

Abstract
The term excitation-coupled Ca2+ entry (ECCE) designates the entry of extracellular Ca2+ into skeletal muscle cells, which occurs in response to prolonged depolarization or pulse trains and depends on the presence of both the 1,4-dihydropyridine receptor (DHPR) in the plasma membrane and the type 1 ryanodine receptor in the sarcoplasmic reticulum (SR) membrane. The ECCE pathway is blocked by pharmacological agents that also block store-operated Ca2+ entry, is inhibited by dantrolene, is relatively insensitive to the DHP antagonist nifedipine (1 μM), and is permeable to Mn2+. Here, we have examined the effects of these agents on the L-type Ca2+ current conducted via the DHPR. We found that the nonspecific cation channel antagonists (2-APB, SKF 96356, La3+, and Gd3+) and dantrolene all inhibited the L-type Ca2+ current. In addition, complete (>97%) block of the L-type current required concentrations of nifedipine >10 μM. Like ECCE, the L-type Ca2+ channel displays permeability to Mn2+ in the absence of external Ca2+ and produces a Ca2+ current that persists during prolonged (∼10-second) depolarization. This current appears to contribute to the Ca2+ transient observed during prolonged KCl depolarization of intact myotubes because (1) the transients in normal myotubes decayed more rapidly in the absence of external Ca2+; (2) the transients in dysgenic myotubes expressing SkEIIIK (a DHPR α1S pore mutant thought to conduct only monovalent cations) had a time course like that of normal myotubes in Ca2+-free solution and were unaffected by Ca2+ removal; and (3) after block of SR Ca2+ release by 200 μM ryanodine, normal myotubes still displayed a large Ca2+ transient, whereas no transient was detectable in SkEIIIK-expressing dysgenic myotubes. Collectively, these results indicate that the skeletal muscle L-type channel is a major contributor to the Ca2+ entry attributed to ECCE.
INTRODUCTION
The skeletal muscle L-type Ca2+ channel (1,4-dihydropyridine receptor [DHPR]) serves as the voltage sensor for excitation–contraction (EC) coupling (Tanabe et al., 1988). Conformational changes in the DHPR in response to membrane depolarization are coupled to gating of the type 1 RYR (RYR1) of the SR (Beam and Horowicz, 2004). The resultant Ca2+ efflux via RYR1 into the myoplasm initiates contraction (Beam and Horowicz, 2004). It has also been reported that the interaction between the DHPR and RYR1 activates entry of extracellular Ca2+ into the myoplasm via an as yet unidentified pathway (Cherednichenko et al., 2004, 2008; Hurne et al., 2005; Yang et al., 2007; Gach et al., 2008; Lyfenko and Dirksen, 2008). This form of Ca2+ entry, termed excitation-coupled Ca2+ entry (ECCE), requires expression of both the DHPR and RYR1, as ECCE is absent in dysgenic (DHPR α1S subunit–null) and dyspedic (RYR1-null) myotubes, respectively. Cherednichenko et al. (2004) proposed that ECCE is independent of L-type Ca2+ current via the DHPR because ECCE (as assessed by Mn2+ quench of the ratiometric Ca2+ indicator Fura-2) persists in dysgenic myotubes transfected with an α1S pore mutant (SkEIIIK) thought to conduct only monovalent cations (Dirksen and Beam, 1999). Thus, it was proposed that ECCE involved an as yet unidentified channel activated by conformational coupling to both the DHPR and RYR1.
When myotubes are stimulated either by maintained depolarization with KCl or by repetitive, brief electrical stimuli, myoplasmic Ca2+ levels initially rise and then decay as a consequence of declining release from the SR, although this decay is slowed by the entry of external Ca2+ associated with ECCE (Yang et al., 2007; Cherednichenko et al., 2008; Gach et al., 2008). Furthermore, ECCE is accentuated in myotubes expressing RYR1 constructs that carry mutations causing malignant hyperthermia in humans (Yang et al., 2007; Cherednichenko et al., 2008). Thus, it appears that ECCE may be important in normal skeletal muscle for helping to maintain force generation during tetanic stimulation, and that accentuated ECCE may contribute to the pathophysiological increase in myoplasmic Ca2+ that occurs during episodes of malignant hyperthermia.
ECCE is blocked by 2-aminoethyl diphenylborate (2-APB), SKF 96356, La3+, and Gd3+ (Cherednichenko et al., 2004; Hurne et al., 2005) and thus has a pharmacological profile similar to that of store-operated Ca2+ entry (SOCE) and transient receptor potential–like channels. However, ECCE can occur without any store depletion (Cherednichenko et al., 2004, 2008; Hurne et al., 2005; Yang et al., 2007; Gach et al., 2008; Lyfenko and Dirksen, 2008) or in cells in which stores are fully depleted (Cherednichenko et al., 2004; Gach et al., 2008). Furthermore, all attempts to identify transient receptor potential or SOCE channels as the molecular basis of ECCE have failed (Lee et al., 2006; Lyfenko and Dirksen, 2008; Woo et al., 2008).
While attempting to identify the Ca2+ current associated with ECCE, we noticed that 2-APB, SKF 96356, La3+, and Gd3+ each block the skeletal muscle L-type current, as has been reported for ECCE (Cherednichenko et al., 2004; Hurne et al., 2005; Yang et al., 2007). These results prompted us to reexamine the similarities and differences between the skeletal muscle L-type current and ECCE. We found that the L-type current is incompletely (∼50%) blocked by 1 μM nifedipine and partially inhibited by the anti-malignant hyperthermia drug dantrolene, as described previously for ECCE (Yang et al., 2007; Cherednichenko et al., 2008). Also like ECCE (Cherednichenko et al., 2004, 2008; Hurne et al., 2005; Gach et al., 2008; Lyfenko and Dirksen, 2008), we found that the L-type channel can conduct Mn2+ in the absence of external Ca2+.
In addition to examining the pharmacology of the DHPR, we evaluated the contribution of the L-type current to myoplasmic Ca2+ transients. In normal myotubes, the Ca2+ transient decays during maintained depolarization due to inactivation of EC coupling (Beam and Horowicz, 2004). This decay is accelerated by the removal of external Ca2+ or by the addition of Gd3+, consistent with the idea that Ca2+ entry contributes to the transient (Cherednichenko et al., 2004; Yang et al., 2007). In the present study, a substantial contribution of L-type current to this entry was revealed by the observation that the decay of the myoplamsic Ca2+ transient in dysgenic cells expressing SkEIIIK was essentially unchanged by the removal of extracellular Ca2+ and resembled that of normal myotubes in the absence of external Ca2+ or in the presence of Gd3+. Furthermore, when intracellular Ca2+ release was blocked by ryanodine, no Ca2+ transients were detectable in SkEIIIK-expressing dysgenic myotubes. Collectively, our results indicate that the skeletal muscle L-type Ca2+ current is a major contributor to the Ca2+ entry that occurs during depolarization of myotubes.
MATERIALS AND METHODS
Myotube Culture and cDNA Expression
All procedures involving mice were approved by the University of Colorado Denver Institutional Animal Care and Use Committee. Primary cultures of normal (+/+ or +/mdg), dysgenic (mdg/mdg), dyspedic (RYR1−/RYR1−), and β1-null (β1−/β1−) myotubes were prepared as described previously (Beam and Franzini-Armstrong, 1997). Cultures were grown for 6–7 d in a humidified 37°C incubator with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Mediatech), supplemented with 10% fetal bovine serum/10% horse serum (Hyclone Laboratories). This medium was then replaced with differentiation medium (DMEM supplemented with 2% horse serum). In experiments that required exogenous expression of SkEIIIK in dysgenic myotubes, single nuclei were microinjected with a solution of plasmid cDNA encoding SkEIIIK (400 ng/μl) (Dirksen and Beam, 1999) and either pEYFP-C1 or pDsRed2-C1 (10 ng/μl; both from Clontech) 2 d after the switch to differentiation medium. Fluorescent myotubes were used in experiments 2 d after microinjection.
