

Abstract
The blockade of CLC-0 chloride channels by p-chlorophenoxy acetate (CPA) has been thought to be state dependent; the conformational change of the channel pore during the “fast gating” alters the CPA binding affinity. Here, we examine the mechanism of CPA blocking in pore-open mutants of CLC-0 in which the residue E166 was replaced by various amino acids. We find that the CPA-blocking affinities depend upon the volume and the hydrophobicity of the side chain of the introduced residue; CPA affinity can vary by three orders of magnitude in these mutants. On the other hand, mutations at the intracellular pore entrance, although affecting the association and dissociation rates of the CPA block, generate only a modest effect on the steady-state blocking affinity. In addition, various amphiphilic compounds, including fatty acids and alkyl sulfonates, can also block the pore-open mutants of CLC-0 through a similar mechanism. The blocking affinity of fatty acids and alkyl sulfonates increases with the length of these amphiphilic blockers, a phenomenon similar to the block of the Shaker K+ channel by long-chain quaternary ammonium (QA) ions. These observations lead us to propose that the CPA block of the open pore of CLC-0 is similar to the blockade of voltage-gated K+ channels by long-chain QAs or by the inactivation ball peptide: the blocker first uses the hydrophilic end to “dock” at the pore entrance, and the hydrophobic part of the blocker then enters the pore to interact with a more hydrophobic region of the pore. This blocking mechanism appears to be very general because the block does not require a precise structural fit between the blocker and the pore, and the blocking mechanism applies to the cation and anion channels with unrelated pore architectures.
INTRODUCTION
Blockers and inhibitors of ion channels inhibit the current flow across the channel protein and thus silence excitable cells critical for many physiological functions. Various animals exploit channel blockers/inhibitors as lethal weapons to capture their prey, while physicians have applied them for therapeutic purposes. In ion channel research, blockers and inhibitors are extremely useful in exploring the mechanistic operation of channel molecules. Good examples include blockades of K+ channels by small venom toxins (MacKinnon and Miller, 1988, 1989) and by organic compounds such as quaternary ammonium (QA) molecules (Armstrong, 1969, 1971; MacKinnon and Yellen, 1990; Yellen et al., 1991). The effects of these blockers on voltage-gated K+ channels have been studied for decades, and understanding the blocking mechanism has provided significant insights into the structural–functional relationship of voltage-gated cation channels (Hille, 2001).
In contrast to the long history of studying cation channel blockers, mechanisms of anion channel blockers just began attracting the attention of investigators in the last 10 years. A group of clofibric acid–derived compounds has recently been discovered to have inhibitory effects on CLC-type chloride (Cl−) channels (Pusch et al., 2000, 2001; Estevez et al., 2003), among which the effect of p-chlorophenoxy acetate (CPA) on the Torpedo CLC-0 channel is the most thoroughly studied at the mechanistic level (Accardi and Pusch, 2003; Moran et al., 2003; Traverso et al., 2003). It was found that the inhibition of CLC-0 by intracellularly applied CPA is voltage dependent, with apparent affinities ranging from low millimolar (∼1 mM) to tens of millimolar (∼30 mM), respectively, at voltages from −150 to +80 mV. Because the opening of the fast gate of CLC-0 is favored by depolarized voltages, the voltage dependence of the apparent affinity of the CPA block was thought to be a state-dependent effect—namely, the CPA affinity is high for the closed-state pore but low for the open-state pore (Accardi and Pusch, 2003).
Because of the proposed state-dependent block, the conformational change of the CLC-0 pore associated with the fast gating was hypothesized to involve more than the swinging of the side chain of E166, which corresponds to E148 of the Escherichia coli CLC (CLCec-1) molecule. In particular, a conformational change of the pore region intracellular to E166 was thought to be responsible for the aforementioned “state-dependent” CPA affinities (Accardi and Pusch, 2003). These hypotheses seemed to be supported by the CPA block of a pore-open mutant, E166A, because of the following observations. First, the fast gate of E166A stayed mostly open, but this mutant had a CPA affinity >100-fold higher than that of the wild-type CLC-0. This finding was unexpected if the open state of CLC-0 had a lower affinity than the closed state, and if the fast gate opening involved only the movement of the side chain of E166. Second, the relationship between the inhibition rate and the blocker concentration in the CPA block of E166A was not linear. Therefore, the overall inhibition mechanism was not a bimolecular reaction. A three-state model was proposed, in which the CPA-bound E166A pore was able to undergo a gating conformational change. Finally, in the presence of the E166A mutation, mutations of two pore residues, S123T and K519E, slowed down the kinetics of the CPA inhibition. This phenomenon was attributed to a slower fast-gating conformational change of these two mutant channels (Traverso et al., 2003).
Although the E166A mutant revealed a high CPA-blocking affinity, we recently discovered that the CPA affinity for another pore-open mutant of CLC-0, E166Q, was quite low (Fig. 1). Previous studies have shown that the structures of the corresponding E148A and E148Q mutants in CLC-ec1 are nearly identical, and the functional recordings of the E166A and E166Q mutants of CLC-0 are the same (Dutzler et al., 2003). Yet, these two pore-open mutants exhibit very different affinities for the CPA block. Unless one assumes that the conformational changes of the E166A and E166Q pores are different, it seems difficult to explain the dramatic difference in the CPA affinities between these two mutants. After this intriguing discovery, we have reexamined the CPA-blocking mechanism in the pore-open mutants of CLC-0. We here report that the apparent CPA affinity depends upon the side chain of the amino acid residue placed at position 166, and the mutation of the E166 residue strongly affects the dissociation rate and the steady-state affinity of the CPA block. In contrast, the mutation at the intracellular pore entrance alters the association (on) and dissociation (off) rates of the CPA block, but the change in the steady-state CPA-blocking affinity is only modest. These results together suggest the possibility that the CPA block in the pore-open mutants of CLC-0 is similar to the three-state pore-blocking model previously proposed for the blockade of the voltage-gated K+ channel by the inactivation ball peptide (Murrell-Lagnado and Aldrich, 1993a,b), or by the long-chain QA compounds (Zhou et al., 2001). The three-state pore-blocking mechanism also led to a discovery of a variety of amphiphilic blockers for CLC-0.
Figure 1.

Inhibitions of the E166A and E166Q mutants by CPA. (A and B) Macroscopic current recordings in the absence (left) and presence (middle and right) of the indicated concentration of CPA for E166A and E166Q mutants. (Inset) Voltage protocol used to obtain the recording traces. Horizontal scale bars represent 50 ms. (C and D) Percentage of current inhibitions at different voltages in various concentrations of CPA. For the E166A mutant, the current inhibition curves from left to right were obtained in the presence of 3, 10, 30, 100, and 300 μM CPA. For the E166Q mutant, the inhibition curves from left to right were in the presence of 0.3, 1, 3, 10, and 30 mM CPA.
MATERIALS AND METHODS
Mutagenesis and Channel Expression
The cDNAs of various mutants were all constructed in the pcDNA3 vector for mammalian cell line expressions. The slow gating of CLC-0, which was not of concern in this study, was suppressed by a point mutation, C212S (Lin et al., 1999). This background construct will be hereby referred to as the “wild-type channel,” and all tested mutants were made in this C212S background. Point mutations were made by standard PCR mutagenesis methods, and the DNA sequence of the inserted DNA segment was fully sequenced through commercially available DNA sequencing services to avoid unexpected mutations. For mutations of the E166 residue, we replaced the wild-type glutamate residue by 16 other amino acids, except for proline, methionine, and threonine. To examine the effects of mutating the residues at the intracellular pore entrance, the mutations of E127/K519 were made in a high CPA affinity mutant, E166C. All mutants were expressed in human embryonic kidney (HEK) 293 cells. Transfections of cDNA into HEK293 cells followed a previously described procedure using a lipofectamine kit (Zhang et al., 2006). Normally, the recording was performed 1–3 d after cDNA was transfected into HEK 293 cells.
