

1. Structure
The mechanisms leading to activation of pleoiotropic transcription factor nuclear factor-kappa binding protein (NF-κB) are the subject of current research in many disease pathologies including ocular. NF-κB controls the expression of a large number of genes involved in apoptosis, cell growth, survival, differentiation and immune response. Alterations in NF-κB activity are associated with a large number of diseases such as autoimmune, cancer and inflammatory. NF-κB proteins (also known as Rel family of proteins) include subunits of p50 (derived from p105; NF-κB1), p52 (derived from p100; NF-κB2), p65 (RelA), c-Rel and RelB. These subunits are ubiquitously expressed in mammalian cells and are highly conserved across the species. They can homodimerize or heterodimerize to form an active molecule of NF-κB. The NF-κB proteins are characterized by their Rel homology domains, which contain crucial functional regions for DNA binding, dimerization, interactions with inhibitory subunits known as IκB proteins in the cytoplasm (IκBα, IκBβ, and IκBε) and nuclear internalization. NF-κB specifically recognizes consensus sequence of 5′-GGGRNYYYCC-3′ (R is an unspecified purine; Y is an unspecified pyrimidine; and N is any nucleotide) that is also known as κB DNA binding sequence (Chen et al., 1998).
2. Function
NF-κB plays an important role in regulation of immune and inflammatory responses. Pathogens, oxidants, cytokines, chemokines, and growth factors associated with oxidative stress trigger specific receptors and cause oxidative stress signaling cascades that lead to activation of NF-κB. Therefore NF-κB is recognized as a redox-sensitive transcription factor. There are two major signaling pathways that lead to activation of NF-κB (Figure-1). In the canonical (or classical) pathway, the ligand binds to its cognate receptor and activates the IκB kinase (IKK) complex that contains the catalytic kinase subunits (IKKα, IKKβ) and the regulatory non-enzymatic scaffold protein called NEMO (NF-κB essential modulator or IKKγ) that leads to phosphorylation of IκB. Proteosomal degradation of the phosphorylated-IκB subunit enables nuclear translocation of active NF-κB resulting in changes in transcription of target genes. This pathway is generic in the sense that the IKK complex is capable of activating NF-κB dimers comprised of P50, P65, c-Rel and RelB subunits. In the non-canonical (or alternative) pathway, receptor activation leads to activation of NF-κB-inducing kinase (NIK) that then activates the IKK complex. Activation of the IKK complex leads to phosphorylation of the inactive NF-κB p100/RelB dimer that is then processed further forming the active p52/RelB dimer. The active NF-κB dimer then translocates into the nucleus and alters gene transcription. This NF-κB activation pathway is triggered by a small number of stimuli and in involved generation of B and T lymphocytes from lymphoid organs (Kumar et al., 2002).
