

Abstract
The canonical sequence LSGGQ, also known as the signature sequence, defines the adenosine triphosphate (ATP)-binding cassette transporter superfamily. Crystallographic studies reveal that the signature sequence, together with the Walker A and Walker B motifs, forms the ATP-binding pocket upon dimerization of the two nucleotide-binding domains (NBDs) in a head-to-tail configuration. The importance of the signature sequence is attested by the fact that a glycine to aspartate mutation (i.e., G551D) in cystic fibrosis transmembrane conductance regulator (CFTR) results in a severe phenotype of cystic fibrosis. We previously showed that the G551D mutation completely eliminates ATP-dependent gating of the CFTR chloride channel. Here, we report that micromolar [Cd2+] can dramatically increase the activity of G551D-CFTR in the absence of ATP. This effect of Cd2+ is not seen in wild-type channels or in G551A. Pretreatment of G551D-CFTR with the cysteine modification reagent 2-aminoethyl methane thiosulfonate hydrobromide protects the channel from Cd2+ activation, suggesting an involvement of endogenous cysteine residue(s) in mediating this effect of Cd2+. The mutants G551C, L548C, and S549C, all in the signature sequence of CFTR's NBD1, show robust response to Cd2+. On the other hand, negligible effects of Cd2+ were seen with T547C, Q552C, and R553C, indicating that a specific region of the signature sequence is involved in transmitting the signal of Cd2+ binding to the gate. Collectively, these results suggest that the effect of Cd2+ is mediated by a metal bridge formation between yet to be identified cysteine residue(s) and the engineered aspartate or cysteine in the signature sequence. We propose that the signature sequence serves as a switch that transduces the signal of ligand binding to the channel gate.
INTRODUCTION
The CFTR is an ATP-gated chloride channel, whose malfunction leads to cystic fibrosis, the most common lethal genetic disease among Caucasians. Being a member of the ATP-binding cassette (ABC) transporter superfamily (Riordan et al., 1989), CFTR consists of two membrane-spanning domains (MSD1 and MSD2) and two nucleotide-binding domains (NBDs; NBD1 and NBD2). Like other members of this family, two NBDs dimerize in a head-to-tail configuration upon ATP binding, and these dimerized NBDs represent the open-channel conformation of CFTR (Vergani et al., 2005). Recently, we showed that ATP opens the channel by binding to the binding pocket formed by the Walker A sequence of NBD2 and the signature sequence of NBD1 (Zhou et al., 2006). Under normal conditions, hydrolysis of this bound ATP closes the channel presumably by breaking the dimer apart (for review see Chen and Hwang, 2008).
Although operationally CFTR can be classified as a ligand-gated channel, unlike classical ligand-gated channels, CFTR's ligand (ATP) is consumed during the gating cycle. ATP hydrolysis at NBD2 provides an input of free energy so that CFTR's gating is not a process in thermodynamic equilibrium (Chen and Hwang, 2008). However, CFTR may work as a classical ligand-gated ion channel when ATP hydrolysis is abolished (either by mutations or by using nonhydrolyzable ATP analogues). In this case, binding and unbinding of the ligand gates the CFTR channel. Recently we found that the disease-associated mutation G551D (the single–amino acid substitution of glycine to aspartate in the signature sequence of NBD1) abolishes the ATP-dependent gating of the channel (Bompadre et al., 2007). The importance of the signature sequence is attested by how drastically the G551D mutation affects the gating of the channel and by the severity of the disease phenotype associated with this mutation, but little is known about its specific role in CFTR gating. Although the signature sequence is part of the ATP-binding pocket in a dimerized configuration, the crystal structure of isolated NBD1 of CFTR shows that ATP binds to the Walker A region (Lewis et al., 2004), and it is presumed that the signature sequence of the partner NBD may contact the bound ATP molecule when the two NBDs approach each other to form a dimer (Fig. 1).
Figure 1.

Schematic presentation of the interactions of ATP with key residues in the ATP-binding pocket of an NBD dimer. The crystal structure of Escherichia coli Malk NBD dimer (Protein Data Bank code 1Q12) was used to demonstrate these interactions. The ATP molecule is represented by the stick model. Selective residues are shown and represented by thin lines, including residues of the signature sequence, the Walker A lysine, the D-loop aspartate, and the aromatic residue that strongly interacts with the adenine ring of ATP. These residues are colored by their atom types and labeled by their residue numbers as they appear in the amino acid sequence of E. coli Malk. The corresponding residues in CFTR are labeled in parentheses.