Electrophysiological Recordings
Electrophysiological recording methods are similar to those described previously (Chen and Chen, 2003; Lin and Chen, 2003; Zhang et al., 2006). Macroscopic current recordings were performed from excised, inside-out membrane patches of HEK 293 cells. Recording pipettes were pulled from borosilicate glass (World Precision Instruments) using a vertical electrode puller (pp830; Narashige). Except for those experiments in which the Cl− concentration ([Cl−]) is changed, both the pipette and the bath solutions contained (in mM): 130 NaCl, 5 MgCl2, 10 HEPES, and 1 EGTA, pH 7.4. For solutions with lower Cl− concentrations, sodium glutamate was used to replace NaCl. The electrode resistance is normally within the range of 1–3 MΩ. Under such recording conditions, all the constructed 16 mutants of E166 expressed well in HEK 293 cells, and except for E166R and E166D, they did not show a prominent current deactivation at negative voltages as that shown in the wild-type channel; they were functionally defined as the “pore-open” channel. Because E166R and E166D (Traverso et al., 2006) did not fit the functionally defined criterion of “open pore,” these two mutants were not studied. The recorded current was amplified using the Axopatch 200B patch clamp amplifier (MDS Analytical Technologies), digitally filtered at 1 kHz, and digitized at 2 kHz using Digidata 1320 digitizing board and pClamp8 software. In some experiments where the current relaxation process was only a few ms (for example, the recordings shown in Figs. 8 A and 9 B), higher sampling and filtered frequency (10 and 5 kHz, respectively) were used.
Figure 8.

Effects of the mutation at the pore entrance on the CPA-unblock rate. (A) Recovery of the current from the CPA-blocked state when the voltage was jumped from −60 to +80 mV. [CPA] = 1 mM. (B) Current recovery rate (1/τ) as a function of [CPA] in various E127/K519 combinatorial mutants. The time constant, τ, was obtained from the current-recovery process at +80 mV in those experiments shown in A.
Figure 9.

Trans-ion effects on the CPA block of E166A. (A) Reduction of [Cl−]o increases the steady-state CPA-blocking affinity in E166A. (B) Recovery of the current from the CPA block. The excised patch was voltage clamped at a prepulse voltage at −160 mV to allow a CPA block to reach a steady state, followed by a voltage step to a positive voltage (from +80 to +20 mV). Intracellular [CPA] was 1 mM in all three experiments. (C) Comparison of the averaged current recovery time constants in the three ionic conditions shown in B. The color of the symbols are the same as those in B: black, symmetrical 140 mM Cl−; green, [Cl−]o = 140 mM, [Cl−]i = 15 mM; red, [Cl−]o = 15 mM, [Cl−]i = 140 mM. Notice that altering [Cl−]o significantly changed the recovery time constants, whereas altering the [Cl−]i had little effect. In the condition of [Cl−]o = 15 mM and [Cl−]i = 140 mM, the current relaxation was not prominent at +40 mV because the voltage was closed to the reversal potential. The time constant of the current recovery was therefore not analyzed.
Single-channel recordings were performed on channels expressed in Xenopus oocytes. The RNA of the expressed channel was made from the cDNA construct subcloned to a pBluescript vector using mMessage mMachine kit (Applied Biosystems), as described previously (Chen, 1998). The single-channel current was recorded via an Axopatch 200B amplifier. The signal was filtered through a four-pole Bessel filter at 200 Hz cutoff and digitized at 1 kHz using a Microstar 800 digitizing board. Analysis of the averaged dwell time of the nonconducting events in the single-channel trace was achieved by a homemade analysis program described previously (Chen and Chen, 2003; Zhang et al., 2006).
Blocker Applications
Reagent-grade CPA and other amphiphilic blockers were purchased from Sigma-Aldrich. A stock solution of 100 mM CPA was first made in the standard bath solution, and the pH of this stock solution was adjusted back to 7.4. Two major series of amphiphilic blockers were also used: saturated fatty acids with various numbers of carbons in the aliphatic chain, and the sulfonate derivatives of these fatty acids. With the carbon number equal to or less than eight, the amphiphilic compounds can be directly dissolved into the working solution. For compounds with 10–12 carbons (decanoate, dodecanoate, and their sulfonate derivatives), stock solutions of 10 mM in DMSO were first made. Working solutions containing various concentrations of blockers were then made by diluting the stock solution into the bath solution. Application of blockers to the cytoplasmic side of the membrane patch was achieved using SF-77 solution exchanger (Warner Instrument), and the patch was constantly immersed in a continuously flowing solution containing the indicated concentration of these blockers.
To examine the CPA-blocking effect from macroscopic current recordings, a voltage protocol similar to that used to evaluate the fast-gating transition of CLC-0 was used (Zhang et al., 2006). This voltage protocol started with a positive prepulse (at +80 mV) voltage step, followed by different test voltages from +80 to −160 mV in −20-mV steps (Fig. 1 A, inset). The positive prepulse ensured that the CPA in the pore was cleared by the inward Cl− flux, while the various levels of the test voltage allowed CPA to enter the pore. The duration of each voltage step was adjusted so that it was long enough for the current relaxation to reach a steady state. After the test voltage pulse, a voltage step at +80 or −100 mV was applied to measure the tail current. In some instances, the relaxation process of the tail current at +80 mV was fitted to a single-exponential function to study the dissociation rate of CPA.
Data Analysis
Measuring the current in the presence of CPA was bracketed by two recordings in the absence of CPA: one before and one after the CPA application. The average of these two recordings in the absence of CPA served as the control. Steady-state current amplitude was normally measured at the last 10 ms of the test voltage step. Various concentrations of CPA were applied to the same patch to assess the dose-dependent inhibition. Because the E166 mutants of CLC-0 studied did not exhibit prominent current relaxation from the channel's gating process, we relied on the inhibition of the current by high concentrations of CPA to differentiate the leak current from the channel current. Only patches in which the inward current can be inhibited by >90% in the presence of the saturated concentration of blocker were used for data analysis. The steady-state current in the presence of certain concentrations of CPA was normalized to that of the current in the absence of CPA. The normalized value, which represents the unblocked fraction (FUB), was then fitted to a Langmüir function (Eq. 1):
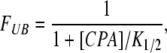 |
(1) |
where K1/2 is the CPA concentration ([CPA]) that effectively blocks 50% of the recorded current. To examine the kinetics of the CPA block, we assumed a three-state blocking model (Model B) similar to that proposed in the blockade of voltage-gated K+ channels by the inactivation ball peptide or by long-chain QA compounds (Zhou et al., 2001). For those mutants with relatively high blocker sensitivities, the current inhibition process by the blocker upon hyperpolarization of membrane voltages was fitted to a single-exponential function. The initial 1-ms recording upon membrane hyperpolarization was mostly the capacitance current and was excluded for the curve fitting. Based upon the three-state model (Zhou et al., 2001), the inverse of the time constant (τ) as a function of [CPA] should follow a hyperbolic equation (Eq. 2):
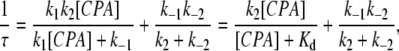 |
(2) |
where Kd (= k−1/k1) reflects the binding dissociation constant of CPA of the docking step (see Discussion). When [CPA] << Kd, Eq. 2 will reduce to:
 |
(3) |
where
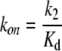 |
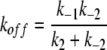 |
From Fig. 4 B, the Kd was estimated to be in mM range. Therefore, we used data points obtained at [CPA] ≤ 300 μM for evaluating the apparent on (kon) and off rates (koff) of the CPA block. All data points were presented as mean ± SEM averaged from approximately four to six patches.
Figure 4.

Kinetics of the CPA block in the high CPA-sensitive mutants. (A) Single-exponential time course of the CPA inhibition. The recording trace was made from the E166A mutant in the presence of 300 μM of intracellular CPA. (B) Dependence of the CPA inhibition rate on [CPA]. The inverse of the time constant of the current inhibition upon stepping the voltage from +80 to −160 mV (as shown in A) was plotted against [CPA]. The solid curve was a best fit to a hyperbolic equation with Kd = 4.3 mM, and the saturated value of 1/τ = 1561 s−1. (C and D) Analysis of the on and off rates of the CPA block in the E166C and E166L mutants. The current inhibition time courses at −140 mV in these two mutants were fitted to a single-exponential function, and the inverse of the time constant (1/τ) was plotted against [CPA]. Only data points obtained in the presence of 300 μM or less were used to fit to a linear equation (Eq. 3). (E) Dependence of the CPA inhibition rate on [CPA] in various mutants of E166 that show relatively high CPA affinities.