Measurement of L-type Ca2+ Currents
Pipettes were fabricated from borosilicate glass and had resistances of ∼1.5 MΩ when filled with internal solution, which consisted of (in mM): 140 Cs-aspartate, 10 Cs2-EGTA, 5 MgCl2, and 10 HEPES, pH 7.4, with CsOH. The standard external solution contained (in mM): 145 TEA-Cl, 10 CaCl2, 0.003 tetrodotoxin, and 10 HEPES, pH 7.4, with TEA-OH. In some experiments, 2 mM CaCl2, 10 mM MnCl2, or 0.5 mM MnCl2 was substituted for 10 mM CaCl2. For generation of I-V relationships, linear capacitive and leakage currents were determined by averaging the currents elicited by 11 30-mV hyperpolarizing pulses from the holding potential of −80 mV. Test currents were corrected for linear components of leak and capacitive current by digital scaling and subtraction of this average control current. In all other experiments, −P/4 subtraction was used. Electronic compensation was used to reduce the effective series resistance (usually to <1 MΩ) and the time constant for charging the linear cell capacitance (usually to <0.5 ms). L-type currents were filtered at 2 kHz and digitized at 5–10 kHz. In most cases, a 1-s prepulse to −20 mV followed by a 50-ms repolarization to −50 mV was administered before the test pulse (prepulse protocol; see Adams et al., 1990) to inactivate T-type Ca2+ channels. Cell capacitance was determined by integration of a transient from −80 to −70 mV using Clampex 8.0 (MDS Analytical Technologies) and was used to normalize current amplitudes (pA/pF). I-V curves were fitted using the following equation:
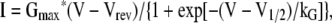 |
(1) |
where I is the peak current for the test potential V, Vrev is the reversal potential, Gmax is the maximum Ca2+ channel conductance, V1/2 is the half-maximal activation potential, and kG is the slope factor. The deactivation phase of SkEIIIK tail currents was fitted as:
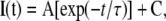 |
(2) |
where I(t) is the current at time t after the repolarization, A is the peak tail current amplitude, τ is the deactivation time constant, and C represents the steady current (see Wilkens et al., 2001). All electrophysiological experiments were performed at room temperature (∼25°C).
Ca2+ Imaging
Myotubes were washed with Ca2+/Mg2+-free Ringer's solution (in mM: 146 NaCl, 5 KCl, 10 HEPES, and 11 glucose, pH 7.4, with NaOH) twice and loaded with 5 μM Fluo-3-AM (Invitrogen) dissolved in Rodent Ringer's solution (in mM: 146 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4, with NaOH) for 20 min. Myotubes were then washed three times in Rodent Ringer's solution with gentle agitation. Fluo-3-AM–loaded myotubes bathed in either Rodent Ringer's solution or 0 Ca2+ Rodent Ringer's solution (in mM: 146 NaCl, 5 KCl, 3 MgSO4, and 10 HEPES, pH 7.4, with NaOH) were then placed on the stage of an LSM META scanning laser confocal microscope (Carl Zeiss, Inc.) and viewed under either 10 or 40× magnification. 10–100 μM N-benzyl-p-toluensulfonamide (Sigma-Aldrich) was continuously present in the bath solution (∼25°C) to prevent myotube contractions. Fluo-3 was excited with the 488-nm line of an argon laser (30-milliwatt maximum output, operated at 50% or 6.3A, attenuated to 1–2%). The emitted fluorescence was directed to a photomultiplier equipped with either a 505-nm long-pass filter or a 505–530-nm band-pass filter. Confocal fluorescence intensity data were digitized at 8 bits, with the photomultiplier gain and offset adjusted such that maximum pixel intensities were no more than ∼70% saturated and cell-free areas had close to zero intensity. Myoplasmic Ca2+ transients were elicited by application of high KCl Ringer's solution (in mM: 71 NaCl, 80 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4, with NaOH) or 0 Ca2+/high KCl Ringer's solution (in mM: 71 NaCl, 80 KCl, 3 MgSO4, and 10 HEPES, pH 7.4, with NaOH) via an extracellular pipette placed near the myotube of interest or a manually operated, gravity-driven global perfusion system that produced complete exchange within ∼5 s. Although slower, the latter method had the advantage of producing an exposure to high KCl that was more uniform for entire myotubes and allowed the sampling of responses from many myotubes simultaneously, which was especially useful for myotubes expressing the SkEIIIK construct. Fluorescence amplitude data are expressed as ΔF/F, where F represents the baseline fluorescence before the application of high KCl Ringer's solution, and ΔF represents the change in peak fluorescence during the application of high KCl Ringer's solution. In addition to peak ΔF/F, the following parameters were assessed: (1) tpeak = [time at peak ΔF] − [time at onset of the upswing of the transient], (2) t1/2 decay = time of decay from peak to 50% of peak ΔF/2, and (3) r20 = [ΔF at the end of a 20-s KCl depolarization]/[peak ΔF] × 100.
Pharmacology
Ryanodine (Sigma-Aldrich) was reconstituted in 40% EtOH and diluted to 200 μM in differentiation medium. GdCl3 (Sigma-Aldrich) or LaCl3 (Sigma-Aldrich) was dissolved in water to make 0.1-M stock solutions and was then added to standard external recording solution for a final trivalent cation concentration of 100 μM. 2-APB (Sigma-Aldrich) and SKF 96356 (provided by O. Delbono, Wake Forest University, Winston-Salem, NC) were dissolved in DMSO (Sigma-Aldrich) to make 1-M and 50-mM stock solutions, respectively. Nifedipine (EMD) was dissolved in 50% EtOH and diluted to various concentrations just before experiments. In some experiments, Ca2+ currents were recorded in the continuous presence of 5 μM of racemic Bay K 8644 (provided by A. Scriabine, Miles Laboratories Inc., New Haven, CT) in the bath solution. Racemic Bay K 8644 was stored as a 20-mM stock in 50% EtOH. Dantrolene (Sigma-Aldrich) was dissolved in dry DMSO to make a 20-mM stock solution and diluted to 10 μM just before use. Dihydropyridines, 2-APB, SKF 96356, and dantrolene were stored and used in the dark.
Analysis
Figures were made using the software program SigmaPlot (version 7.0; SSPS Inc.). All data are presented as mean ± SEM. Statistical comparisons were by ANOVA or by unpaired two-tailed t test (as appropriate), with P < 0.05 considered significant.
Online Supplemental Material
Fig. S1 shows that SKF 96356 accelerated the inactivation kinetics of the L-type current sufficiently so that block was essentially complete at test pulses slightly longer than the test pulse shown in Fig. 1 D. Fig. S2 shows the I-V relationships for dysgenic myotubes expressing SkEIIIK in the presence and absence of 5 μM ± Bay K 8644. Fig. S3 shows that the anti-malignant hyperthermia drug dantrolene partially inhibits Ca2+ entry during long exposures to elevated KCl. Figs. S1–S3 are available at http://www.jgp.org/cgi/content/full/jgp.200810105/DC1.
Figure 1.