Here, we found that “soft” metal ions such as Cd2+ and Zn2+ can dramatically increase the activity of G551D-CFTR with a micromolar-apparent affinity. This effect of Cd2+ was not seen with the wild-type (WT) channels or the corresponding mutation, G1349D, at the signature sequence of NBD2. The apparent affinity for Cd2+ was further increased when glycine 551 was converted to cysteine, but the effect of Cd2+ was mostly abolished when the G551 residue was substituted by an alanine. Engineering a cysteine residue at position 548 or 549, but not at 547, 552, and 553, also creates channels that can be effectively gated by Cd2+. These data strongly support the notion that, like in ATP-dependent gating of CFTR, the signature sequence of genetically modified NBD1 also plays a key role in mediating Cd2+-dependent gating, presumably because the mutations craft a multi-dentate Cd2+-binding site. Consistent with this idea, pretreatment of the G551D-CFTR with the sulhydryl reagent 2-aminoethyl methane thiosulfonate hydrobromide (MTSEA) abolished the effect of Cd2+, suggesting that Cd2+ ions are coordinated by 551C/D and some other partner cysteine(s). Identifying the responsible cysteine residue(s) will provide molecular insight into the gating mechanism of CFTR.
MATERIALS AND METHODS
Site-directed Mutagenesis and Cell Culture
Mutations were introduced into WT-CFTR, as described previously (Bompadre et al., 2007), using a QuickChange XL kit (Agilent Technologies) according to the manufacturer's instructions. All mutations were confirmed by sequencing (DNA core, University of Missouri-Columbia). The cDNA of WT or mutant CFTR was cotransfected with pEGFP-C3 (Clontech Laboratories, Inc.) to Chinese hamster ovary cells using Superfect transfection reagent (QIAGEN) according to the manufacturer's instructions. Cells were used for patch clamp experiments at least 2 d after transfection.
Electrophysiological Experiments and Data Analysis
All data were recorded at room temperature (23–25°C) using an EPC10 patch clamp amplifier (HEKA). Inside-out membrane patches were excised from the transfected cells and held at −50 mV. The currents were filtered at 100 Hz with a built-in four-pole Bessel filter and digitized online at 500 Hz. A 50/60-Hz noise eliminator (Quest Scientific) was used to reduce 60-Hz noise. Cells were perfused with a bath solution containing (in mM): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 HEPES, and 20 sucrose (pH 7.4 with NaOH). The pipette solution contained (in mM): 140 NMDG-Cl, 2 MgCl2, 5 CaCl2, and 10 HEPES (pH 7.4 with NMDG). After establishing the inside-out configuration, the patch was perfused with a standard perfusion solution containing (in mM): 150 NMDG-Cl, 2 MgCl2, 10 EGTA, and 8 Tris (pH 7.4 with NMDG). The perfusion solution with the metal ions contained (in mM): 150 NMDG-Cl, 2 MgCl2, and 8 Tris (pH 7.4 with ∼18 mM HEPES). Measurements of the steady-state mean current amplitude and the fits to the dose–response relationships were performed using Igor software (Wavemetrics).
Recordings with up to five channel-opening steps were used for single-channel kinetic analysis. Mean open times were calculated using a program developed by L. Csanady (Csanady, 2000), as described previously (Zhou et al., 2006). All averaged data are presented as mean ± SEM.
The Cd2+ dose–response relationships were calculated as the ratio between the steady-state current in the presence of different [Cd2+] to the current under control conditions. For the G551D mutant, because it does not respond to ATP, we used the current in the absence of ATP (which is the same as in the presence of 1 mM ATP) as control. For the G551C and S549C mutants, because they are ATP dependent, we used the current in the presence of 1 mM ATP as control. The fold increase of the current in the presence of Cd2+ was normalized to the maximal fold increase for each mutant (100 μM for G551D, 10 μM G551D, and 5 μM for S549C).
Reagents
Mg-ATP was purchased from Sigma-Aldrich, PKA was purchased from Promega and Sigma-Aldrich, and MTSEA was purchased from Toronto Research Chemicals.
Online Supplemental Material
In the supplemental figures we show the comparison between the Cd2+-induced currents and the basal current (i.e., current in the absence of ATP) for all the tested mutants. The figures also point to the 548–551 amino acids as the critical region for mediating Cd2+ effects when they are converted to cysteine. The online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.200810049/DC1.