RESULTS
Various Pore-open Mutants of CLC-0 Have Different CPA-blocking Affinities
Previous studies by other investigators showed that the CPA-blocking affinity of the E166A mutant is >100-fold higher than that of the wild-type channel; the CPA affinities of E166A at negative voltages are in the μM concentration range (Traverso et al., 2003). Results shown in Fig. 1 A indeed confirm this mutation effect. 300 μM CPA inhibits nearly 95% of the E166A current at −160 mV. However, in another mutant, E166Q, the CPA-blocking affinity is very low. 300 μM CPA generates an effect of <10% inhibition at −160 mV (Fig. 1 B). The overall percentage of inhibitions of CPA in the E166A and E166Q mutants at different voltages is shown in Fig. 1 (C and D, respectively). Although CPA produces a significant inhibition of E166A in the concentration range of 3–300 μM (Fig. 1 C), the inhibition of E166Q requires higher [CPA] by two orders of magnitude (Fig. 1 D). This initial finding of different CPA affinities in E166A and E166Q motivated us to examine more E166 mutants of CLC-0.
The original recording trace and the percentage of CPA inhibition of the E166C, E166L, and E166K mutants are shown in Fig. 2 (A and B, respectively). Again, these mutants show different sensitivities to the CPA inhibition. At −160 mV, while 0.3 mM CPA blocks >90% of the E166C current, the same concentration of CPA blocks ∼60% of the E166L current and only ∼10% of the E166K current. To evaluate the apparent CPA-blocking affinity in various mutants of E166, we plotted the fraction of the unblocked current as a function of [CPA], and the half-blocking concentration (K1/2) was obtained by fitting the data points with a Langmüir function (Eq. 1). The K1/2's of the CPA block at various voltages in different mutants are summarized in Table I, and Fig. 3 A shows the dose-dependent inhibition curves of several mutants at −140 mV. It can be seen that all mutants show voltage-dependent block—the more hyperpolarized the membrane potential, the higher the CPA-blocking affinity (Table I). However, different mutants have very different sensitivities for the CPA block. Fig. 3 B shows a plot of the apparent CPA affinities as a function of the side chain volume of the residue placed at position 166. It appears that two factors determine the steady-state blocking affinity: the side chain volume and the hydrophobicity of the introduced residue. Roughly speaking, the larger the side chain volume is, the lower the blocking affinity is. Moreover, a hydrophobic residue at position 166 (such as in E166A, E166V, and E166I mutants) renders the channel sensitive to the CPA block. The numbers associated with the data points in Fig. 3 B are the hydrophobic index of the amino acid (Kyte and Doolittle, 1982)—the more positive the number, the higher the hydrophobicity of the amino acid. At −140 mV, the mutant with a hydrophobic side chain (positive hydrophobic index) shows a K1/2 of <200 μM. On the other hand, placing a hydrophilic residue with a bulky side chain (such as E166, E166K, and E166Q) renders the channel insensitive to the CPA block; the K1/2 is on the order of several mM for these three channels (Fig. 3 B). A hydrophilic residue with a small side chain (such as serine) can still make the mutant sensitive to the CPA block. Overall, these mutants can be roughly classified into two groups (encompassed by the magenta and cyan circles, respectively, in Fig. 3 B), except for the E166G mutant, for which the introduced glycine has only a hydrogen atom on the side chain. The block of E166G by CPA is unique and is described in this issue (see p. 59) by Zhang and Chen (2008). However, this classification of the CPA block for various mutants into two groups has exceptions. The amino acids histidine, tyrosine, and tryptophan have a negative hydrophobic index (hydrophilic side chains), yet the CPA affinities of E166H, E166Y, and E166W are relatively higher than the other mutants in the hydrophilic category. As will be shown later, the ring structure on the side chain of these three residues, and the other amino acid phenylalanine (shown by red solid squares in Fig. 3 B), may cause this anomaly. The observation that the blocking affinity depends upon the side chain of the residue suggests that perhaps the CPA molecule directly interacts with the introduced residue at position 166. That CPA interacts with the E166 side chain was also suggested in a previous mutation study (Estevez et al., 2003).
Figure 2.

Inhibition of the E166C, E166L, and E166K mutants by CPA. (A) Macroscopic current recordings of these mutants in the absence (top) and presence (bottom) of 0.3 mM CPA. The horizontal and vertical scale bars in each mutant are 50 ms and 1 nA, respectively. (B) Percentage of current inhibitions at different voltages in various concentrations of CPA. [CPA] was (in mM): E166C: 0.03, 0.1, 0.3, 1, and 3; E166L: 0.03, 0.1, 0.3, 1, and 3; and E166K: 0.3, 1, 3, 10, and 30.
TABLE I.
Steady-state Apparent CPA Affinity (K1/2) of Various Mutants of E166
| −160 mV | −140 mV | −120 mV | −100 mV | −80 mV | −60 mV | −40 mV | |
|---|---|---|---|---|---|---|---|
| E166G | 0.32 ± 0.03a | 0.24 ± 0.03 | 0.18 ± 0.02 | 0.15 ± 0.01 | 0.14 ± 0.01 | 0.18 ± 0.01 | 0.30 ± 0.02 |
| E166A | 0.010 ± 0.001 | 0.015 ± 0.001 | 0.025 ± 0.002 | 0.046 ± 0.002 | 0.082 ± 0.004 | 0.162 ± 0.009 | 0.337 ± 0.019 |
| E166S | 0.021 ± 0.001 | 0.035 ± 0.002 | 0.058 ± 0.003 | 0.104 ± 0.004 | 0.179 ± 0.007 | 0.342 ± 0.015 | 0.641 ± 0.036 |
| E166C | 0.026 ± 0.001 | 0.038 ± 0.001 | 0.061 ± 0.002 | 0.104 ± 0.003 | 0.186 ± 0.007 | 0.349 ± 0.015 | 0.581 ± 0.094 |
| E166N | 0.38 ± 0.01 | 0.62 ± 0.02 | 1.06 ± 0.02 | 1.85 ± 0.04 | 3.27 ± 0.09 | 5.78 ± 0.22 | 9.75 ± 0.57 |
| E166 | 1.34 ± 0.25 | 1.17 ± 0.19 | 1.57 ± 0.19 | 1.96 ± 0.21 | 2.87 ± 0.20 | 5.22 ± 0.23 | 9.37 ± 0.33 |
| E166V | 0.026 ± 0.003 | 0.040 ± 0.004 | 0.065 ± 0.007 | 0.113 ± 0.010 | 0.198 ± 0.015 | 0.373 ± 0.037 | 0.676 ± 0.078 |
| E166Q | 3.34 ± 0.08 | 4.71 ± 0.15 | 6.84 ± 0.31 | 9.71 ± 0.48 | 14.0 ± 0.7 | 19.7 ± 0.9 | ND |
| E166H | 0.21 ± 0.04 | 0.39 ± 0.05 | 0.74 ± 0.08 | 1.45 ± 0.11 | 2.87 ± 0.15 | 5.86 ± 0.26 | 12.37 ± 0.52 |
| E166I | 0.039 ± 0.004 | 0.068 ± 0.008 | 0.117 ± 0.011 | 0.220 ± 0.024 | 0.384 ± 0.041 | 0.683 ± 0.085 | 1.25 ± 0.23 |
| E166L | 0.18 ± 0.01 | 0.28 ± 0.01 | 0.47 ± 0.01 | 0.80 ± 0.02 | 1.46 ± 0.03 | 2.61 ± 0.08 | 4.64 ± 0.19 |
| E166K | 5.62 ± 0.27 | 7.99 ± 0.35 | 11.3 ± 0.5 | 15.6 ± 0.7 | 21.3 ± 0.9 | ND | ND |
| E166F | 0.047 ± 0.007 | 0.093 ± 0.012 | 0.17 ± 0.02 | 0.35 ± 0.03 | 0.73 ± 0.09 | 2.06 ± 0.30 | 5.12 ± 1.72 |
| E166Y | 0.13 ± 0.01 | 0.22 ± 0.01 | 0.38 ± 0.01 | 0.70 ± 0.01 | 1.37 ± 0.05 | 2.79 ± 0.15 | 6.18 ± 0.67 |
| E166W | 0.55 ± 0.07 | 0.97 ± 0.13 | 1.86 ± 0.22 | 3.49 ± 0.41 | 6.66 ± 0.98 | 13.2 ± 2.1 | ND |
The K1/2 values are presented as mean ± SEM (in mM).
Figure 3.

Steady-state CPA-blocking affinities of various mutants of E166. (A) CPA dose-dependent inhibition curves of the wild-type (E166) and seven mutants. (B) Relationship between the apparent K1/2 of CPA and the side chain volume of the amino acid placed at position 166. The K1/2 value in each mutant was obtained by fitting the dose-dependent curve shown in A to a Langmüir equation (Eq. 1). The x axis represents the side chain volume of amino acids (Zamyatnin, 1972). The number associated with each data point in the plot is the hydrophobicity index of the introduced amino acid (Kyte and Doolittle, 1982). Except for the E166G mutant, all data points were classified into two groups as judged by eyes, and the two groups were enclosed in cyan and magenta circles, respectively. Notice that in comparison with the octanoate affinities shown in Fig. 12, the K1/2's of E166H, E166Y, E166W, and E166F (red squares) are disproportionally smaller than those of the other mutants.
