acta Physiol Scand 66: 337–345, 1966 [DOI] [PubMed] [Google Scholar]
- 21.Samra JS, Frayn KN, Giddings JA, Clark ML, MacDonald IA: Modification and validation of a commercially available portable detector for measurement of adipose tissue blood flow. Clin Physiol 15: 241–248, 1995 [DOI] [PubMed] [Google Scholar]
- 22.Burns E, Triger DR, Tucker GT, Bax ND: Indocyanine green elimination in patients with liver disease and in normal subjects. Clin Sci (Lond) 80: 155–160, 1991 [DOI] [PubMed] [Google Scholar]
- 23.Wolfe RL, Chinkes DL: Arterial-venous balance technique to measure amino acid kinetics. In Isotope Tracers in Metabolic Research. Principles and Practice of Kinetic Analysis. 2nd ed. New York, John Wiley and Sons, 2005, p. 381–420
- 24.Leevy CM, Mendenhall CL, Lesko W, Howard MM: Estimation of hepatic blood flow with indocyanine green. J Clin Invest 41: 1169–1177, 1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoli M, Magalotti D, Bianchi G, Gueli C, Orlandini C, Grimaldi M, Marchesini G: Total and functional hepatic blood flow decrease in parallel with ageing. Age Ageing 28: 29–33, 1999 [DOI] [PubMed] [Google Scholar]
- 26.Richter S, Mucke I, Menger MD, Vollmar B: Impact of intrinsic blood flow regulation in cirrhosis: maintenance of hepatic arterial buffer response. Am J Physiol Gastrointest Liver Physiol 279: G454–G462, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Lautt WW: Regulatory processes interacting to maintain hepatic blood flow constancy: vascular compliance, hepatic arterial buffer response, hepatorenal reflex, liver regeneration, escape from vasoconstriction. Hepatol Res 37: 891–903, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menzel J, Schober O, Reimer P, Domschke W: Scintigraphic evaluation of hepatic blood flow after intrahepatic portosystemic shunt (TIPS). Eur J Nucl Med 24: 635–641, 1997 [DOI] [PubMed] [Google Scholar]
- 29.Wake DJ, Strand M, Rask E, Westerbacka J, Livingstone DE, Soderberg S, Andrew R, Yki-Jarvinen H, Olsson T, Walker BR: Intra-adipose sex steroid metabolism and body fat distribution in idiopathic human obesity. Clin Endocrinol (Oxf) 66: 440–446, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Tyler FH: The effect of liver disease on adrenal cortical function. Am J Clin Nutr 5: 377–380, 1957 [DOI] [PubMed] [Google Scholar]
- 31.Kawai S, Ichikawa Y, Homma M: Differences in metabolic properties among cortisol, prednisolone, and dexamethasone in liver and renal diseases: accelerated metabolism of dexamethasone in renal failure. J Clin Endocrinol Metab 60: 848–854, 1985 [DOI] [PubMed] [Google Scholar]
- 32.Stewart PM, Burra P, Shackleton CHL, Sheppard MC, Elias E: 11β-Hydroxysteroid dehydrogenase deficiency and glucocorticoid status in patients with alcoholic and non-alcoholic liver disease. J Clin Endocrinol Metab 76: 748–751, 1993 [DOI] [PubMed] [Google Scholar]
- 33.Basu R, Singh RJ, Basu A, Chittilapilly EG, Johnson MC, Toffolo G, Cobelli C, Rizza RA: Obesity and type 2 diabetes do not alter splanchnic cortisol production in humans. J Clin Endocrinol Metab 90: 3919–3926, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Laiz JM, Banares R, Echenagusia A, Casado M, Camunez F, Perez-Roldan F, de Diego A, Cos E, Clemente G: Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension: preliminary results. Dig Dis Sci 40: 2121–2127, 1995 [DOI] [PubMed] [Google Scholar]
- 35.Rector WG Jr, Hoefs JC, Hossack KF, Everson GT: Hepatofugal portal flow in cirrhosis: observations on hepatic hemodynamics and the nature of the arterioportal communications. Hepatology 8: 16–20, 1988 [DOI] [PubMed] [Google Scholar]
- 36.Bosch J, Pizcueta P, Feu F, Fernandez M, Garcia-Pagan JC: Pathophysiology of portal hypertension. Gastroenterol Clin North Am 21: 1–14, 1992 [PubMed] [Google Scholar]
- 37.Westerbacka J, Yki-Jarvinen H, Vehkavaara S, Hakkinen A-M, Andrew R, Wake DJ, Seckl JR, Walker BR: Body fat distribution and cortisol metabolism in healthy men: enhanced 5β-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver. J Clin Endocrinol Metab 88: 4924–4931, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Bujalska IJ, Walker EA, Tomlinson JW, Hewison M, Stewart PM: 11Beta-hydroxysteroid dehydrogenase type 1 in differentiating omental human preadipocytes: from de-activation to generation of cortisol. Endocr Res 28: 449–461, 2002 [DOI] [PubMed] [Google Scholar]
- 39.Rask E, Walker BR, Soderberg S, Livingstone DE, Eliasson M, Johnson O, Andrew R, Olsson T: Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11beta-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab 87: 3330–3336, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Shafqat N, Elleby B, Svensson S, Shafqat J, Jornvall H, Abrahmsen L, Oppermann U: Comparative enzymology of 11beta-hydroxysteroid dehydrogenase type 1 from glucocorticoid resistant (guinea pig) versus sensitive (human) species. J Biol Chem 278: 2030–2035, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Stewart PM, Boulton A, Kumar S, Clark PMS, Shackleton CHL: Cortisol metabolism in human obesity: impaired cortisone: cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab 84: 1022–1027, 1999 [DOI] [PubMed] [Google Scholar]