RESULTS
Cd2+ Increases the Activity of G551D-CFTR
We studied the effect of different divalent cations, including Cd2+, Ca2+, Ni2+, and Zn2+, on G551D-CFTR channels in excised inside-out membrane patches from transiently transfected Chinese hamster ovary cells. Fig. 2 A shows a real-time recording of G551D-CFTR channels in an excised patch that have been activated with 1 mM ATP and PKA (not depicted) before being exposed to different metal ions. Application of 10 μM Zn2+ or 10 μM Cd2+ in the absence of ATP increased the channel activity by 5.0 ± 0.4-fold (n = 6) and 12.2 ± 1.2-fold (n = 6), respectively, but neither 1 mM Ca2+ nor 10 μM Ni2+ had significant effects (n = 10). As a control, we applied the same metal ions to patches containing WT-CFTR channels. None of these cations, when applied in the absence of ATP, had any effect on WT-CFTR (n = 10) (Fig. 2 B). Because CFTR channels can only be opened by ATP after they have been phosphorylated by PKA, we tested whether the effect of Cd2+ on G551D-CFTR is also phosphorylation dependent. We applied the same [Cd2+] before and after PKA-dependent phosphorylation. As shown in Fig. 3 A, Cd2+ only increased the current of G551D-CFTR after the channels had been pre-phosphorylated by PKA (n = 5).
Figure 2.

Effect of different metal ions on G551D-CFTR and WT-CFTR currents. (A) 10 μM Cd2+ and 10 μM Zn2+ potentiate G551D-CFTR ATP-independent currents, but 1 mM Ca2+ and 10 μM Ni2+ have little effect on the currents. (B) WT-CFTR currents are not affected by any of these cations. Dashed lines in all figures represent the baseline.
Figure 3.

Functional characterization of the Cd2+-dependent effect in G551D-CFTR. (A) Activation of G551D-CFTR by Cd2+ is phosphorylation dependent. Note that in the same patch, Cd2+ only increases the activity of G551D-CFTR after the channels are activated by PKA and ATP. (B) A CFTR-specific blocker, inh-172, can inhibit the G551D-CFTR current induced by 200 μM Cd2+.
Previously, we estimated the Po of G551D channels to be 100-fold smaller than the maximal Po of WT channels, ∼0.004 ± 0.001 with a mean open time of 367 ± 42 ms (Bompadre et al., 2007). A quick calculation gives us an opening rate of 0.010 ± 0.003 s−1 (from Po = τo/(τo+τc), where τo is the open time of the channel, τc = 1/rco is the closed time of the channel, and rco is the opening rate). Because 100 μM Cd2+ potentiates G551D activity by ∼20-fold, we estimate that the Po of G551D-CFTR is 0.08 ± 0.03 under this condition. From patches with fewer than five simultaneous channel opening steps, we calculated the open time of G551D channels in the presence of 100 μM Cd2+ to be 2.4 ± 0.4 s (n = 10). Then the opening rate of G551D-CFTR in the presence of 100 μM Cd2+ is ∼0.036 ± 0.015 s−1, a near fourfold increase (3.6 ± 1.8) of the opening rate of G551D-CFTR in the absence of Cd2+.
Although Cd2+ increases the activity of G551D-CFTR, Cd2+ shows little effect on G1349D-CFTR (n = 5) (not depicted), a mutation of the corresponding glycine residue in the signature sequence of NBD2, indicating that this effect of Cd2+ is specific for the glycine-to-aspartate mutation at NBD1. To ensure that the channels opened by Cd2+ were indeed CFTR, we tested Cd2+ in patches excised from nontransfected cells and did not observe any channel activation (n = 5). In addition, the channels opened by 200 μM Cd2+ could be inhibited by inhibitor-172, a specific CFTR inhibitor that has been shown to modulate gating of WT-CFTR (Ma et al., 2002; Caci et al., 2008). As shown in Fig. 3 B, the current induced by 200 μM Cd2+ was nearly abolished by 5 μM inh-172 (97.2 ± 0.8% inhibition; n = 7). Thus, like ATP-dependent gating for WT-CFTR, activation of G551D mutant channels by Cd2+ requires pre-phosphorylation of the channel by PKA, and the Cd2+-induced currents can be readily and reversibly inhibited by inh-172. Because the activation of G551D-CFTR channels by Cd2+ is more effective than Zn2+, we focused our studies on this particular cation.