Skeletal muscle L-type current is blocked by nonspecific cation channel antagonists. Acute block of L-type currents evoked by step depolarizations to +30 mV by 100 μM Gd3+ (A), 100 μM La3+ (B), 100 μM 2-APB (C), and 20 μM SKF 96356 (D). The test pulse was applied after a prepulse protocol (see Materials and methods) (Adams et al., 1990). Results of these experiments and application of DMSO vehicle (0.01%) control experiments are summarized in E. For trivalent cations, maximal block was generally achieved in <30 s after gravity perfusion of the 30-mm dish, whereas longer perfusion times were required for maximal block by 2-APB (1–2 min) and SKF 96356 (2–3 min). The asterisk indicates a significant difference for current (measured at the end of a 200-ms test pulse) remaining in myotubes exposed to blockers relative to DMSO-treated control cells (P < 0.0001, ANOVA). Although not indicated, block by SKF 96356 measured at the end of a 500-ms test pulse was also significantly (P < 0.00008, t test) different than DMSO controls. Throughout, the error bars represent ± SEM.
RESULTS
L-type Current Is Blocked by Nonspecific Cation Channel Blockers
ECCE can be blocked by exposure to trivalent metal ions (Gd3+ and La3+) and to the nonspecific cation channel blockers 2-APB and SKF 96356 (Cherednichenko et al., 2004; Hurne et al., 2005; Yang et al., 2007). Fig. 1 (A and B) shows that Gd3+ and La3+ (both 100 μM) potently blocked the skeletal L-type current by 96 ± 1% (n = 7) and 97 ± 1% (n = 4), respectively. Substantial block of the L-type current was also produced by exposure of normal myotubes to 100 μM 2-APB (87 ± 2%; n = 6; Fig. 1 C) and to 20 μM SKF 96356 (at the end of 200-ms test pulses: 81 ± 3%; n = 4; Fig. 1 D). Moreover, SKF 96356 accelerated the inactivation kinetics of the L-type current sufficiently so that block was essentially complete (98 ± 2% block; n = 3) at 500 ms (Fig. S1). The L-type current was not appreciably affected by exposure to DMSO vehicle (7 ± 5%; n = 3; P < 0.0001, ANOVA; Fig. 1 E).
Complete Block of Skeletal L-type Current Requires a High Concentration of Nifedipine
A small component of ECCE is blocked by the 1,4-dihydropyridine L-type Ca2+ channel antagonist, nifedipine (1 μM) (Yang et al., 2007), whereas ECCE is completely blocked by 50 μM nifedipine (see Fig. 7 C). Block of the L-type current showed a similar dependence on nifedipine dose; only partial block was produced by either 1 or 10 μM nifedipine (Fig. 2, A and B), whereas nearly complete block required 50 μM nifedipine (Fig. 2 C). The dose–response curve for nifedipine is shown in Fig. 2 D.
Figure 7.

SkEIIIK supports minimal Ca2+ entry during long global depolarizations. Representative Ca2+ transients evoked by global perfusion of 80 mM KCl Ringer's solution for normal myotubes (A), normal myotubes exposed to 100 μM Gd3+ (B), normal myotubes exposed to 50 μM nifedipine (C), and dysgenic myotubes expressing SkEIIIK (D). In each case, myotubes were exposed to 200 μM ryanodine for >1 h at 37°C before experiments to block the contribution of RYR1 to the Ca2+ transient. In experiments with SkEIIIK, a slower sampling rate was used to minimize any bleaching of the Fluo-3 dye. Expression of SkEIIIK was confirmed by electrically evoked (100 V, 5 ms) contractions (17 of 20 myotubes tested) before ryanodine treatment (see Papadopoulos et al., 2004).
Figure 2.

Block of skeletal L-type current by nifedipine. (A) Incomplete block of L-type current by 1 μM nifedipine. (B) Incomplete block of L-type current by 10 μM nifedipine. (C) Complete block of L-type current by 50 μM nifedipine. Currents evoked by step depolarizations to +30 mV from −50 mV after a prepulse protocol (see Materials and methods). (D) Dose–response relationship for the effects of nifedipine, which were measured ∼1 min after addition. Note that the minimal apparent block (at 1 nM) was ∼80%, most likely as a consequence of current rundown. Each symbol represents data collected from 2–10 cells.
Dantrolene Inhibits L-type Currents
The anti-malignant hyperthermia drug dantrolene at 10 μM blocks ∼65% of ECCE as measured by Mn2+ quench (Cherednichenko et al., 2008). We found that 10 μM dantrolene also inhibited the L-type current in myotubes (Fig. 3 and Table I). At +30 mV (near the peak of the I-V relationship), dantrolene inhibited L-type current by ∼19% compared with untreated control myotubes (−9.7 ± 0.6 pA/pF; n = 12 vs. −11.9 ± 0.5 pA/pF; n = 10, respectively; P < 0.05). Dantrolene also caused an ∼5-mV depolarizing shift in the I-V relationship (Fig. 3 C). Collectively, the data presented in Figs. 1–3 indicate that the skeletal L-type Ca2+ current and ECCE have very similar pharmacological profiles.
Figure 3.

Dantrolene inhibits skeletal L-type Ca2+ currents. Recordings of L-type currents elicited by 200-ms depolarizations from −50 mV to the indicated test potentials are shown for an untreated normal myotube (A) or a normal myotube exposed to 10 μM dantrolene for >10 min at ∼25°C (B). (C) Comparison of I-V relationships for untreated (•; n = 10) and dantrolene-treated myotubes (○; n = 12). Currents were evoked at 0.1 Hz in 10-mV increments after a prepulse protocol (see Materials and methods). Current amplitudes were normalized by linear cell capacitance (pA/pF). The smooth curves are plotted according to Eq. 1, with best-fit parameters presented in Table I.
TABLE I.
Conductance Fits
| G-V | |||
|---|---|---|---|
|
Gmax (nS/nF) |
V1/2 (mV) |
kG (mV) |
|
| normal (10 mM Ca2+ external) | 244 ± 12 (10) |
18.1 ± 1.4 | 4.4 ± 0.2 |
| normal (2 mM Ca2+ external) | 115 ± 16a (4) |
7.8 ± 2.4b | 4.7 ± 0.5 |
| normal (10 mM Ca2+ external) + 10 μM dantrolene |
228 ± 10 (12) |
22.4 ± 0.8b | 5.2 ± 0.2b |
| normal (10 mM Mn2+ external) | 135 ± 6b (4) |
16.7 ± 1.9 | 5.6 ± 0.2b |
Data are given as mean ± SEM, with the numbers in parentheses indicating the number of myotubes tested. Data were fit according to Eq. 1 (see Materials and Methods). Asterisks indicate significant differences compared to L-type currents recorded from normal, untreated myotubes in 10 mM external Ca2+
P < 10−4, t test.
P < 0.01, t test.
L-type Channels Inactivate Slowly and Conduct Substantial Current at Weak Test Potentials in Physiological Ca2+
The pharmacological data described above were obtained using 200-ms test pulses and 10 mM Ca2+ in the external recording solution to allow comparison with previous data from our laboratory. Under these conditions, no L-type current was observable at test potentials below +10 mV. In contrast, ECCE can be strongly evoked by ∼80 mM KCl (Hurne et al., 2005), which results in depolarizations to between −10 and 0 mV (Allen, P.D., and J.R. López, personal communication). To determine whether L-type current can be observed in the same range of potentials as ECCE, we reduced the external Ca2+ to the more physiological 2 mM used for the ECCE measurements (Cherednichenko et al., 2004, 2008; Hurne et al., 2005; Yang et al., 2007; Gach et al., 2008; Lyfenko and Dirksen, 2008). This maneuver reduced L-type current amplitude and caused an ∼11-mV hyperpolarizing shift in the peak I-V relationship measured with 200-ms test pulses (P = 0.0011; Fig. 4 A and Table I). However, the currents were much larger with ∼10-s depolarizations like those used to elicit ECCE (Cherednichenko et al., 2004; Hurne et al., 2005) because activation was so slow (Fig. 4 B). In particular, L-type currents evoked at −10 mV in 2 mM Ca2+ had a time-to-peak of 4.02 ± 1.02 s (n = 6; Table II). Furthermore, the current inactivated very little under these conditions (36% in 9 s; Table II). Collectively, the data in Fig. 4 indicate that long, weak depolarizations elicit substantial, sustained L-type current in the presence of physiological Ca2+.