Figure 1. NF-κB signaling pathways.
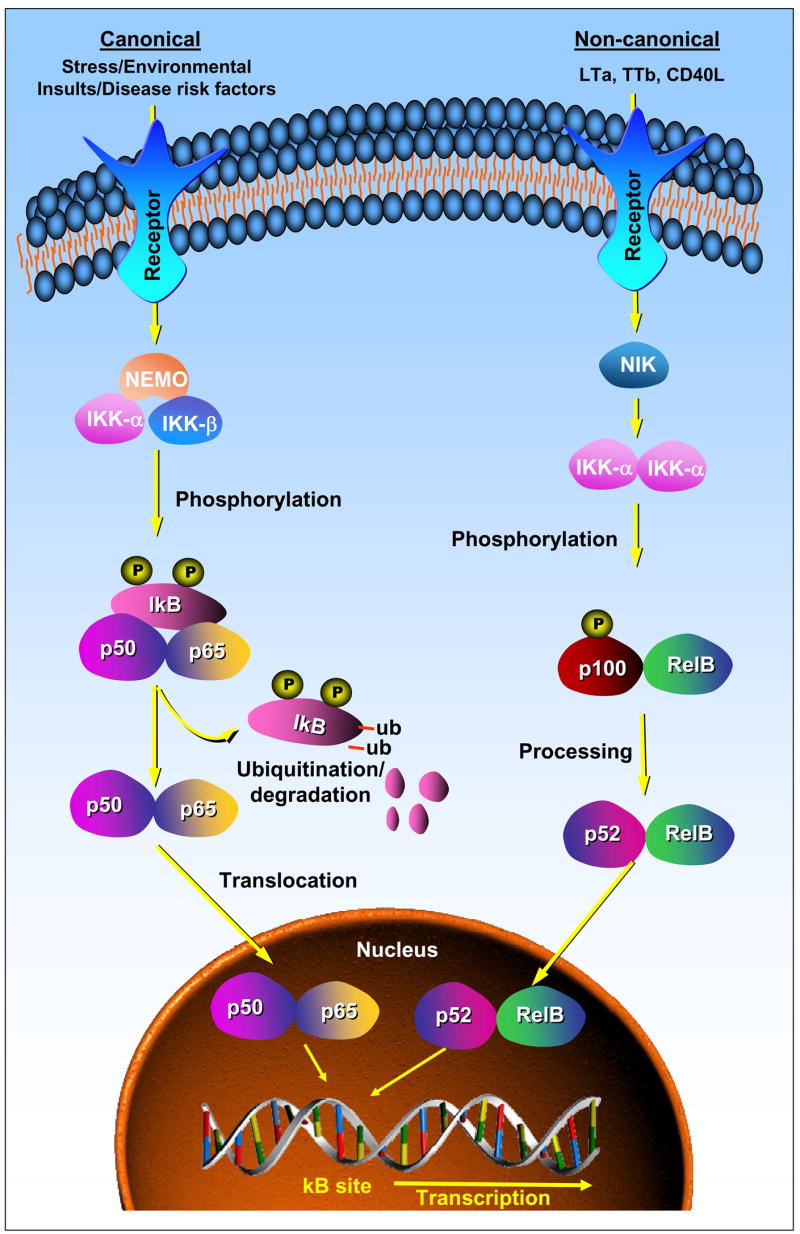
In canonical pathway (left), the binding of specific ligand to a receptor leads to the phosphorylation of IκB by IKK complex comprising of IKKα, IKKβ and NEMO which releases active NF-κB dimer that translocates in to nucleus to transcribe target genes. In non-canonical pathway (right), the binding of ligand to receptor leads to processing of p100/RelB via NIK mediated activation of IKK complex comprising of two units of IKKα. The processed p52/RelB translocates in to the nucleus to transcribe target genes.
NF-κB activation is responsible for the expression of a wide variety of genes that encode cytokines (TNF, IL-1, IL-6), chemokines (MIP-1, MCP-1), adhesion molecules (ICAM, VCAM, E-Selectin), iNOS and Cox-2. In the innate immune response, NF-κB facilitates T-cell activation by up-regulating expression of MHC proteins and CD80/86 in antigen - presenting cells. In the adaptive phase of an immune system, NF-κB activates T and B lymphocytes. Further, activation of NF-κB increases transcription of genes encoding anti-apoptotic, cell cycle regulator, and survival proteins. Therefore, activation of NF-κB is required for the differentiation, survival, proliferation and apoptosis of a wide variety of cells including immune, cancer, and normal cells. Studies using knockout animals have revealed both specific and redundant functions of NF-κB. For example, deletion of p65 in mice is embryonic lethal and mice lacking p50 or RelB develop immunodeficiency.
3. Disease Involvement
Increased expression of cytokines and chemokines associated with activation of NF-κB is a key feature of inflammation. Inflammation triggers distinct intracellular signaling cascades that lead to both acute physiological effects and long-term changes in gene expression. Hence elucidation of NF-κB signaling is critical for understanding multiple diseases, including infections, atherosclerosis, uveitis and cancer, and for developing therapeutic interventions for minimizing their inflammatory components. Oxidative stress –induced abnormal activation of NF-κB has been observed in a number of diseases such as heart failure, atherosclerosis, diabetes, asthma, cancer, sepsis and autoimmune (Kumar et al, 2004). Further, activation of NF-κB under various oxidative stress conditions has been observed in almost all the eye tissues including lens and retina, cornea and iris. However, detailed studies on the involvement of NF-κB in ocular diseases such as uveitis, age-related macular degeneration (AMD), cataracts, glaucoma, retinopathy and iritis are lacking.