Cd2+ Is More Potent on G551C than on G551D
We considered two possible mechanisms for the effect of Cd2+ on G551D-CFTR. First, Cd2+ may directly interact with the side chain of the aspartate at position 551 and form a metal bridge between D551 and some other amino acids. Alternatively, the G551D mutation may induce protein conformational changes, and these structural changes enable Cd2+ to enhance the activity of G551D-CFTR. In this case, Cd2+ may exert its effects by binding to somewhere else and opening the channel in a nonspecific manner. To differentiate these two possibilities, we first mutated the glycine at position 551 to cysteine. We reasoned that if Cd2+ directly interacts with the side chain of the aspartate at 551, it will do so more effectively with a cysteine at that position because the thiol group of cysteines can better coordinate soft metal ions like Cd2+ (Rothberg et al., 2003). Fig. 4 shows representative traces of G551D (A) and G551C (B) in the presence of different [Cd2+]. Note that 5 μM Cd2+ induces a higher G551C-CFTR current than 1 mM ATP, despite that this mutation retains responsiveness to ATP. The normalized dose–response relationships of Cd2+ for these two mutants are shown in Fig. 4 (C and D). For G551D-CFTR, 100 μM Cd2+ increases the current by 21.38 ± 4.19-fold (n = 6), but the current response is not quite saturated. On the other hand, 10 μM Cd2+ already generates a maximal response for G551C-CFTR, with a maximal fold increase of 7.4 ± 0.3 (n = 5) compared with the currents generated by 1 mM ATP. Fitting the dose–response relationships with the Hill equation yields a K1/2 of 14.6 ± 6.3 μM and 3.29 ± 0.66 μM for G551D and G551C, respectively.
Figure 4.

Representative current traces of G551D-CFTR (A) and G551C-CFTR (B) in the presence of different [Cd2+]. The Cd2+ dose–response relationships for G551D-CFTR (C) and G551C-CFTR (D) were fitted with the Hill equation, y = min + (max−min)/ [1+ (K1/2/[x])n)] (smooth curves). The fold increase of the current in the presence of Cd2+ was normalized to the maximal fold increase for each mutant (G551D: 21.38 ± 4.19-fold, 100 μM Cd2+; G551C: 7.4 ± 0.3, 10 μM Cd2+). G551D: K1/2 = 14.6 ± 6.3 μM and n = 1.03 ± 0.34; G551C: K1/2 = 3.29 ± 0.66 μM and n = 1.89 ± 0.52.
To further test our hypothesis that the engineered aspartate or cysteine at position 551 is directly involved in coordinating Cd2+, we mutated G551 to alanine, which Cd2+ should not be able to bind effectively. Like the G551C mutant, G551A-CFTR remains responsive to ATP. However, the effect of Cd2+ on G551A-CFTR is negligibly small compared with that of G551C-CFTR (Fig. 5 A vs. Fig. 4 B). This difference between G551A and G551C was quantified in Fig. 5 B, where we compared the current generated by 1 mM ATP with the current generated by 10 μM Cd2+ for these two mutants. Because the side chains of aspartate and cysteine, but not alanine, are found in the multi-dentate coordinating geometries of metalloproteins that bind Cd2+ (Rulisek and Vondrasek, 1998), these results support the notion that Cd2+ interacts directly with the side chain of engineered aspartate or cysteine at position 551 despite the different apparent affinities.
Figure 5.

Comparison of Cd2+ and ATP-induced currents between G551A and G551C mutants. (A) Representative current trace of G551A-CFTR. Although G551A-CFTR remains ATP dependent, Cd2+ fails to increase the activity of the channels. (B) The ratio of currents induced by 10 μM Cd2+ and those with 1 mM ATP for G551C-CFTR and G551A-CFTR.
Cd2+ Increases the Activity of Other Signature Sequence Mutants
To further probe the Cd2+-binding position and specificity, we introduced a cysteine at different positions in the signature sequence of NBD1 (LSGGQ), as well as the two amino acids framing the signature sequence. Specifically, we mutated amino acids T547, L548, S549, G551, Q552, and R553, one at a time, to cysteine. A representative S549C-CFTR current recording is shown in Fig. 6 A. Cd2+ elicited macroscopic current even at sub-micromolar [Cd2+]. At a concentration as low as 5 μM, Cd2+ already induced a maximal response that is 4.6 ± 0.4-fold (n = 6) larger than the currents generated by 1 mM ATP. Fitting the dose–response relationship with the Hill equation yields a K1/2 value of 2.4 ± 0.8 μM (Fig. 6 C).
Figure 6.

Comparison of Cd2+ and ATP-induced currents for different mutants in the signature sequence region of NBD1. (A) Representative S549C-CFTR current recording in the presence of different [Cd2+]. (B) Representative T547C current recording in the presence of 1 mM ATP or 10 μM Cd2+. (C) Dose–response relationship for S549C-CFTR fitted with the Hill equation (solid line); K1/2 = 2.4 ± 0.8 μM and n = 1.12 ± 0.39. The maximum fold increase is 4.6 ± 0.4. (D) Summary of the current ratios for different mutants with cysteine-substituting amino acids in or around the signature sequence of NBD1.