Figure 4.

In 2 mM of external Ca2+, L-type currents are activated by weak depolarizations and inactivate very slowly. (A) Comparison of I-V relationships for normal myotubes in 10 mM of external Ca2+ (•; n = 10) or 2 mM of external Ca2+ (○; n = 4) elicited at 0.1 Hz by test potentials ranging from −20 through +80 mV in 10-mV increments after a prepulse protocol. The smooth curves are plotted according to Eq. 1. The best-fit parameters for each plot are presented in Table I. (B) Recordings of L-type currents in 2 mM of external Ca2+ elicited by 9,800-ms depolarizations from −80 mV to the indicated test potentials.
TABLE II.
Activation and Decay of L-type Current during Extended Depolarizations
| Ipeak (pA/pF) |
tpeak (s) |
t1/2 decay (s) |
r9 (%) |
|
|---|---|---|---|---|
| normal (−10 mV) | −0.7 ± 0.2 (5) |
4.02 ± 1.02 (6) |
ND | 0.64 ± 0.10 (6) |
| normal (0 mV) | −2.3 ± 0.6 (5) |
2.54 ± 1.14 (6) |
2.65 ± 0.34 (6) |
0.52 ± 0.10 (6) |
Data presented here were obtained from 9.8-s test potentials from −80 to −10 and 0 mV with 2 mM Ca2+ in the external recording solution. r9 is ratio of the current remaining at 9 s to the peak current. t1/2 decay at −10 mV was not determined (ND) because the test potential was too short to accurately assess this parameter in three of six cells. Data are given as mean ± SEM, with the numbers in parentheses indicating the number of myotubes tested.
Skeletal Muscle L-type Channels Pass Mn2+
In addition to producing Ca2+ entry measurable as a component of myoplasmic Ca2+ transients (compare with Yang et al., 2007), ECCE can be assayed by quench of Fura-2 fluorescence resulting from Mn2+ influx in the absence of extracellular Ca2+ (Cherednichenko et al., 2004, 2008; Hurne et al., 2005; Gach et al., 2008; Lyfenko and Dirksen, 2008). Fig. 5 A shows that the L-type channel also conducts Mn2+ ions in the absence of external Ca2+ (peak current of −4.1 ± 0.3 pA/pF at +30 mV; n = 4). Under standard recording conditions (10 mM external Ca2+), large L-type Ca2+ currents were observed at depolarizations to +30 mV (Fig. 5 B, bottom trace). A substantial current remained upon the replacement of 10 mM Ca2+ by 10 mM Mn2+ (Fig. 5 B, middle trace). This Mn2+ current was blocked nearly completely by 10 μM nifedipine (95 ± 2% channel block; n = 4), confirming that the Mn2+ entry was indeed via L-type channels (Fig. 5 B, top trace).
Figure 5.

The skeletal muscle L-type channel conducts Mn2+. (A) I-V relationship for normal myotubes in 10 mM of external Mn2+ (○; n = 4) for currents elicited at 0.1 Hz by test potentials ranging from −20 through +80 mV in 10-mV increments after a prepulse protocol. The smooth curves are plotted according to Eq. 1. The best-fit parameters for each plot are presented in Table I. (B) Representative currents recorded at +30 mV from a normal myotube in 10 mM Ca2+ (bottom trace), in 10 mM Mn2+ (middle trace), and in 10 mM Mn2+ after acute application of 10 μM nifedipine (top trace). (C) L-type current evoked by steps from −50 to 0 mV (top) and to +30 mV (bottom) in 0.5 mM Mn2+. (D) Average peak currents in 10 mM of divalent cation at a test potential of +30 mV (left) and for 0.5 mM Mn2+ at the indicated test potentials (right).
To approximate the conditions that have been previously used to measure ECCE via Mn2+ quench, we also recorded L-type currents in the presence of 0.5 mM Mn2+ (Fig. 5 C). Under these conditions, the average current amplitude was reduced (Fig. 5 D), and there was a substantial hyperpolarizing shift in the activation of the Mn2+ current (i.e., peaking at ∼0 mV; Fig. 5, C and D).
Ca2+ Influx via the L-type Channel Accounts for the Majority of Ca2+ Influx in Response to Extended Depolarization
Cherednichenko et al. (2004) found that ECCE could be detected in dysgenic myotubes after the expression of a DHPR α1S subunit in which a conserved glutamate in the pore-forming region of repeat III was mutated to lysine (SkEIIIK) (Dirksen and Beam, 1999). Because SkEIIIK produces no obvious inward Ca2+ current during 200-ms depolarizations or subsequent repolarization (Dirksen and Beam, 1999) (Fig. S2), it was concluded that ECCE was independent of cation flux via the L-type channel. To assess a potential contribution of the L-type current to ECCE, we compared myoplasmic Ca2+ transients that were elicited locally by 20-s exposures to elevated (80 mM) KCl in both untransfected normal myotubes and dysgenic myotubes expressing SkEIIIK. Ca2+ transients of normal myotubes elicited by locally applied 80 mM KCl (Fig. 6 A) rose to a peak and decayed slowly (t1/2 decay = 10.64 ± 0.98 s; n = 24) and incompletely (r20 = 31.92 ± 5.03%). As shown in Fig. 6 B, transients of normal myotubes obtained in nominally 0 mM Ca2+ decayed much faster (t1/2 decay = 4.98 ± 0.45 s; n = 12; P < 0.0005, t test) and more completely (r20 = 4.36 ± 2.38%; P < 0.001, t test) than transients measured in 2 mM Ca2+ (Fig. 6, B–D, superimposed gray trace). An accelerated rate of decay was also observed in experiments in which 100 μM Gd3+ was added to the 80 KCl Ringer's solution in the micropipette (t1/2 decay = 3.49 ± 0.47 s; r20 = 2.23 ± 0.74%; n = 14; Table III), supporting the idea that the acceleration of decay in 0 mM Ca2+ resulted from a loss of Ca2+ entry rather than from some other effect of removing Ca2+ from the bath.
Figure 6.

Ca2+ influx via the L-type channel accounts for the bulk of Ca2+ influx in response to depolarization. Representative Ca2+ transients are shown for a normal myotube in 2 mM of bath Ca2+ (A), a normal myotube in nominal 0 mM of bath Ca2+ (B), a dysgenic myotube expressing SkEIIIK in 2 mM Ca2+ (C), and a dysgenic myotube expressing SkEIIIK in 0 mM Ca2+ (D). Summary of t1/2 decay (E) and r20 (F) values for normal myotubes and dysgenic myotubes expressing SkEIIIK in the presence (solid bars) and absence (hatched bars) of 2 mM Ca2+. In B–D, a scaled version of the transient shown in A has been included for comparison of the rates of transient decay (gray traces). Transients were elicited locally by 20-s applications of 80 mM KCl via micropipette placed close to the myotube of interest. See Materials and methods for definitions of t1/2 decay and r20. Asterisks indicate a significant difference (P < 0.0005, ANOVA).
TABLE III.