On and Off Rates of the CPA Block on the Pore-open Mutants of CLC-0
To explore the mechanism underlying the difference in the CPA affinity among various mutants of E166, we examined the kinetics of the CPA block. For those mutants that have a relatively high CPA affinity (such as E166A, E166C, and E166L), the block was associated with a prominent current relaxation when the voltage was perturbed by jumping from +80 mV to various hyperpolarized voltages (Figs. 1 and 2). This current relaxation can be well fitted to a single-exponential function (Fig. 4 A). As shown previously (Traverso et al., 2003), the inverse of the time constant of this CPA inhibition process in the E166A mutant depends upon [CPA] in a nonlinear way (Fig. 4 B) according to Eq. 2. However, when the blocker concentration is low (for example, [CPA] ≤ 300 μM), the inverse of the current inhibition time constant (1/τ) is linearly related to [CPA], and the apparent on and off rates of the block can be evaluated by Eq. 3 (Fig. 4, C and D). The slope and the y-intercept of the linear plot represent the kon and koff of the CPA block on the E166C and the E166L mutants, respectively. The analysis for more E166 mutants shown in Fig. 4 E further supports that the major effect of the E166 mutation is to change the y-intercept (koff) of the plot, whereas the slope (kon) is only slightly changed (Fig. 4, D and E). Thus, the underlying reason for the different steady-state CPA affinity in these high CPA-sensitive mutants is mostly a change in the koff of the CPA block.
The kinetics of the CPA block in the low-affinity mutants, such as E166Q and E166K, cannot be examined using the above method because blocking these mutants requires a high concentration (several mM) of CPA, which results in a large kon[CPA] value, and therefore a very fast current inhibition that is inseparable from the capacitance spike upon hyperpolarization of the membrane voltage (Fig. 1 B, right). To evaluate the kinetics of the CPA block in E166Q and E166K, we compare the CPA effect between E166A and these two mutants at the single-channel level (Fig. 5). Fig. 5 A shows the effect of 0.1 mM CPA on E166A at −100 mV. In the absence of CPA, the E166A mutant is mostly open (the fast gate open probability, Po ≈ 1). In the presence of 0.1 mM CPA in the cytoplasmic side, E166A exhibits prominent, nonconducting events (indicated by arrows), with an averaged time of ∼55 ms (Fig. 5 A). These nonconducting events most likely correspond to the “blocked” state due to the obstruction of the ionic flow by CPA. For the E166Q mutant, however, the long blocked state is absent with 0.1 mM CPA (Fig. 5 B). Even when [CPA] is increased to 5 mM, the current of E166Q does not show similarly long, nonconducting events as those in E166A. Instead, the trace becomes much noisier compared with the control trace, indicating that the blocked state is not adequately resolved by the sampling and filtering frequency used in the recording (1 and 0.2 kHz, respectively). Although the very flickery trace renders the dwell-time analysis impossible, these experiments at the single-channel level suggest a qualitative picture that the koff of the CPA block in E166Q is larger than that in E166A by orders of magnitude. The block of E166K at the single-channel level is similar to that of the E166Q mutant shown in Fig. 5 B (not depicted). These experiments together indicate that the very low affinities of the CPA block in the E166Q and E166K mutants are most likely due to very high off rates of the CPA block.
Figure 5.

The CPA block of E166A and E166Q mutants at the single-channel level. Membrane potential: −100 mV. (A) Effect of 100 μM CPA on the E166A mutant. (B) Effect of 100 μM and 5 mM CPA on the E166Q mutant.
Effects of the Mutations at the Pore Entrance on the CPA Block
Although mutations of E166 alter the steady-state CPA blocking affinity by three orders of magnitude, the mutations of two residues, E127 and K519, at the intracellular pore entrance only affect the CPA affinity slightly. The CPA dose-dependent inhibition curves of E166C (with a lysine residue at position 519), E166C/K519M, and E166C/K519E mutants are compared in Fig. 6. By replacing K519 with a glutamate residue, the K1/2 is increased from ∼40 to ∼160 μM. The fourfold lower CPA affinity in the E166C/K519E mutant (compared with that of the E166C mutant) generated by an alteration of two unit charges at position 519 is small compared with the several hundred–fold change in K1/2 when E166 is mutated to alanine. The E127Q mutation likewise has a small effect on the steady-state CPA-blocking affinity (see Table II for the apparent CPA affinities of various E127/K519 combinatorial mutants).
Figure 6.

Effects of the charged mutations, K519M and K519E, on the apparent K1/2 of the CPA block. Data points were obtained at a membrane voltage of −140 mV. The apparent K1/2's for various E127/K519 combinatorial mutants in the background of the E166C mutation are given in Table II.
TABLE II.
CPA Affinity (K1/2) of E127/K519 Combinatorial Mutants
| −160 mV | −140 mV | −120 mV | −100 mV | −80 mV | −60 mV | −40 mV | |
|---|---|---|---|---|---|---|---|
| E127/K519a | 0.026 ± 0.001b | 0.038 ± 0.001 | 0.061 ± 0.002 | 0.104 ± 0.003 | 0.186 ± 0.007 | 0.349 ± 0.015 | 0.581 ± 0.094 |
| E127Q/K519 | 0.015 ± 0.002 | 0.021 ± 0.002 | 0.033 ± 0.003 | 0.057 ± 0.004 | 0.113 ± 0.010 | 0.228 ± 0.023 | 0.491 ± 0.058 |
| E127/K519M | 0.051 ± 0.004 | 0.069 ± 0.004 | 0.098 ± 0.004 | 0.141 ± 0.005 | 0.194 ± 0.004 | 0.271 ± 0.005 | 0.378 ± 0.007 |
| E127/K519E | 0.115 ± 0.025 | 0.158 ± 0.031 | 0.203 ± 0.034 | 0.277 ± 0.037 | 0.377 ± 0.039 | 0.516 ± 0.046 | 0.730 ± 0.059 |
| E127Q/K519M | 0.022 ± 0.002 | 0.031 ± 0.002 | 0.047 ± 0.003 | 0.075 ± 0.003 | 0.132 ± 0.006 | 0.240 ± 0.013 | 0.457 ± 0.030 |
| E127Q/K519E | 0.044 ± 0.005 | 0.062 ± 0.005 | 0.087 ± 0.006 | 0.123 ± 0.009 | 0.198 ± 0.014 | 0.327 ± 0.024 | 0.586 ± 0.052 |
The mutations in this table are all made in the presence of the E166C mutation.
The K1/2 values are presented as mean ± SEM (in mM).
Previous experiments showed that the K519E mutation in the background of E166A mutant renders the inhibition process slower (Traverso et al., 2003). Therefore, we also examined the effects of the K519/E127 combinatorial mutations on the CPA inhibition kinetics. Consistent with the previous report, the K519E mutation indeed slows down the CPA inhibition kinetics (Fig. 7 A, top). Analysis of the CPA inhibition rate (1/τ) between E166C and E166C/K519E mutants (Fig. 7 A, bottom) shows that the K519E mutation reduces the apparent kon and koff by approximately six- and twofold, respectively, at −160 mV. Fig. 7 (B and C) displays similar comparisons among various combinatorial mutations of K519 and E127, all in the background of the E166C mutation. It can be seen that the major effect caused by the mutations of E127 or K519 is a change of kon. These mutations also change the koff of CPA block, although such a change is not easily discerned at the very negative voltage of −160 mV (Fig. 7, B and C).
Figure 7.

Effects of mutations at the intracellular pore entrance on the kon and koff of the CPA block. All mutants were with the E166C background mutation. (A) Recording traces of the E166C (black trace) and the E166C/K519E (red trace) mutants. On the left are the original current traces of these two mutants obtained by stepping the membrane voltage from +80 to −160 mV. On the right, the initial current amplitude (after the capacitance spike) of the E166C/K519E mutant at −160 mV is scaled to the same initial value as that of the E166C mutant. The inverse of the time constant of the current inhibition course is plotted against [CPA] in the bottom panel to evaluate kon and koff according to Eq. 3. (B) Analysis of the apparent kon and koff of the CPA block at −160 mV among three channels in which K, M, and E are at position 519 of the E166C mutant, respectively. The fitted kon and koff values were: E166C/K519: 6.3 × 105 M−1s−1 and 17.2 s−1; E166C/K519M: 1.7 × 105 M−1s−1 and 10.5 s−1; and E166C/K519E: 1.1 × 105 M−1s−1 and 9.5 s−1. Notice that both kon and koff are reduced. Thus, the effects of these mutations on the apparent blocker affinity (shown in Fig. 6) are small. (C) Comparison of the apparent kon and koff of the CPA block at −160 mV among four E127Q/K519M combinatorial mutations in the E166C background. The kon and koff values were: E166C/E127Q: 7.6 × 105 M−1s−1 and 17.9 s−1; and E166C/E127Q/K519M: 4.6 × 105 M−1s−1 and 11.4 s−1.