In contrast, 10 μM Cd2+ shows negligible effect on the T547C mutant (Fig. 6 B). Fig. 6 D, in which the relative efficacy of ATP (1 mM) versus Cd2+ (10 μM) is compared, summarizes data for all these mutations in or around the signature sequence. It appears that when cysteine is engineered outside the signature sequence (i.e., T547C and R553C) or at the C-terminal end of the signature sequence (i.e., Q552C), ATP remains a much better ligand than Cd2+ (e.g., Fig. 6 B). However, for L548C, S549C, and G551C, the specificity of the ligand is altered so that Cd2+ becomes more effective at gating the channels than ATP. These results set apart amino acids 548–551 in the signature sequence in mediating the effect of Cd2+. It is interesting to note that S549 and G551 residues are involved in forming hydrogen bonds with the γ-phosphate of the bound ATP in several crystal structures of NBD dimers (e.g., Fig. 1) in the ABC transporters (Hopfner et al., 2000; Smith et al., 2002; Chen et al., 2003; Zaitseva et al., 2005). The significance of these overlapping regions of the signature sequence between ATP-dependent gating for WT-CFTR and Cd2+-dependent gating of mutant channels is discussed below.
The Binding Partner of Cd2+ Is Likely To Be a Cysteine Residue
The micromolar affinity of Cd2+ in activating G551C or S549C mutants raises the possibility that some endogenous cysteine(s) or histidine(s) may participate in forming a high-affinity binding site for Cd2+. To test this idea, we used the thiol-specific reagent MTSEA. Fig. 7 shows a representative recording. The G551D-CFTR channels were exposed to 1 mM MTSEA for a short period of time (<1 min), and MTSEA was subsequently washed out. Cd2+ no longer increased the channel activity after MTSEA pretreatment (n = 5). However, this effect of MTSEA can be reversed by 5 mM DTT. This result suggests that the binding partner of Cd2+ is likely a cysteine residue. To further test this hypothesis, we made a G551D-CFTR construct with 16 out of the 18 endogenous cysteine residues replaced with serines (the 2 cysteine residues, C590 and C592, left unchanged are essential for protein expression). Cd2+ can no longer activate this G551D/16-Cys–less channel (not depicted). We therefore conclude that Cd2+ opens these CFTR mutants likely by forming a metal bridge between the introduced cysteine/aspartate and the endogenous cysteine(s) that has yet to be identified.
Figure 7.

MTSEA abolishes the effect of Cd2+ on G551D-CFTR. G551D-CFTR channels were activated with 1 mM ATP plus PKA (not depicted). Pretreatment of the patch with the thiol-specific reagent MTSEA abolished the potentiation effect of Cd2+. 5 mM DTT can reverse the effect of MTSEA. Note that neither MTSEA nor DTT by itself had discernable effects on the channel activity.
DISCUSSION
Here, we show that micromolar concentrations of Cd2+ can dramatically increase the activity of G551D-CFTR, a disease-associated mutant, as well as G551C-CFTR, L548C, and S549C-CFTR, in the absence of ATP. We speculate that this effect of Cd2+ is mediated by forming a metal ion bridge between the engineered aspartate or cysteine residue in the signature sequence of NBD1 (LSGGQ) and the yet to be identified cysteine residue(s) in another part of the CFTR protein.
Possible Kinetic Mechanism for the Action of Cd2+
Before we discuss the possible biochemical/structural implications of our results, we will first look into the possible kinetic mechanism for the action of Cd2+. It should be noted that this discussion on kinetics is meant to facilitate a better understanding of the action of Cd2+ and by no means suggest a definitive kinetic mechanism for Cd2+, which will require extensive single-channel studies. The simplest interpretation of Cd2+-dependent activation of CFTR mutants shown here is to consider Cd2+ as a ligand that increases the open probability (Po) of the channels. Two basic kinetic mechanisms are considered. Scheme 1 dictates that Cd2+, by binding to the open state, induces another open state to increase the Po. This scenario can be envisioned if the Cd2+ binding site is exposed when the channel is in the open state.
| SCHEME 1 |
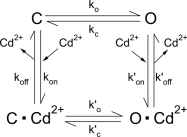
Scheme 2, a more generalized scheme for ligand-gated channels, proposes that Cd2+ can bind to both the open state and the closed state, but the Cd2+-bound channel assumes a more favorable gating transition for the open state than the ligand-free channel.