Activation and Decay of Ca2+ Transients during Local Depolarizations
| ΔF/F (ΔF/F) |
tpeak (s) |
t1/2 decay (s) |
r20 (%) |
||||
|---|---|---|---|---|---|---|---|
| normal (2 mM Ca2+) | 3.25 ± 0.35 (24) |
3.76 ± 0.34 | 10.64 ± 0.98 | 31.92 ± 5.03 | |||
| normal (0 mM Ca2+) | 1.31 ± 0.15a (12) |
1.97 ± 0.22b | 4.98 ± 0.45a | 4.36 ± 2.38a | |||
| normal + 100 μM Gd3+(2 mM Ca2+) | 1.77 ± 0.18b (14) |
1.94 ± 0.13a | 3.49 ± 0.47a | 2.23 ± 0.74a | |||
| dysgenic + SkEIIIK (2 mM Ca2+) | 0.90 ± 0.18a (9) |
3.51 ± 0.50 | 4.47 ± 0.52a | 10.41 ± 2.70c | |||
| dysgenic + SkEIIIK (0 mM Ca2+) | 0.25 ± 0.09a (5) |
2.70 ± 0.45 | 5.68 ± 1.12c | 4.97 ± 2.17c | |||
| dysgenic (2 mM Ca2+) | no response (12) | ||||||
| dyspedic (2 mM Ca2+) | no response (14) | ||||||
| β1 null (2 mM Ca2+) | no response (10) | ||||||
| dysgenic + SkEIIIK + ryanodine (2 mM Ca2+) |
no response (20) | ||||||
Data presented here were obtained with depolarizations evoked by 20-s applications of 80 mM KCl Ringer's solution via a micropipette placed near the cell of interest. Where indicated, 100 μM Gd3+ was added to the 80 mM KCl Ringer's solution in the micropipette. See Materials and methods for definitions of tpeak, t1/2 decay, and r20. Data are given as mean ± SEM, with the numbers in parentheses indicating the number of myotubes tested. Footnotes indicate significant differences compared to normal myotubes in 2 mM Ca2+ by unpaired t test:
P < 0.001.
P < 0.005.
P < 0.05.
In 2 mM Ca2+, high KCl-evoked Ca2+ transients of dysgenic myotubes expressing SkEIIIK also decayed more rapidly than Ca2+ transients of normal myotubes (Fig. 6 C), having decay kinetics (t1/2 decay = 4.47 ± 0.52 s; r20 = 10.41 ± 2.70 s; n = 9; Fig. 6, E and F) more similar to those of normal myotubes in nominally 0 mM Ca2+ than in 2 mM Ca2+ (Table III). Furthermore, the decay kinetics of Ca2+ transients in myotubes expressing SkEIIIK were little affected by the removal of extracellular Ca2+ (t1/2 decay = 5.68 ± 1.16 s; r20 = 4.97± 2.17%; n = 5; Fig. 6, D–F, and Table III). The fact that Ca2+ removal accelerated the decay of Ca2+ transients in normal myotubes but not in dysgenic myotubes expressing SkEIIIK is consistent with the idea that Ca2+ entry slows the decay of transients in normal myotubes bathed in 2 mM Ca2+, and that this entry does not occur for SkEIIIK.
In control experiments, untransfected dysgenic and dyspedic myotubes failed to respond to locally applied 80 mM KCl (n = 12 and 14, respectively; Table III). Myotubes harvested from mice null for the DHPR β1 subunit also failed to respond to locally applied 80 mM KCl Ringer's solution (n = 10; Table III).
SkEIIIK Supports Minimal Ca2+ Entry during Long, Global Depolarizations
To assess changes in myoplasmic Ca2+ attributable solely to Ca2+ influx, we applied 80 mM KCl globally to myotubes after block of intracellular Ca2+ release by ryanodine pretreatment (200 μM; > 1 h; 37°C). Under these experimental conditions, 40 of 42 normal myotubes showed a large, slowly activating Ca2+ entry from the extracellular space in response to global application of 80 mM KCl (ΔF/F = 2.11 ± 0.16, n = 40; Fig. 7 A). The presence of either 100 μM Gd3+ or 50 μM nifedipine, which both block the L-type current (Figs. 1 and 2), completely eliminated this slow Ca2+ entry (n = 6 and n = 10, respectively; Fig. 7, B and C). In contrast to the robust Ca2+ transients observed in ryanodine-treated normal myotubes, no Ca2+ transients were detectable (ΔF/F < 0.05) in ryanodine-treated dysgenic myotubes expressing SkEIIIK (n = 31; Fig. 7 D). Collectively, these results support the idea that L-type Ca2+ current is a major contributor to Ca2+ entry in response to prolonged, weak depolarization.
The SkEIIIK Pore Mutant Allows Inward Divalent Current
Previously, it was reported that in dysgenic myotubes expressing SkEIIIK, entry of divalent cations in response to depolarization could be measured with the indicator Fura-2 (Cherednichenko et al., 2004), whereas we were unable to detect such entry with the indicator Fluo-3 (Figs. 6 and 7). Thus, we sought to determine if SkEIIIK could permit a small influx of divalent cations that might have accounted for the signal measured with Fura-2 by Cherednichenko et al. (2004). In this regard, it is important to note that L-type Ca2+ channels display at least two distinct open states of either brief or long duration (i.e., Mode 1 and Mode 2 gating, respectively) (Hess et al., 1984; Pietrobon and Hess, 1990; Dirksen and Beam, 1996). Compared with the short-lived open state, the long-duration open state is relatively unpopulated during 200-ms depolarizations to potentials below about +50 mV (Dirksen and Beam, 1996). However, entry into the long-duration open state does occur during longer depolarizations (e.g., as during extended exposure to elevated KCl) and is accelerated by stronger depolarizations as well as by exposure to dihydropyridine agonists (Hess et al., 1984; Pietrobon and Hess, 1990; Dirksen and Beam, 1996). Importantly, the long duration open state of the wild-type channel preferentially allows inward divalent current (Leuranguer et al., 2003). Thus, we tested whether strong depolarization in the presence of ±Bay K 8644 would cause SkEIIIK to enter a long-lived open state that would produce inward Ca2+ tail current upon repolarization.
Fig. 8 A compares currents for depolarizations to either 0 or +80 mV recorded from a dysgenic myotube expressing SkEIIIK and bathed in 5 μM ±Bay K 8644. The depolarization to 0 mV, which is near threshold for activation of ionic current, evoked little outward current and resulted in only a small inward current upon the subsequent repolarization to −50 mV. However, the depolarization to 80 mV evoked a robust, slowly activating outward current (see peak I-V in Fig. S2) and resulted in an appreciable, slowly decaying inward current upon repolarization to −50 mV. As shown in Fig. 8 B, this slowly decaying inward current did not appear to be a consequence of the potentiation by ±Bay K 8644 of the endogenous L-type current that can be present at low levels in dysgenic myotubes (n = 14; Idys) (Adams and Beam, 1989).
Figure 8.

Potentiated SkEIIIK conducts inward Ca2+ current. Representative currents are shown for a ±Bay K 8644–treated (5 μM) dysgenic myotube expressing SkEIIIK (A) and a ±Bay K 8644–treated, uninjected dysgenic myotube (B). The inset illustrates the tail currents shown in A on an expanded time base. (C) Summary of amplitudes (left) and deactivation time constants (right) of SkEIIIK tail currents recorded in the absence (n = 9) and presence (n = 10) of ±Bay K 8644. (D) Total charge moved obtained by integration of tail currents recorded in the absence (n = 9) and presence (n = 10) of ±Bay K 8644 from dysgenic myotubes expressing SkEIIIK. Asterisks indicate significant differences between untreated and ±Bay K 8644–treated cells (*, P < 0.05; **, P < 0.01, t test).