To examine the effect of the E127/K519 mutations on the koff of the CPA block more directly, we compare the current recovery from the CPA block at +80 mV (Fig. 8 A). The time constant of the current recovery is nearly independent of [CPA] (Fig. 8 B), indicating that the relaxation rate of the current at this positive voltage (which is carried by inward Cl− movement) is dominated by the koff of the CPA block. Both K519M and K519E mutations reduce the current recovery rate (koff) at +80 mV (Fig. 8, A and B). On the other hand, the E127Q mutation can reverse the effects of the K519M and K519E mutations on koff (Fig. 8 B). The fact that the E127Q mutation can recover the reduction of kon (Fig. 7) and koff (Fig. 8) caused by K519 mutations recapitulates the collaborating control of E127 and K519 on the single-channel conductance of CLC-0 (Chen and Chen, 2003). These results, together with those obtained in mutants of E166, are reminiscent of the blocking mechanism shown in the Shaker K+ channel block by long-chain QA compounds or by the inactivation ball peptide (see Discussion for details).
Effects of Extracellular Anions on the CPA Block
The K+ channel block by the intracellularly applied inactivation ball peptide and long-chain QA compounds has been extensively studied (Armstrong, 1971; Zagotta et al., 1990; Foster et al., 1992; Choi et al., 1993; Murrell-Lagnado and Aldrich, 1993a,b; Zhou et al., 2001). One hallmark of this blocking mechanism is that the block can be reduced by permeant ions on the other side of the membrane (Armstrong, 1971). Reducing the extracellular Cl− concentration ([Cl−]o) has been known to increase the CPA affinity for wild-type CLC-0 (Accardi and Pusch, 2003). In pore-open mutants, reducing [Cl−]o also increases the CPA block. Fig. 9 A shows the steady-state CPA-blocking affinities of the E166A mutant at various negative voltages. The reduction of [Cl−]o from 140 to 15 mM leads to an approximately twofold increase of the apparent affinity (Fig. 9 A). We further evaluated the effect of extracellular Cl− on the current recovery rate at positive voltages, which is dominated by koff. Fig. 9 B shows that lowering [Cl−]o slows down the recovery from the CPA block. In contrast, reducing intracellular Cl− concentration ([Cl−]i) has no effect. These results (also see the averaged data in Fig. 9 C) are supportive of the trans-ion effect on the CPA block of the CLC-0 pore.
Blocking Pore-open Mutants of CLC-0 by Fatty Acids
The mechanism of the Shaker K+ channel block by long-chain QA molecules or by the inactivation ball peptide was proposed to consist of two blocking steps: an initial step for the hydrophilic end of the blocker to “dock” at the intracellular pore entrance, followed by an interaction of the hydrophobic part of the blocker with the pore (Zhou et al., 2001). Like long-chain QA compounds and the inactivation ball peptide, CPA is an amphiphilic molecule. This blocker may use its negatively charged end to dock at the superficial site, and the more hydrophobic part of the molecule (the benzene ring) then enters the pore to interact with deep pore residues. We suspect that such a general blocking mechanism does not require a specific structural fit between the blocker and the binding site because long-chain QAs and the inactivation ball peptide differ in their detailed molecular structures. Accordingly, we tested whether other amphiphilic molecules, such as fatty acids, can block the pore of CLC-0. Fig. 10 (A and B) shows a comparison of the block of the E166A mutant by intracellular octanoate (Fig. 10 A) and butyrate (Fig. 10 B), each at 300 μM. The dose-dependent inhibition curves of various fatty acids on the E166A mutant are shown in Fig. 10 C. Fatty acids with a long aliphatic chain, such as octanoate, decanoate, and dodecanoate, have their K1/2's in the low μM range, whereas the K1/2's of shorter compounds, such as propionate and butyrate, are in the mM range. In contrast, the blocking affinities of these fatty acids for the E166Q mutant are much lower; even the highest-affinity compound, octanoate, has a K1/2 near 1 mM (Fig. 10 D). However, CPA and various fatty acids produce a block with similar voltage dependence in E166A and E166Q, with a slope of ∼0.5–0.9 (Fig. 10 E) and ∼0.4∼0.6 (Fig. 10 F), respectively.
Figure 10.

Inhibitions of the E166A and E166Q mutants by fatty acids. (A and B) Inhibitions of E166A by 300 μM octanoate (OA) and butyrate (BA). (C) Dose-dependent inhibition curves of the E166A mutant by various fatty acids at −140 mV. The color-coded symbols are as follows: black, proprionate (PA); red, butyrate (BA); green, hexanoate (HA); blue, octanoate (OA); cyan, decanoate (DeA); magenta, dodecanoate (DoA). (D) Dose-dependent inhibition curves of the E166Q mutant by various fatty acids. The color representations are the same as in C. (E and F) Voltage dependence of the K1/2 of the E166A and E166Q mutants by various amphiphilic blockers. Data points are connected by straight lines. The color representations are the same as those in C and D. For comparison, yellow symbols represent the apparent K1/2 of CPA. The slopes of the fitted straight lines (not shown) are as follows: E166A: PA, 0.56; BA, 0.61; HA, 0.74; OA, 0.90; DeA, 0.67; DoA, 0.51; CPA, 0.88; and E166Q: PA, 0.57; BA, 0.42; HA, 0.61; OA, 0.61; DeA, 0.41; DoA, 0.36; CPA, 0.45.
Plotting the half-blocking concentrations as a function of the carbon number reveals that the length of these compounds is critical for the blocking affinity. For both E166A and E166Q mutants, the affinity increases with the length of the aliphatic chain (Fig. 11 A), a phenomenon similar to that reported in the block of Shaker K+ channel by long-chain QA compounds (Choi et al., 1993). However, when the fatty acid is longer than octanoate, the effect seems to be saturated.
Figure 11.

Blockade of E166A and E166Q by various amphiphilic reagents. (A) Comparison of the steady-state K1/2 of various fatty acids in the E166A and E166Q mutants. Vm = −140 mV. The horizontal axis represents the carbon number (or the length of the hydrophobic tail) in these compounds: 3 carbons (PA); 4 carbons (BA); 6 carbons (HA); 8 carbons (OA); 10 carbons (DeA); 12 carbons (DoA). (B) Comparison of the steady-state blocking affinities of fatty acids and alkyl sulfonates in the E166A mutant. Vm = −140 mV. The alkyl sulfonate compounds are structurally similar to fatty acids except that the charged end is a SO3− group. (C) The E166A mutant is also sensitive to SDS. Both recordings were obtained from the same patch.
Blocking Pore-open Mutants with Various Amphiphilic Compounds
To further examine whether an exact structural fit between the blocker and the pore is required, we tested the blocking effect of another group of amphiphilic molecules, the alkyl sulfonic acids. Alkyl sulfonates have the same aliphatic chain as that of the corresponding fatty acids, but they differ from the fatty acid molecules in the negatively charged end: a sulfonate versus a carboxylate. These alkyl sulfonate compounds block the E166A mutant with similar affinities to those of the fatty acids of similar lengths (Fig. 11 B). Another amphiphilic molecule, dodecyl sulfate, also blocks the E166A mutant with a high affinity. 5 μM SDS is able to block the current at −160 mV by ∼50% (Fig. 11 C). On the other hand, the same concentration of SDS has a very limited effect on E166Q (not depicted). Octanol, a molecule similar to octanoate and octanosulfonate except for the absence of the negatively charged end, blocks E166A with a low affinity, and the block has no voltage dependence (not depicted). This indicates that the charge of the blocker molecule is also important to block the pore-open mutants of CLC-0, similar to the previous finding on the wild-type CLC-1 channel (Liantonio et al., 2003).