 |
SCHEME 2 |
The major difference between these two schemes is that Scheme 1 predicts an unaltered opening rate in the presence of Cd2+. As described above, the opening rate of G551D-CFTR is increased by Cd2+. For G551C-CFTR, this effect of Cd2+ on the opening rate likely also occurs. The open time for G551C-CFTR in the absence of ATP is 202.6 ± 29.6 ms (n = 3). In the presence of a saturating concentration of Cd2+, the open time, estimated from the current relaxation upon removal of Cd2+, is increased to 1.58 ± 0.21 s (n = 7). This approximately eightfold increase of the open time can hardly explain an ∼40-fold (40.3 ± 2.5; n = 3) increase of the macroscopic current unless the opening rate is also increased significantly by Cd2+. Although more complicated schemes are necessary to explain the full effect of Cd2+, we consider Scheme 2, a generalized allosteric mechanism for the modulation of protein function by ligand binding (Monod et al., 1965), as a simplistic model merely to aid this discussion.
Structure/Function Implications of the Effects of Cd2+ on CFTR Mutants
Although the signature sequence of ABC transporters is highly conserved, its functional role remains unclear despite numerous reports that mutations in this region perturb the function of the ABC proteins (Browne et al., 1996; Schmees et al., 1999; Chen et al., 2004; Ren et al., 2004; Szentpetery et al., 2004; Cai et al., 2006). Our previous studies have suggested that the ATP-binding pocket (i.e., ABP2 in Bompadre et al., 2007), formed by the Walker A region of NBD2 and the signature sequence of NBD1, plays a key role in the ATP-dependent opening of CFTR. Mutations of a conserved tyrosine residue (Y1219) at NBD2 significantly reduce the potency of ATP to increase the opening rate of CFTR, likely because the mutations decrease ATP-binding affinity at ABP2 (Zhou et al., 2006). That a mutation in the signature sequence of NBD1 (i.e., G551D) renders a channel completely irresponsive to ATP further supports the critical role of ABP2 in catalyzing channel opening (Bompadre et al., 2007). The current finding that Cd2+ can gate CFTR channels through an interaction with 551D/C, 548C, or 549C not only reinforces the role of ABP2 in channel gating, but also provides mechanistic insights into the functional role of the signature sequence in NBD1. We propose that the signature sequence of NBD1 may serve as a “switch” that, when activated by a ligand (ATP or Cd2+), transmits the signal to the channel gate presumably located in the membrane-spanning domains that form the anion-selective pore.
The idea that ATP and Cd2+ gate the channel through a common structural motif of the signature sequence is supported by several pieces of evidence. First, as shown in Fig. 3 A, gating of G551D-CFTR by Cd2+, like ATP-dependent gating of WT-CFTR, requires prior phosphorylation of the channels by PKA. Second, G551D-CFTR currents induced by Cd2+ can be inhibited by inhibitor-172, a gating modifier that has been shown to inhibit ATP-dependent gating of WT-CFTR (Ma et al., 2002; Caci et al., 2008). Third, normal gating of WT-CFTR channels involves interactions of ATP with the signature sequence of NBD1, as exemplified by the G551D mutation that completely eliminates the ATP-dependent gating (Bompadre et al. 2007). Fig. 6 demonstrates that the Cd2+-dependent gating also involves the signature sequence because engineering cysteine residues framing the signature sequence (i.e., T547C and R553C) did not confer this effect of Cd2+. It should be noted that crystal structures of NBD dimers (e.g., Fig. 1) reveal that γ-phosphate, a critical component for ATP being a successful ligand for CFTR gating, forms hydrogen bonds with the side chain of serine (S549 in CFTR) and the main chain of nitrogen of glycine (G551 in CFTR). These two positions coincide with the region involved in Cd2+-dependent gating (Fig. 6 D). Although we cannot definitively rule out the possibility that Cd2+-dependent gating of CFTR mutants bears no relationship to normal gating of WT channels by ATP, collectively, our data support the idea that a defined region of the signature sequence transduces the signal of ligand (ATP or Cd2+) binding to the gate of CFTR.