In dysgenic cells expressing SkEIIIK, the presence of ±Bay K 8644 caused the inward current elicited by repolarization from +80 to −50 mV to be both larger and more slowly decaying than in the absence of the DHP agonist (Fig. 8 C), consistent with the idea that the inward current represents Ca2+ influx through channels relaxing from the long open state to closed. The total charge transported during the inward current elicited by repolarization to −50 mV was obtained by integration and is plotted in Fig. 8 D. The total charge for repolarization from 0 to −50 mV (3.6 ± 0.7 nC/μF; n = 9) was similar to that previously measured (see Fig. 4 of Dirksen and Beam, 1999). This charge was likely to have been nonionic and to have arisen from closed–closed gating transitions of the DHPR because a 200-ms depolarization to 0 mV activates little ionic conductance via SkEIIIK (Dirksen and Beam, 1999). Compared with 0 mV, the total charge was substantially larger for repolarization from +80 to −50 mV (10.5 ± 1.1 nC/μF; n = 9). A fraction of this additional charge movement (∼66% of the total 300% increase) was also likely to have been nonionic and to have arisen from gating transitions that would have occurred for repolarization to −50 mV after a 200-ms depolarization to +80 mV, but not after a 200-ms depolarization to 0 mV. Possibly, the remaining portion (∼33% of the 300% increase) of additional charge movement was a consequence of inward Ca2+ current that resulted from the entry of a small fraction of the SkEIIIK channels into the long-duration open state that supports such current. In the presence of ±Bay K 8644 (Fig. 8 D, gray bars), the total charge after repolarization from +80 to −50 mV was so large (33.5 ± 8.0 nC/μF; n = 10) compared with that after repolarization from 0 to −50 mV (4.4 ± 1.0 nC/μF; n = 10) that it clearly indicated the presence of inward Ca2+ current via the mutant L-type channel. Thus, these results strongly support the hypothesis that SkEIIIK can enter into a long-duration open state that permits inward divalent cation flux. However, the similarity of Ca2+ transient decay in SkEIIIK-expressing dysgenic myotubes in 2 mM Ca2+ and normal myotubes in 0 mM Ca2+ (Fig. 6 and Table III) and the lack of detectable SkEIIIK-mediated transients in the presence of ryanodine (Fig. 7 D) argues that such a mode of Ca2+ permeation via SkEIIIK must be quite small compared with Ca2+ entry via the native L-type channel.
DISCUSSION
Here, we demonstrate that the skeletal muscle L-type Ca2+ current and ECCE share many common attributes (summarized in Table IV). Thus, the skeletal muscle L-type channel is blocked by nonspecific cation channel antagonists (Fig. 1), requires nearly 50 μM nifedpine for complete channel block (Fig. 2), and is inhibited by dantrolene (Fig. 3). We also show that in 2 mM of external Ca2+, the L-type Ca2+ current, like ECCE, is persistently activated by weak voltage steps (Fig. 4). In addition, we demonstrate that the L-type channel is permeable to Mn2+ in the absence of bath Ca2+ (Fig. 5). During prolonged (20-s) KCl depolarization, Ca2+ transients in intact, normal myotubes rise to a peak and then decay, and this decay is more rapid after the removal of external Ca2+ or in the presence of Gd3+; the decay of transients in dysgenic myotubes expressing the DHPR α1S subunit pore mutant (SkEIIIK), which has a low permeability to Ca2+, is similar to that of transients in normal myotubes in a Ca2+-free external medium and is little affected by the removal of extracellular Ca2+ (Fig. 6 and Table III). Collectively, these results suggest that Ca2+ entry via the L-type channel slows the decay of Ca2+ transients during prolonged depolarization of normal myotubes. After block of SR Ca2+ release by pretreatment with 200 μM ryanodine, depolarization of normal myotubes still resulted in large myoplasmic Ca2+ transients, whereas depolarization did not elicit a detectable transient in dysgenic myotubes expressing SkEIIIK (Fig. 7), providing strong support to the idea that the L-type channel is the predominant source of Ca2+ entry. Although SkEIIIK has very low permeability to Ca2+, we did find that after strong depolarization in the presence of ±Bay K 8644, the channel could conduct inward Ca2+ tail currents upon repolarization (Fig. 8).
TABLE IV.
Common attributes of ECCE and the skeletal muscle L-type current
| ECCE | IL | |
|---|---|---|
| Is voltage-dependent | yesab | yescde |
| Conducts Ca2+ | yesabcf | yescde |
| Requires α1S subunit | yesabe | yescd |
| Requires β1a subunit | yese | increased expressionghij |
| Reduced by siRNA against α2δ1 subunit | yesk | faster inactivationkl |
| Requires RYR1 | yesabef | greatly potentiatedmn |
| Blocked by 100 μM La3+ | substantialo | completee |
| Blocked by 100 μM Gd3+ | completeape | completee |
| Blocked by 20 μM SKF 96356 | completeabo | completee |
| Blocked by 100 μM 2-APB | ∼80%ab | ∼80%e |
| Blocked by 1 μM nifedipine | incompleteo | incompletee |
| Blocked by 50 μM nifedipine | completee | completee |
| Reduced by ≥10 μM dantrolene | partialeq | modester |
| Conducts Mn2+ | yesabfkq | yese |
| Divalent entry via SkEIIIK | yesa | yese |
| Mediated by Orai1/STIM1 | nof | nocd |
References:
This paper.
Gregg et al., 1995.
Cherednichenko et al., 2004, no concentration reported.
One very clear conclusion from our results is that any of the manipulations that have been used to evaluate the magnitude of ECCE will necessarily produce changes in current via the L-type Ca2+ channel (Table IV) and thus the contribution of the L-type current to the myoplasmic Ca2+ transient. For example, the addition of Gd3+, 20 μM SKF 96356, and 100 μM 2-APB and the removal of external Ca2+ block 80–100% of ECCE (Cherednichenko et al., 2004; Hurne et al., 2005), and we find they also block L-type Ca2+ current to a similar extent. Conversely, the relative insensitivity of ECCE to 1 μM nifedipine has been taken as an indication that it is independent of L-type current (Yang et al., 2007). In particular, Yang et al. (2007) reported that 1 μM nifedipine causes only a 5% decrease in the decay of the sustained phase of the KCl-evoked transient for wild-type RYR1 (Fig. 6 B of Yang et al., 2007), whereas 100 μM La3+ causes a 14% decrease (Fig. 4 B of Yang et al., 2007). If it is assumed that the altered decay rate for 100 μM La3+ reflected a complete block of ECCE, then 1 μM nifedipine caused a 36% block, close to the ∼50% block of L-type current that we found for 1 μM nifedipine (Fig. 2 D).
We found that maximal L-type Ca2+ currents in myotubes were reduced ∼15% by 10 μM dantrolene (Fig. 3), a similar finding to that of Szentesi et al. (2001), who observed that L-type currents are reduced ∼25% in adult rodent fibers by 25 μM dantrolene. These electrophysiological results are in good agreement with the observation that Ca2+ transients evoked by 40 mM KCl were reduced ∼20% in 10 μM of dantrolene-pretreated myotubes compared with matched control myotubes (Fig. S3). However, an earlier report indicated that 10 μM dantrolene caused a more substantial (∼65%) reduction in quench of Fura-2 fluorescence by Mn2+ entry in response to 40 mM KCl or electrical stimulation (Cherednichenko et al., 2008). In evaluating the difference in dantrolene sensitivity found in the two sets of experiments, it should be noted that the response of the fluorescent indicator (Fluo-3 to Ca2+, Fura-2 to Mn2+) depends not only on the magnitude of the entry flux, but also on the divalent affinity of the indicator and on the buffering and removal of the divalent cations that have entered the myoplasm. Consequently, the change in fluorescence might well not depend linearly on the entry flux, making it difficult to directly compare these measurements.