The eye may be affected as a target of immune inflammatory attack due to over-activity of NF-κB in autoimmune diseases. Systemic autoimmune diseases such as Behcet’s disease and juvenile chronic arthritis often attack a variety of organs, including various parts of the eye. However, in some autoimmune diseases the eye may be the only target affected. Such diseases include ocular cicatricial pemphigoid and Mooren’s corneal ulcers. Uveitis, another inflammatory eye disease caused by increased activation of NF-κB-dependent inflammatory signals initiated due to infections and autoimmune diseases. Although activation of NF-κB plays a central and crucial role in these infection and autoimmune-initiated inflammatory processes, the mechanisms are not clearly understood. Activation of NF-κB is associated with the overexpression of inflammatory cytokines, iNOS and Cox-2 enzymes and increasing levels of NO and PGE2. TNF-α, a major NF-κB dependent inflammatory cytokine, is recognized as a central mediator in the pathophysiology of many chronic inflammatory diseases which cause increased risk of ocular inflammation.
Recent studies have suggested the use of anti-TNF-α and anti-NF-κB therapies to treat ocular inflammation specially uveitis. Our recent reports show that inhibition of polyol pathway enzyme aldose reductase (AR) could prevent endotoxin- and TNF-α-induced activation of NF-κB in human lens epithelial cells demonstrating a major role of AR in mediating inflammatory signals in lens. This should lead to the use of AR inhibitors as potential anti-inflammatory drugs to prevent ocular inflammation, especially uveitis. In deed, using endotoxin- induced uveitis in Lewis rats, we have recently (Yadav et al., 2007) shown that inhibition of AR prevents activation of NF-κB and significantly reduces inflammatory cell infiltration and increase in inflammatory markers. This highlights the potential clinical use of AR inhibitors in therapy for uveitis and other inflammatory disorders.
4. Future Studies
It is well established that increased activation of NF-κB transcription factor elicits the cytotoxic actions of bacterial pathogens, bacterial toxins and oxidative stress induced by various oxidants and xenobiotics. Over 100,000 deaths in the U.S. each year can be attributed to an excessive inflammatory response alone. Activation of NF-κB plays a critical role in several cardiovascular and neurological degenerative diseases as well as cancer. However, it is still not clearly understood how the activation of NF-κB in various ocular tissues plays a critical role in the ocular inflammation leading to blindness. Hence detailed studies are required to elucidate the mechanisms that regulate NF-κB signals in ocular tissues. There has been very little work done to delineate the mechanisms leading to canonical NF-κB activation in uveitis, glaucoma and AMD and no reports are available on non-canonical activation of NF-κB in ocular tissues. Furthermore, NF-κB activation is likely to have wide implications not only for ocular immunology and inflammatory diseases but also for conditions such as corneal transplantation, posterior capsular opacification and intraocular tumors. Novel modulatory components in NF-κB signaling pathways in ocular inflammation need to be explored. One of such modulatory component could be the AR inhibitors that could prevent NF-κB activation and inflammatory signaling (Srivastava et al, 2005). The use of AR inhibitors could represent a substantial change in the current clinical approach in ocular inflammatory diseases such as uveitis. It is also potentially important to examine the roles of NF-κB family members in the development of eye and to uncover the relationship of these processes to NF-κB function. A better understanding of NF-κB regulation is of fundamental significance in escalating our knowledge of ocular inflammation would be of major clinical importance, as it may identify potential therapeutic options that may be more effective and safer than currently available treatments for various sight threatening eye diseases.
Acknowledgments
NIH grants DK36118 (SKS) and GM71036 (KVR).
References
- Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- Kumar A, Takada Y, Aggarwal BB. Nuclear Factor-kB: its role in health and disease. J Mol Med. 2004;82:432–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- Yadav UC, Srivastava SK, Ramana KV. Aldose reductase inhibition prevents endotoxin-induced uveitis in rats. Invest Ophthalmol Vis Sci. 2007;48:4634–4642. doi: 10.1167/iovs.07-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–92. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]