Because the fatty acid block appeared to be similar to the CPA block, we further compared octanoate affinities with CPA affinities in various mutants of E166. Table III summarizes K1/2's of the octanoate block at various voltages in different mutants. The relationships between K1/2 and the side chain volume of the replacing amino acid (Fig. 12) are similar to those shown in Fig. 3 B. Overall, octanoate affinities are approximately threefold higher than CPA affinities in various mutants. However, for E166H, E166F, E166Y, and E166W, in which the side chain of the introduced residue contains a ring structure, the octanoate affinities are significantly lower than the CPA affinities (compare red squares in Fig. 12 with those in Fig. 3 B). Consequently, the K1/2's of octanoate in various mutants perfectly fit into two categories: hydrophobic and hydrophilic residues, respectively. We speculate that the interaction between the aromatic ring on the CPA molecule and the ring structure of the side chain of histidine, phenylalanine, tyrosine, and tryptophan may provide extra interaction energy. This possibility further strengthens the argument for a direct interaction of the side chain of the residue at position 166 with the benzene ring of the blocker molecule.
TABLE III.
Apparent Octanoate Affinity (K1/2) of Various Mutants of E166
| −160 mV | −140 mV | −120 mV | −100 mV | −80 mV | −60 mV | −40 mV | |
|---|---|---|---|---|---|---|---|
| E166G | 0.175 ± 0.019a | 0.115 ± 0.014 | 0.082 ± 0.008 | 0.075 ± 0.006 | 0.085 ± 0.006 | 0.127 ± 0.008 | 0.225 ± 0.013 |
| E166A | 0.0019 ± 0.0001 | 0.0028 ± 0.0002 | 0.0047 ± 0.0002 | 0.0082 ± 0.0002 | 0.0157 ± 0.0007 | 0.0297 ± 0.0014 | 0.0643 ± 0.0037 |
| E166S | 0.016 ± 0.002 | 0.026 ± 0.003 | 0.050 ± 0.003 | 0.100 ± 0.009 | 0.198 ± 0.011 | 0.403 ± 0.022 | 0.855 ± 0.037 |
| E166C | 0.0020 ± 0.0005 | 0.0035 ± 0.0008 | 0.0056 ± 0.0011 | 0.0102 ± 0.0018 | 0.0207 ± 0.0034 | 0.0431 ± 0.0064 | 0.0885 ± 0.0171 |
| E166N | 0.125 ± 0.002 | 0.212 ± 0.004 | 0.380 ± 0.009 | 0.694 ± 0.018 | 1.335 ± 0.029 | 2.551 ± 0.047 | 4.93 ± 0.18 |
| E166 | 0.19 ± 0.04 | 0.26 ± 0.03 | 0.30 ± 0.02 | 0.39 ± 0.01 | 0.67 ± 0.05 | 1.29 ± 0.13 | 2.61 ± 0.30 |
| E166V | 0.0060 ± 0.0004 | 0.0102 ± 0.0008 | 0.0184 ± 0.0015 | 0.0353 ± 0.0028 | 0.0672 ± 0.0037 | 0.1258 ± 0.0065 | 0.231 ± 0.014 |
| E166Q | 0.88 ± 0.09 | 1.45 ± 0.10 | 2.53 ± 0.16 | 4.21 ± 0.24 | 6.94 ± 0.36 | 11.41 ± 0.69 | 17.8 ± 1.5 |
| E166H | 4.57 ± 0.28 | 7.02 ± 0.40 | 11.47 ± 0.57 | 18.39 ± 0.81 | 29.55 ± 1.30 | ND | ND |
| E166I | 0.009 ± 0.001 | 0.015 ± 0.002 | 0.026 ± 0.003 | 0.045 ± 0.007 | 0.079 ± 0.013 | 0.136 ± 0.026 | 0.218 ± 0.049 |
| E166L | 0.011 ± 0.001 | 0.018 ± 0.002 | 0.032 ± 0.003 | 0.057 ± 0.005 | 0.106 ± 0.010 | 0.192 ± 0.022 | 0.342 ± 0.053 |
| E166K | 7.24 ± 0.33 | 10.62 ± 0.45 | 15.36 ± 0.50 | 21.65 ± 0.54 | 29.24 ± 0.53 | ND | ND |
| E166F | 0.091 ± 0.001 | 0.150 ± 0.004 | 0.254 ± 0.007 | 0.455 ± 0.015 | 0.803 ± 0.050 | 1.63 ± 0.16 | 3.09 ± 0.56 |
| E166Y | 3.36 ± 0.18 | 4.99 ± 0.31 | 7.43 ± 0.41 | 11.16 ± 0.54 | 16.70 ± 0.65 | ND | ND |
| E166W | 3.50 ± 0.59 | 5.53 ± 0.87 | 8.6 ± 1.3 | 13.0 ± 2.0 | 19.2 ± 3.3 | ND | ND |
The K1/2 values are presented as mean ± SEM (in mM).
Figure 12.

Steady-state octanoate blocking affinities (at −140 mV) of various pore-open mutants of CLC-0. The octanoate affinities of various mutants are approximately threefold higher than the CPA affinities in Fig. 3 B, except for the four mutants labeled by red solid squares. Also notice that in this plot, the hydrophobic (with a positive hydrophobic index) and the hydrophilic (with a negative hydrophobic index) residues are easily separated into two distinct groups.
Octanoate Also Shifts the Fast Gate Po-V Curve of Wild-type CLC-0
Because the octanoate block of pore-open mutants of CLC-0 appears to be similar to the CPA block, we investigated if octanoate, like CPA, can shift the fast gate Po-V curve of wild-type CLC-0. The fast gate Po-V curves of wild-type CLC-0 in the absence and presence of various concentrations of octanoate were constructed from measuring the initial tail current following various tested voltages as described in previous studies (Chen and Chen, 2001; Zhang et al., 2006). Fig. 13 shows that octanoate indeed shifts the fast gate Po-V curve toward more depolarized voltages, and the degree of shift is roughly similar to that obtained by using the same concentration of CPA. These results suggest that the effect of shifting the fast gate Po-V curve of the wild-type CLC-0 is likely related to the pore-blocking mechanism.
Figure 13.

The effect of octanoate in shifting the fast gate Po-V curve of the wild-type CLC-0 channel.
DISCUSSION
The CPA block on the wild-type CLC-0 and the E166A mutant has been extensively studied (Accardi and Pusch, 2003; Traverso et al., 2003), but the block on other mutants of E166 has not been examined. The results of the E166A mutant and the wild-type CLC-0 channel shown in the current study are in agreement with those in the previous study; E166A has a CPA-blocking affinity several hundred–fold higher than that of the wild-type channel, and the rate of the CPA inhibition shows saturation at high [CPA] (Fig. 4 B), suggesting that the block cannot be a two-state bimolecular reaction.
Three-state CPA-blocking Models
Previous studies have already suggested that a model with at least three states is required to explain both the steady state and the kinetic properties of the CPA block (Traverso et al., 2003). It was thought that the fast gating of the wild-type CLC-0 includes, besides the swing of the side chain of E166, a conformational change in the pore, and that CPA binds to the intracellular pore region in a state-dependent way (Accardi and Pusch, 2003; Traverso et al., 2003). Although the negatively charged side chain is removed in the E166A mutant, the conformational change in the pore region intracellular to E166 might be retained, and the CPA block of E166A could still be a state-dependent block described by the model shown in Scheme 1 (Traverso et al., 2003):
 |
(SCHEME 1) |
This model (Model A) suggests that CPA first binds to the pore region intracellular to the gate, and the CPA-bound pore undergoes a conformational change between the open and blocked (OB) state and the closed and blocked (CB) state, so that the channel has a much higher affinity for CPA in the CB state than in the OB state. When the membrane potential is hyperpolarized, the reaction in Scheme 1 is tilted to the right, leading to a higher apparent CPA-blocking affinity.
An alternative model (Model B) proposed for the CPA effect on E166A mutant can be a mechanism similar to that found in the block of voltage-gated K+ channels by the inactivation ball peptide or by long-chain QA compounds (Zhou et al., 2001). The essence of this model is that the blocker first binds to the protein surface (or the pore entrance), producing a “pre-blocked” state. The blocker then inserts its hydrophobic region to interact with a deeper site in the pore:
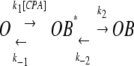 |
(SCHEME 2) |
In Scheme 2, the binding of the blocker to the protein surface or the pore entrance (the transition between O and OB* states) can be called a “docking” step. Results from the K+ channel-blocking experiments showed that alterations in the hydrophobic region in either the channel or the blocker had a large energetic effect, expressed as the change in the apparent dissociation rate (1/τoff), whereas those in the hydrophilic region, although altering both the association and dissociation rates, only modestly change the apparent affinity (Murrell-Lagnado and Aldrich, 1993a,b; Zhou et al., 2001).