Chemical Mechanism for the Action of Cd2+
Several lines of evidence suggest that the effect of Cd2+ reported here is mediated though a metal bridge formation between at least two coordinating residues. First, the apparent affinities for G551C and S549C are at low micromolar range, supporting the idea that multiple cysteines are involved in coordinating Cd2+. Second, a thiol-specific reagent, MTSEA, can abolish the effect of Cd2+ on G551D. Third, based on the Monod-Wyman-Changeux model (Scheme 2) for allosteric modulation of protein function by ligand binding (Monod et al., 1965), we can conclude that when Cd2+ binding yields an ∼30-fold increase of the gating constant (i.e., K′/K = 30, where K′ = k′o/k′c and K = ko/kc), as in the case of G551D, thermodynamics dictates that the binding affinity for the open state has to be 30-fold higher than that for the closed state (i.e., Kd/Kd′ = 30, where Kd = koff/kon and Kd′ = k′off/k′on). One can imagine that in the closed state, the coordinating residues (aspartate or cysteine) are too far apart to form an ideal coordinate for Cd2+ binding. However, if the side chains of these residues move closer to each other after the conformational changes during channel opening, the open state could assume a higher affinity for Cd2+ simply because of a better coordination.
Although our data have pointed to an involvement of partner cysteine(s) in mediating the effect of Cd2+, the exact position of the cysteine residue(s) remains unclear. There are 18 cysteines in human CFTR (14 cysteines in the cytoplasmic domains). We are currently in the process of identifying the partner cysteine by systematically removing cysteines in different parts of CFTR. Although our top candidates will be cysteine(s) in NBD2 because NBD dimerization is coupled to ATP-dependent opening of CFTR (Vergani et al., 2005), we do not exclude the possibility that cysteine(s) in other domains may be involved. In fact, demonstrating an involvement of cysteines outside of NBD2 could bring up the possibility of opening CFTR channels independently of NBD dimerization. Our preliminary data (not depicted) indeed suggest that this may be the case because removing all six cysteines in NBD2 does not seem to affect the action of Cd2+ on S549C-CFTR. More thorough studies are currently underway to test this interesting possibility.
Pathophysiological Implications
The disease-associated mutant G551D has an ∼100-fold smaller Po than WT channels. Many compounds (Amaral and Kunzelmann, 2007) and ATP analogues (Cai et al., 2006; Bompadre et al., 2007) have been found to potentiate the activity of G551D-CFTR channels, but none of them could increase the activity of G551D-CFTR to WT levels. The binding site for only a very limited number of these CFTR “potentiatiors” has been identified (Bompadre et al., 2008). Interestingly, the 20-fold increase of the G551D-CFTR current by Cd2+ is by far the most effective potentiation demonstrated for this disease-associated mutant. Unfortunately, because of its toxicity, Cd2+ cannot be used therapeutically. Although Zn2+ may be an interesting alternative, its low potency and efficacy also prohibit a possible therapeutic application. Nevertheless, our findings open the door for rational drug design that can greatly benefit cystic fibrosis patients carrying the G551D mutation. These studies suggest that the signature sequence of NBD1 can be a drug target for rescuing the dysfunctional G551D channels. It is worth noting that the significance of our results in future drug design should hold even if this Cd2+-dependent gating for G551D-CFTR and normal ATP-dependent gating of WT channels turn out to use different gating machinery.
Supplementary Material
Acknowledgments
We thank Dr. Xiaoqin Zou for the critical reading of the manuscript and Dr. Hao-Yang Liu for helpful discussions.
This work was supported by NIHR01DK55835, NIHR01HL53455, and CFFT CLARKE06XX0 (T.-C. Hwang). S.G. Bompadre is currently supported by a research grant from the Cystic Fibrosis Foundation (BOMPAD06G0), and by National Institutes of Health (K01 DK075408). This investigation was conducted in a facility constructed with support from the Research Facilities Improvement Program (C06 RR-016489-01) from the National Center for Research Resources, National Institutes of Health.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: ABC, ATP-binding cassette; MTSEA, 2-aminoethyl methane thiosulfonate hydrobromide; NBD, nucleotide-binding domain; WT, wild-type.