The dependence of ECCE on the presence of both the DHPR α1S subunit and RYR1 (Cherednichenko et al., 2004; Hurne et al., 2005; Lyfenko and Dirksen, 2008) is also a feature of the slow L-type Ca2+ current, which is totally absent in dysgenic muscle cells lacking α1S (Beam et al., 1986; Tanabe et al., 1988), and greatly reduced in dyspedic muscle cells lacking RYR1 (Nakai et al., 1996; Avila and Dirksen, 2000). This distinguishes ECCE and the slow L-type current from SOCE, which functions normally in dysgenic and dyspedic myotubes (Cherednichenko et al., 2004; Hurne et al., 2005; but see Lyfenko and Dirksen, 2008). Another parallel between ECCE and the slow L-type Ca2+ current is that myotubes null for the DHPR β1a subunit lack ECCE (Table III) and have greatly reduced expression of L-type current (Gregg et al., 1996; Strube et al., 1996; Papadopoulos et al., 2004; Sheridan et al., 2004). Interestingly, knockdown of the DHPR α2δ1 subunit in myotubes by siRNA causes a reduction in ECCE (Gach et al., 2008) and an accelerated inactivation of the L-type Ca2+ current measured with 200-ms depolarizations (Obermair et al., 2005; Gach et al., 2008; Obermair et al., 2008). During prolonged depolarizations, the accelerated inactivation might greatly reduce Ca2+ entry via the L-type channel.
Previously, Hurne et al. (2005) attempted unsuccessfully to measure a current associated with ECCE by the application of 6-s test pulses to −20 mV. In retrospect, this result is consistent with the hypothesis that the L-type Ca2+ current is a major contributor to the ECCE signal because depolarizations to −20 mV would not have activated L-type current in 10 mM of extracellular Ca2+ (Fig. 4 A), as used in the measurements of Hurne et al. (2005). In 2 mM Ca2+, however, the leftward shift of activation has the result that long (∼10-s) depolarizations to −10 or 0 mV evoke sustained L-type currents (Fig. 4 B) that would support substantial Ca2+ entry under the conditions used for measuring ECCE. In addition, relatively weak depolarizations activate currents carried by Mn2+ in the absence of external Ca2+ (Fig. 5), which means that the L-type channel would produce Mn2+ entry under the conditions used to measure Mn2+ quench attributed to ECCE (Table IV).
During prolonged depolarization of normal myotubes, the entry of Ca2+ appears to slow the decay of the myoplasmic Ca2+ transient inasmuch as removal of external Ca2+ causes the decay to become more rapid (Fig. 6). This same manipulation does not greatly affect the time course of transients in dysgenic myotubes expressing SkEIIIK, which decay much like those of normal myotubes in a Ca2+-free medium. In principle, removing extracellular Ca2+ could not only have eliminated Ca2+ entry, but also increased inactivation of the voltage sensor for EC coupling (Brum et al., 1988a,1988b). Indeed, the amplitude of the Ca2+ transients in Ca2+-free medium was smaller for both normal myotubes and dysgenic myotubes expressing SkEIIIK (Table III), consistent with the possibility that removal of Ca2+ increased steady-state inactivation of voltage sensors in resting myotubes. Cherednichenko et al. (2004) also observed a similar reduction of the KCl-evoked Ca2+ transient in a Ca2+-free medium. However, altered inactivation during the KCl application does not seem to have been a major factor in causing more rapid decay because: (1) an acceleration of decay like that seen with Ca2+ removal was caused by the administration of Gd3+ concurrently with the high KCl, and (2) removal of external Ca2+ had little effect on the time course of Ca2+ transients for SkEIIIK.
Our measurements of Ca2+ transients in intact myotubes strongly support the idea that the L channel is a major contributor to ECCE. These transients were characterized both in myotubes where intracellular release occurred via RYR1 (Fig. 6) and in myotubes in which this release had been blocked by prior exposure to ryanodine (Fig. 7). Under the latter situation, depolarization of normal myotubes, but not dysgenic myotubes expressing SkEIIIK, elicits large Ca2+ entry transients. Thus, any Ca2+ entry via a pathway other than the L-type channel must be too small to be detected by our experiments (myotubes loaded with Fluo-3-AM). This result raises the obvious question of why previous work found expression of SkEIIIK in dysgenic myotubes restored ECCE as indicated by the entry of either Mn2+ or Ca2+ (Cherednichenko et al., 2004; their Fig. 4 and not depicted, respectively). One possibility is that SkEIIIK has sufficient permeability to divalent cations to produce a signal detectable by Fura-2 as used by Cherednichenko et al. (2004), but not by Fluo-3 as used in our current experiments. Indeed, our electrophysiological data (Fig. 8) demonstrate that after a brief (200-ms), strong (+80 mV) depolarization in the presence of the DHP agonist ±Bay K 8644, repolarization of cells expressing SkEIIIK results in an inward tail current carried by Ca2+ (the only external cation other than TEA). This inward tail current seems likely to be a consequence of the entry of SkEIIIK into a long-duration open state, which has been shown to be promoted in L-type channels by both strong depolarization and DHP agonists (see, for example, Wilkens et al., 2001). Entry into this long-duration open state also occurs for much weaker depolarizations in the absence of DHP agonist as long as these depolarizations are sustained (Pietrobon and Hess, 1990). The long-duration open state of both the wild-type cardiac L-type channel and the pore mutant “CEIIIK” appears to be rectifying and to permit the flow of inward divalent current, but not that of outward current (Leuranguer et al., 2003). Thus, it seems a reasonable possibility that SkEIIIK could support a very small inward divalent current during long depolarizations and that this small current might account for the detection of Mn2+ entry by means of Fura-2 in myotubes expressing SkEIIIK (Cherednichenko et al., 2004).
In skeletal muscle, the DHPR has dual functions as an L-type Ca2+ channel and as the voltage sensor for EC coupling. The physiological role of the L-type Ca2+ current has remained obscure because the voltage sensor function of the DHPR does not require the entry of external Ca2+ (Fig. 6) (Armstrong et al., 1972; Tanabe et al., 1990; Dirksen and Beam, 1999). However, the results described here support the hypothesis that the L-type Ca2+ current mediated by the DHPR is the major (and perhaps sole) component of ECCE. Previously, the Ca2+ entry ascribed to ECCE was shown to help maintain myoplasmic Ca2+ levels during either sustained depolarizations or during trains of repetitive brief stimuli (Cherednichenko et al., 2008; Gach et al., 2008). It now appears that this role can be reassigned to the L-type Ca2+ current and perhaps represents one of its physiological functions in skeletal muscle.
Supplementary Material
Acknowledgments
We are grateful to Dr. O. Delbono (Wake Forest University) for the gift of SKF 96356. We thank Drs. N.M. Lorenzon, A.M. Payne, and D.C. Sheridan, and Mr. J.D. Ohrtman for insightful discussion.
This work was supported in part by grants from the National Institutes of Health (AR44750 to K.G. Beam and AR43140 to I.N. Pessah) and from the Muscular Dystrophy Association (MDA4155 to R.A. Bannister and MDA4319 to K.G. Beam).
Edward N. Pugh Jr. served as editor.
Abbreviations used in this paper: 2-APB, 2-aminoethyl diphenylborate; DHPR, 1,4-dihydropyridine receptor; EC, excitation–contraction; ECCE, excitation-coupled Ca2+ entry; SOCE, store-operated Ca2+ entry.