Both Schemes 1 and 2 predict identical apparent association rates (kon), dissociation rates (koff), and the apparent half-blocking concentration (K1/2) for the block:
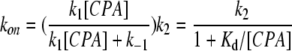 |
(4) |
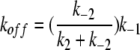 |
(5) |
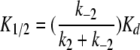 |
(6) |
In these equations, k1, k−1, k2, and k−2 represent the kinetic parameters shown in Schemes 1 and 2. K1/2 is the experimentally determined steady-state half-inhibition concentration, and Kd = k−1/k1 can be viewed as the binding affinity in Model A or as the docking affinity in Model B. Although kon, koff, and K1/2 have identical expressions for Schemes 1 and 2, these two schemes represent very different blocking mechanisms. The major difference is that the state dependence in Scheme 1 results from a gating-conformational change between OB and CB states (Traverso et al., 2003), whereas that in Scheme 2 is due to a movement of the blocker from a docking site at the pore entrance to a deeper hydrophobic site (Zhou et al., 2001). These two models had been compared in the previous study of the CPA block of E166A, with a final conclusion that Scheme 1 was a favored CPA-blocking model (Traverso et al., 2003).
The CPA Block of Pore-open Mutants of CLC-0 Is More Consistent with Model B
Our experimental results show that the blockers used in this study, which are amphiphilic molecules, appear to directly interact with the side-chain of the residue introduced at position 166. This conclusion comes from several observations. First, the steady-state K1/2 of the block in various mutants of E166 appears to correlate with the side chain volume and the side chain hydrophobicity of the introduced amino acid; the apparent affinity increases with a smaller and more hydrophobic side chain. Second, the disproportional reduction of the octanoate-blocking affinities, compared with the CPA-blocking affinities, in E166H, E166Y, E166W, and E166F may suggest that π-electron interaction between CPA and the four amino acids, histidine, tyrosine, tryptophan, and phenylalanine, provides extra interaction energy, thus increasing the CPA-blocking affinity. Finally, we found that if the side chain of residue 166 is removed (such as in E166G), CPA and octanoate can easily punch through the pore (Zhang and Chen, 2008). This punch-through effect suggests that CPA and fatty acid can indeed reach the deep region of the CLC-0 pore.
Thus, placing different amino acids at position 166 likely changes a hydrophobic interaction between the blocker and the pore, therefore altering the apparent affinity of the blocker significantly. In comparison, the charged mutations at the intracellular pore entrance (i.e., mutations of E127/K519 residues) also affect the block, but these mutations have a very different effect: the kinetics of the block is changed, although the alteration of the steady-state blocking affinity is only modest. It was also observed that mutations of K585 of CLC-1 (corresponding to K519 of CLC-0) had only a small effect on the blocking affinity of a related charged blocker, 9-anthracene carboxylic acid, although the block was examined in the wild-type CLC-1 channel (Estevez et al., 2003). In light of this characteristic pattern from mutating deep and superficial pore residues, we propose that the CPA block of the pore is more consistent with Model B. On the other hand, with the finding that various mutants of the E166 residue have different CPA affinities, Model A appears to be more difficult to apply because one might have to speculate that each mutant has a different conformational change in the intracellular pore region to explain the change of CPA affinity by three orders of magnitude.
Besides the comparison of the steady-state affinity of various mutants, kinetic evidence also suggests that Model B explains the block better. When residue 166 that likely contributes to the hydrophobic interaction is altered, the mutation significantly changes koff (Figs. 4 and 5), probably due to altering k−2 of Eq. 5. Eq. 6 shows that K1/2 can be altered by changing k−2, even if Kd is kept constant. The change of K1/2 by mutations of E166 is mostly due to the change of koff (Fig. 4), a situation similar to altering the hydrophobic interaction between the K+ channel pore and its blockers (Zhou et al., 2001). On the other hand, the mutation at the pore entrance, the site for the initial blocker docking, alters both kon and koff. The mutation effect at the docking site appears to have some electrostatic components because E127Q can reverse the effect of K519M or K519E mutations, similar to the control of E127 and K519 on the channel conductance (Chen and Chen, 2003). Thus, the interaction between the docking site and the blocker appears to be more hydrophilic than that between the blocker and the deep blocking site. The modest effect on the apparent blocker affinity by the mutations at the hydrophilic docking site is similar to the results from the K+ channel–blocking experiments as well (Zhou et al., 2001).
Additional evidence supporting Model B is the effect of permeant ions on the block. The trans-ion effect to reduce the affinity of the blocker is well documented in the K+ channel field (Armstrong, 1971; Demo and Yellen, 1991). It has also been reported in the intracellular CPA inhibition of the wild-type CLC-0 channel (Accardi and Pusch, 2003). In our study we demonstrated that increasing [Cl−]o decreased the affinity of the CPA block. This effect results from the increase of koff, as can be directly seen from the faster recovery rate of the CPA block (Fig. 9). In contrast, altering [Cl−]i did not significantly alter the recovery rate (Fig. 9). The lack of a significant effect of [Cl−]i on the CPA-blocking affinity of E166A had been noticed previously and was considered to be evidence against Model A (Traverso et al., 2003). On the other hand, Model B is consistent with the lack of a significant effect by changing [Cl−]i because the access of intracellular Cl− to the pore is controlled by E127 and K519 (Chen and Chen, 2003), and mutations of these two residues generate only small effects on the on and off rates, which tend to cancel out each other in altering the blocker affinity (Fig. 7).
Finally, the finding that other amphiphilic compounds can also block the pore-open mutants of CLC-0 with a similar mechanism is also consistent with Model B. We have systemically tested two groups of amphiphilic molecules: fatty acids and alkyl sulfonates, both of which are linear molecules with a negative charge on one end of a hydrophobic aliphatic chain. These various compounds all block pore-open mutants with similar voltage dependence, and the overall blocking patterns of these compounds are similar to those of the CPA block. For example, these compounds block the mutants with a hydrophobic residue at position 166 (such as E166A) quite well but have a much weaker effect on the mutants with a hydrophilic residue (such as E166Q) (Fig. 11 A).
A General Blocking Mechanism without a Precise Structural Fit between the Blocker and the Pore
Besides fatty acids and alkyl sulfonates, we have tested other amphiphilic compounds. The commonly used detergent, SDS, blocks E166A with a high affinity (Fig. 11C), comparable to the affinity of dodecanoate. Negatively charged methanethiolsulfonate (MTS) reagents, such as 2-sulfonatoethyl MTS and 5-sulfonatopentyl MTS can also reversibly block E166A (not depicted), although with lower affinities than that observed with SDS. These results suggest that the block may not require a precise structural fit between the blocker and the pore, an idea consistent with the fact that long-chain QAs and the inactivation ball peptide have similar affinities in the Shaker K+ channel (Zagotta et al., 1990; Choi et al., 1993).
Another characteristic property of the blocking mechanism represented by Model B is that the blocker affinity depends upon the length of the hydrophobic part of the blocker. Although octanoate, decanoate, and dodecanoate are quite potent to block E166A, shorter compounds, such as propionate and butyrate, are less potent. In general, the longer the aliphatic chain of the blocker, the higher the apparent blocking affinity (Fig. 11). Similar phenomena were documented in the long-chain QA block of voltage-gated K+ channels (Armstrong, 1971; Choi et al., 1993). The increase of the inhibition effect with blockers of higher hydrophobicity has also been observed in previous studies on the wild-type CLC-1 channel (Rychkov et al., 2001; Liantonio et al., 2003). Unlike the experiment with the Shaker K+ channel (Choi et al., 1993), however, the relation between K1/2 and the length of the aliphatic chain shown in this study appears to saturate at a length of 8–10 carbons (Fig. 11). We speculate that this may reflect the limited space in the CLC-0 pore to accommodate a molecule larger than octanoate or decanoate, and the saturation of the apparent affinity in longer blockers may result from a hydrophobic penalty in extending the molecule into a linear form to snake into the pore.