References
- Amaral, M.D., and K. Kunzelmann. 2007. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol. Sci. 28:334–341. [DOI] [PubMed] [Google Scholar]
- Bompadre, S.G., Y. Sohma, M. Li, and T.-C. Hwang. 2007. G551D and G1349D, two CF-associated mutations in the signature sequence of CFTR, exhibit distinct gating defects. J. Gen. Physiol. 129:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre, S.G., M. Li, and T.-C. Hwang. 2008. Mechanism of G551D-CFTR potentiation by a high affinity ATP analog. J. Biol. Chem. 283:5364–5369. [DOI] [PubMed] [Google Scholar]
- Browne, B.L., V. McClendon, and D.M. Bedwell. 1996. Mutations within the first LSGGQ motif of Ste6p cause defects in a-factor transport and mating in Saccharomyces cerevisiae. J. Bacteriol. 178:1712–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caci, E., A. Caputo, A. Hinzpeter, N. Arous, P. Fanen, N. Sonawane, A.S. Verkman, R. Ravazzolo, O. Zegarra-Moran, and L.J. Galietta. 2008. Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem. J. 413:135–142. [DOI] [PubMed] [Google Scholar]
- Cai, Z., A. Taddei, and D.N. Sheppard. 2006. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel. J. Biol. Chem. 281:1970–1977. [DOI] [PubMed] [Google Scholar]
- Chen, J., G. Lu, J. Lin, A.L. Davidson, and F.A. Quiocho. 2003. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol. Cell. 12:651–661. [DOI] [PubMed] [Google Scholar]
- Chen, M., R. Abele, and R. Tampe. 2004. Functional non-equivalence of ATP-binding cassette signature motifs in the transporter associated with antigen processing (TAP). J. Biol. Chem. 279:46073–46081. [DOI] [PubMed] [Google Scholar]
- Chen, T.Y., and T.C. Hwang. 2008. CLC-0 and CFTR: chloride channels evolved from transporters. Physiol. Rev. 88:351–387. [DOI] [PubMed] [Google Scholar]
- Csanady, L. 2000. Rapid kinetic analysis of multichannel records by a simultaneous fit to all dwell-time histograms. Biophys. J. 78:785–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner, K.P., A. Karcher, D.S. Shin, L. Craig, L.M. Arthur, J.P. Carney, and J.A. Tainer. 2000. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily. Cell. 101:789–800. [DOI] [PubMed] [Google Scholar]
- Lewis, H.A., S.G. Buchanan, S.K. Burley, K. Conners, M. Dickey, M. Dorwart, R. Fowler, X. Gao, W.B. Gruggino, W.A. Hendrickson, et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, T.J., R. Thiagarajah, H. Yang, N.D. Sonawane, C. Folli, L.J. Galietta, and A.S. Verkman. 2002. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 110:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod, J., J. Wyman, and J.P. Changeus. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12:88–118. [DOI] [PubMed] [Google Scholar]
- Ren, X.Q., T. Furukawa, M. Haraguchi, T. Sumizawa, S. Aoki, M. Kobayashi, and S. Akiyama. 2004. Function of the ABC signature sequences in the human multidrug resistance protein 1. Mol. Pharmacol. 65:1536–1542. [DOI] [PubMed] [Google Scholar]
- Riordan, J.R., J.M. Rommens, B. Kerem, N. Alon, R. Rozmahel, Z. Grzelczak, J. Zielenski, S. Lok, N. Plavsic, J.-L. Chou, et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245:1066–1073. [DOI] [PubMed] [Google Scholar]
- Rothberg, B.S., K.S. Shin, and G. Yellen. 2003. Movements near the gate of a hyperpolarization-activated cation channel. J. Gen. Physiol. 122:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulisek, L., and J. Vondrasek. 1998. Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J. Inorg. Biochem. 71:115–127. [DOI] [PubMed] [Google Scholar]
- Schmees, G., A. Stein, S. Hunke, H. Landmesser, and E. Schneider. 1999. Functional consequences of mutations in the conserved ‘signature sequence’ of the ATP-binding-cassette protein MalK. Eur. J. Biochem. 266:420–430. [DOI] [PubMed] [Google Scholar]
- Smith, P.C., N. Karpowich, L. Millen, J.E. Moody, J. Rosen, P.J. Thomas, and J.F. Hunt. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 10:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentpetery, Z., A. Kern, K. Liliom, B. Sarkadi, A. Varadi, and E. Bakos. 2004. The role of the conserved glycines of ATP-binding cassette signature motifs of MRP1 in the communication between the substrate-binding site and the catalytic centers. J. Biol. Chem. 279:41670–41678. [DOI] [PubMed] [Google Scholar]
- Vergani, P., S.W. Lockless, A.C. Nairn, and D.C. Gadsby. 2005. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 433:876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva, J., S. Jenewein, A. Wiedenmann, H. Benabdelhak, I.B. Holland, and L. Schmitt. 2005. Functional characterization and ATP-induced dimerization of the isolated ABC-domain of the haemolysin B transporter. Biochemistry. 44:9680–9690. [DOI] [PubMed] [Google Scholar]
- Zhou, Z., X. Wang, H.-Y. Liu, X. Zou, M. Li, and J.H. Hwang. 2006. The two ATP binding sites of the cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. J. Gen. Physiol. 128:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.