References
- Adams, B.A., and K.G. Beam. 1989. A novel calcium current in dysgenic skeletal muscle. J. Gen. Physiol. 94:429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams, B.A., T. Tanabe, A. Mikami, S. Numa, and K.G. Beam. 1990. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 346:569–572. [DOI] [PubMed] [Google Scholar]
- Armstrong, C.M., F.M. Bezanilla, and P. Horowicz. 1972. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N′-tetraacetic acid. Biochim. Biophys. Acta. 267:605–608. [DOI] [PubMed] [Google Scholar]
- Avila, G., and R.T. Dirksen. 2000. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca2+ channel. J. Gen. Physiol. 115:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beam, K.G., and C. Franzini-Armstrong. 1997. Functional and structural approaches to the study of excitation-contraction coupling. Methods Cell Biol. 52:283–306. [DOI] [PubMed] [Google Scholar]
- Beam, K.G., and P. Horowicz. 2004. Excitation-contraction coupling in skeletal muscle. In Myology. A.G. Engel and C. Franzini-Armstrong, editors. McGraw-Hill, New York. 257–280.
- Beam, K.G., C.M. Knudson, and J.A. Powell. 1986. A lethal mutation in mice eliminates the slow calcium current in skeletal muscle cells. Nature. 320:168–170. [DOI] [PubMed] [Google Scholar]
- Brum, G., R. Fitts, G. Pizarro, and E. Ríos. 1988. a. Voltage sensors of the frog skeletal muscle membrane require calcium to function in excitation-contraction coupling. J. Physiol. 398:475–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum, G., E. Ríos, and E. Stefani. 1988. b. Effects of extracellular calcium on calcium movements of excitation-contraction coupling in frog skeletal muscle fibers. J. Physiol. 398:441–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherednichenko, G., A.M. Hurne, J.D. Fessenden, E.H. Lee, P.D. Allen, K.G. Beam, and I.N. Pessah. 2004. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. USA. 101:15793–15798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherednichenko, G., C.W. Ward, W. Feng, E. Carabales, L. Michaelson, M. Samsó, J.R. López, P.D. Allen, and I.N. Pessah. 2008. Enhanced excitation-coupled calcium entry in myotubes expressing malignant hyperthermia mutation R163C is attenuated by dantrolene. Mol. Pharmacol. 73:1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen, R.T., and K.G. Beam. 1996. Unitary behavior of skeletal, cardiac, and chimeric L-type Ca2+ channels expressed in dysgenic myotubes. J. Gen. Physiol. 107:731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen, R.T., and K.G. Beam. 1999. Role of calcium permeation in dihydropyridine receptor function. Insights into channel gating and excitation–contraction coupling. J. Gen. Physiol. 114:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gach, M., G. Cherednichenko, C. Haarmann, J.R. Lopez, K.G. Beam, I.N. Pessah, C. Franzini- Armstrong, and P.D. Allen. 2008. Alpha2delta1 dihydropyridine receptor subunit is a critical element for excitation-coupled calcium entry but not for formation of tetrads in skeletal myotubes. Biophys. J. 94:3023–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, R.G., A. Messing, C. Strube, M. Beurg, R. Moss, M. Behan, M. Sukhareva, S. Haynes, J.A. Powell, R. Coronado, and P.A. Powers. 1996. Absence of the β1a subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 93:13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, P., J.B. Lansman, and R.W. Tsien. 1984. Different modes of Ca channel gating behaviour favoured by dihydropyridine agonists and antagonists. Nature. 311:538–544. [DOI] [PubMed] [Google Scholar]
- Hurne, A.M., J.J. O'Brien, D. Wingrove, G. Cherednichenko, P.D. Allen, K.G. Beam, and I.N. Pessah. 2005. Ryanodine receptor type I (RyR1) mutations C4958S and C4961S reveal excitation-coupled calcium entry (ECCE) is independent of sarcoplasmic reticulum store depletion. J. Biol. Chem. 280:36994–37004. [DOI] [PubMed] [Google Scholar]
- Lee, E.H., G. Cherednichenko, I.N. Pessah, and P.D. Allen. 2006. Functional coupling between TRPC3 and RyR1 regulates the expressions of key triadic proteins. J. Biol. Chem. 281:10042–10048. [DOI] [PubMed] [Google Scholar]
- Leuranguer, V., R.T. Dirksen, and K.G. Beam. 2003. Potentiated L-type Ca2+ channels rectify. J. Gen. Physiol. 121:541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyfenko, A.D., and R.T. Dirksen. 2008. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J. Physiol. 586:4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, J., R.T. Dirksen, H.T. Nguyen, I.N. Pessah, K.G. Beam, and P.D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Obermair, G.J., G. Kugler, S. Baumgartner, P. Tuluc, M. Grabner, and B.E. Flucher. 2005. The calcium channel α2δ-1 subunit determines Ca2+ current kinetics in skeletal muscle but not targeting of α1S or excitation-contraction coupling. J. Biol. Chem. 280:2229–2237. [DOI] [PubMed] [Google Scholar]
- Obermair, G.J., P. Tuluc, and B.E. Flucher. 2008. Auxiliary Ca2+ channel subunits: lessons learned from muscle. Curr. Opin. Pharmacol. 8:311–318. [DOI] [PubMed] [Google Scholar]
- Papadopoulos, S., V. Leuranguer, R.A. Bannister, and K.G. Beam. 2004. Mapping sites of potential proximity between the DHPR and RyR1 in muscle using a CFP-YFP tandem as a FRET probe. J. Biol. Chem. 279:44046–44056. [DOI] [PubMed] [Google Scholar]
- Pietrobon, D., and P. Hess. 1990. Novel mechanism of voltage-dependent gating in L-type calcium channels. Nature. 346:651–655. [DOI] [PubMed] [Google Scholar]
- Sheridan, D.C., W. Cheng, L. Carbonneau, C.A. Ahern, and R. Coronado. 2004. Involvement of a heptad repeat in the carboxyl terminus of the dihydropyridine receptor β1a subunit in the mechanism of excitation-contraction coupling in skeletal muscle. Biophys. J. 87:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strube, C., M. Beurg, P.A. Powers, R.G. Gregg, and R. Coronado. 1996. Reduced Ca2+ current, charge movement and absence of Ca2+ transients in skeletal muscle deficient in dihydorpyridine receptor β1 subunit. Biophys. J. 75:2531–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentesi, P., C. Collet, S. Sárközi, C. Szegedi, I. Jona, V. Jacquemond, L. Kovács, and L. Csernoch. 2001. Effects of dantrolene on steps of excitation-contraction coupling in mammalian skeletal muscle fibers. J. Gen. Physiol. 118:355–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe, T., K.G. Beam, J.A. Powell, and S. Numa. 1988. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 336:134–139. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., K.G. Beam, B.A. Adams, T. Niidome, and S. Numa. 1990. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 346:567–569. [DOI] [PubMed] [Google Scholar]
- Wilkens, C.M., M. Grabner, and K.G. Beam. 2001. Potentiation of the cardiac L-type Ca2+ channel (α1C) by dihydropyridine agonists and strong depolarization occur via distinct mechanisms. J. Gen. Physiol. 118:495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, J.S., D.H. Kim, P.D. Allen, and E.H. Lee. 2008. TRPC3-interacting triadic proteins in skeletal muscle. Biochem. J. 411:399–405. [DOI] [PubMed] [Google Scholar]
- Yang, T., P.D. Allen, I.N. Pessah, and J.R. López. 2007. Enhanced excitation-coupled calcium entry in myotubes is associated with expression of RyR1 malignant hyperthermia mutations. J. Biol. Chem. 282:37471–37478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.