The observations that a precise structural fit between the blocker and the pore is not necessary and that the block occurs in cation and anion channels with unrelated pore architectures suggest that the blocking mechanism of Model B is quite general. The two determinants, the superficial docking site and the deep, hydrophobic-interaction site, are both important for the block. The deep hydrophobic site provides a sticky interaction for the blocker, resulting in a low off rate, whereas the charged docking site may help increase the on rate of the blocker. The role of the docking step becomes apparent when considering that the concentration of 10 μM corresponds to only a probability of 0.006 for finding a molecule in a 100-Å cube, the approximate volume directly under the channel protein. Although the probability for a blocker close to the pore entrance is small, CPA at this concentration can block the E166A current by ∼50%. The hydrophilic interaction thus may play a role in increasing the number of docking trials for the blocker to find the correct docking site through rapid thermal motions. It is therefore not surprising that the residues important for the CPA docking, namely E127 and K519, also exert an electrostatic effect to help recruit intracellular Cl− ions to the pore entrance (Chen and Chen, 2003; Lin and Chen, 2003).
The voltage dependence of the block is quite consistent among various amphiphilic compounds, even though these blockers have different molecular lengths (Fig. 10, E and F). If these blockers in the pore have an extended linear structure, the charged end should be located at different positions in the membrane electric field given that the hydrophobic end interacts with the side chain of residue 166. The fact that the block by these fatty acids does not show a monotonic change of voltage dependence renders the classical voltage-dependent model (Woodhull, 1973) less attractive. We speculate that at least part of the voltage dependence may come from the interaction of the blocker with the ions in the pore. The voltage dependence of the channel block can be explained by ion-blocker interactions (Heginbotham and Kutluay, 2004), and examples in the cation channel block have suggested voltage-dependent blocking mechanisms different from the Woodhull model (Spassova and Lu, 1998, 1999; Thompson and Begenisich, 2003).
Besides blocking the pore of CLC-0, the present study also shows that octanoate shifts the fast gate Po-V curve toward more depolarized voltages, an effect similar to that of CPA on the fast gating. The results thus suggest that the effect of CPA and octanoate on the fast gating may be closely related to the pore-blocking mechanism. This suggestion comports with the intimate coupling of the fast gating of CLC-0 with the permeant ion in the pore. The exact mechanism for how CPA and octanoate shift the Po-V curve of the CLC-0 fast gate awaits further studies. Although we cannot rule out the possibility that the fast-gating motion of CLC-0 is more complex than the proposed side chain movement of E166, our results argue against the use of the high CPA affinity in blocking E166A as evidence to support a large conformational change associated with the fast gating of CLC-0 because the CPA-blocking mechanism is similar to that of the Shaker K+ channel block by long-chain QAs or by the inactivation ball peptide.
Acknowledgments
We thank Drs. Robert Fairclough, Crina Nimigean, and Gary Yellen for critical readings and comments on the manuscript.
This work was supported by a research grant (GM065447) from National Institutes of Health to T.-Y. Chen. X.-D. Zhang is a postdoctoral fellow of the American Heart Association, Western affiliate.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: CB, closed and blocked; CPA, p-chlorophenoxy acetate; HEK, human embryonic kidney; MTS, methanethiolsulfonate; OB, open and blocked; QA, quaternary ammonium.
References
- Accardi, A., and M. Pusch. 2003. Conformational changes in the pore of CLC-0. J. Gen. Physiol. 122:277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M. 1969. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J. Gen. Physiol. 54:553–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.F., and T.Y. Chen. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.F., and T.Y. Chen. 2003. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 122:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.Y. 1998. Extracellular zinc ion inhibits ClC-0 chloride channels by facilitating slow gating. J. Gen. Physiol. 112:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.L., C. Mossman, J. Aube, and G. Yellen. 1993. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 10:533–541. [DOI] [PubMed] [Google Scholar]
- Demo, S.D., and G. Yellen. 1991. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 7:743–753. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E.B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Estevez, R., B.C. Schroeder, A. Accardi, T.J. Jentsch, and M. Pusch. 2003. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 38:47–59. [DOI] [PubMed] [Google Scholar]
- Foster, C.D., S. Chung, W.N. Zagotta, R.W. Aldrich, and I.B. Levitan. 1992. A peptide derived from the Shaker B K+ channel produces short and long blocks of reconstituted Ca(2+)-dependent K+ channels. Neuron. 9:229–236. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., and E. Kutluay. 2004. Revisiting voltage-dependent relief of block in ion channels: a mechanism independent of punchthrough. Biophys. J. 86:3663–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. 3rd Edition. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Kyte, J., and R.F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132. [DOI] [PubMed] [Google Scholar]
- Liantonio, A., A. De Luca, S. Pierno, M.P. Didonna, F. Loiodice, G. Fracchiolla, P. Tortorella, L. Antonio, E. Bonerba, S. Traverso, et al. 2003. Structural requisites of 2-(p-chlorophenoxy)propionic acid analogues for activity on native rat skeletal muscle chloride conductance and on heterologously expressed CLC-1. Br. J. Pharmacol. 139:1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.W., and T.Y. Chen. 2003. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J. Gen. Physiol. 122:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.W., C.W. Lin, and T.Y. Chen. 1999. Elimination of the slow gating of ClC-0 chloride channel by a point mutation. J. Gen. Physiol. 114:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, R., and C. Miller. 1988. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 91:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, R., and C. Miller. 1989. Mutant potassium channels with altered binding of charybdotoxin, a pore-blocking peptide inhibitor. Science. 245:1382–1385. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and G. Yellen. 1990. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 250:276–279. [DOI] [PubMed] [Google Scholar]
- Moran, O., S. Traverso, L. Elia, and M. Pusch. 2003. Molecular modeling of p-chlorophenoxyacetic acid binding to the CLC-0 channel. Biochemistry. 42:5176–5185. [DOI] [PubMed] [Google Scholar]
- Murrell-Lagnado, R.D., and R.W. Aldrich. 1993. a. Energetics of Shaker K channels block by inactivation peptides. J. Gen. Physiol. 102:977–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado, R.D., and R.W. Aldrich. 1993. b. Interactions of amino terminal domains of Shaker K channels with a pore blocking site studied with synthetic peptides. J. Gen. Physiol. 102:949–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch, M., A. Liantonio, L. Bertorello, A. Accardi, A. De Luca, S. Pierno, V. Tortorella, and D.C. Camerino. 2000. Pharmacological characterization of chloride channels belonging to the ClC family by the use of chiral clofibric acid derivatives. Mol. Pharmacol. 58:498–507. [DOI] [PubMed] [Google Scholar]
- Pusch, M., A. Accardi, A. Liantonio, L. Ferrera, A. De Luca, D.C. Camerino, and F. Conti. 2001. Mechanism of block of single protopores of the Torpedo chloride channel ClC-0 by 2-(p-chlorophenoxy)butyric acid (CPB). J. Gen. Physiol. 118:45–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov, G.Y., M. Pusch, M.L. Roberts, and A.H. Bretag. 2001. Interaction of hydrophobic anions with the rat skeletal muscle chloride channel ClC-1: effects on permeation and gating. J. Physiol. 530:379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassova, M., and Z. Lu. 1998. Coupled ion movement underlies rectification in an inward-rectifier K+ channel. J. Gen. Physiol. 112:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassova, M., and Z. Lu. 1999. Tuning the voltage dependence of tetraethylammonium block with permeant ions in an inward-rectifier K+ channel. J. Gen. Physiol. 114:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J., and T. Begenisich. 2003. External TEA block of shaker K+ channels is coupled to the movement of K+ ions within the selectivity filter. J. Gen. Physiol. 122:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverso, S., L. Elia, and M. Pusch. 2003. Gating competence of constitutively open CLC-0 mutants revealed by the interaction with a small organic inhibitor. J. Gen. Physiol. 122:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverso, S., G. Zifarelli, R. Aiello, and M. Pusch. 2006. Proton sensing of CLC-0 mutant E166D. J. Gen. Physiol. 127:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull, A.M. 1973. Ionic blockage of sodium channels in nerve. J. Gen. Physiol. 61:687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen, G., M.E. Jurman, T. Abramson, and R. MacKinnon. 1991. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 251:939–942. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1990. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 250:568–571. [DOI] [PubMed] [Google Scholar]
- Zamyatnin, A.A. 1972. Protein volume in solution. Prog. Biophys. Mol. Biol. 24:107–123. [DOI] [PubMed] [Google Scholar]
- Zhang, X.D., and T.Y. Chen. 2008. Amphiphilic blockers punch through a mutant CLC-0 pore. J. Gen. Physiol. 133:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.D., Y. Li, W.P. Yu, and T.Y. Chen. 2006. Roles of K149, G352, and H401 in the channel functions of ClC-0: testing the predictions from theoretical calculations. J. Gen. Physiol. 127:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M., J.H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]