

Abstract
Patients with severe infections almost invariably exhibit evidence of activation of the coagulation system. The lungs are amongst the most frequently affected organs during severe infection and sepsis. The abundant presence of intravascular and extravascular fibrin appears to be a specific hallmark of acute lung injury after sepsis. Tissue factor (TF) is regarded to be the primary initiator of coagulation in severe infection. Effective blockade of the TF pathway, either by recombinant TF pathway inhibitor or by anti-TF antibodies in experimental sepsis, attenuates lung injury and partially prevents pulmonary dysfunction. In addition, inhibition of the activity of TF prevents local activation of coagulation in models of pneumonia. The TF pathway can influence inflammatory signaling by activation of protease activated receptor-1 and -2. This review presents the most recent data on the crosstalk between TF-mediated coagulation and inflammation, with a specific emphasis on these processes in the lung.
Introduction
Disseminated intravascular coagulation (DIC) is an acquired syndrome that is characterized by intravascular activation of coagulation and loss of localization, and complications that compromise blood supply to organs. Bacterial infection is the most common cause of DIC, and both Gram-negative and Gram-positive organisms have been identified as causative pathogens, although coagulopathy may be more pronounced in infections caused by Gram-negative bacteria [1].
DIC is commonly seen in sepsis, and particularly in septic shock, in which the incidence is somewhere between 30% and 50% [2]. Understanding the pathogenetic mechanisms of the coagulation imbalance seen in DIC is particularly important, because of the role played by DIC in the development of multiple organ failure. Several concurrent mechanisms contribute to the pathogenesis of DIC. Bacterial lipopolysaccharide (LPS) and other bacterial products, as well as proinflammatory cytokines, all promote fibrin deposition through three main pathways [3,4]: tissue factor (TF)-mediated thrombin generation; dysfunctional physiological anticoagulant mechanisms; and impaired fibrin removal due to depression of the fibrinolytic system by plasminogen activator inhibitor-1. All three of these mechanisms contribute to fibrin deposition in DIC. Severely reduced anticoagulant capacity and inhibited fibrinolysis, in conjunction with a massive activation of coagulation, lead to excessive fibrin formation and consumption of clotting factors and inhibitors.
In recent years it has become clear that tightly regulated interactions exist between coagulation and inflammation during sepsis. In this review we focus on the role played by TF in the activation of coagulation and inflammation that occurs during severe infection, with a particular emphasis on these processes in the lungs.
Roles played by tissue factor and tissue factor pathway inhibitor in coagulation
Coagulation activation in sepsis is primarily driven by the TF pathway. TF, a 47 kDa transmembrane glycoprotein, is a potent stimulator of the extrinsic coagulation cascade and an essential mediator of coagulation [5,6]. It is not exposed to circulating blood in a resting state, but it becomes exposed on the surface of mononuclear cells and endothelial cells when they are stimulated by bacteria or by bacterial products such as LPS and proinflammatory cytokines. Alternatively, TF located at extravascular sites, such as on adventitial fibroblasts and vascular smooth muscle cells, can become exposed to blood at sites of vascular injury or disruption of the endothelium.
TF binds and activates factor VII. The TF-activated factor VII (FVIIa) complex that is generated after exposure of TF presenting cells to blood initiates coagulation activation by activating factor X. Activated factor X (FXa) allows conversion of prothrombin to thrombin, although this reaction occurs to a significant extent only after thrombin-induced feedback activation of factors VIII and V, which are nonenzymatic cofactors in the tenase and prothrombinase complexes, respectively. FXa activates prothrombin more than 105-fold more efficiently in conjunction with activated factor V than in the absence of activated factor V, resulting in abundant formation of fibrin and clotting [6].
Of note, besides in its traditional cell-associated form, TF antigen and procoagulant activity have also been detected in cell-free plasma [7]. Circulating TF resides in microparticles that can be shed from leukocytes, endothelial cells, vascular smooth muscle cells, and platelets [4,6]. Intravenous injection of LPS into healthy humans resulted in an increase in TF-containing microparticles of up to 800% [8]. Moreover, TF and associated procoagulant activity have been detected on microparticles derived from platelets and granulocytes in patients with meningococcal sepsis [9]. Recently, alternatively spliced TF (lacking exon 5) was discovered as a soluble form of TF, which circulates in blood and may exert pro-coagulant activity, expanding the concept of 'circulating TF' by a further element [10]. Alternatively spliced TF is released from endothelial cells upon stimulation with proinflammatory cytokines [10]. The plasma concentration of alternatively spliced TF may be of use as a clinical marker of inflammation-induced coagulation activation. The predominant post-translational mechanism by which TF activity is regulated is through so-called encryption and decryption, reflecting processes of self-association and dissociation, respectively [6]. Although the encrypted form of TF is inactive, the decrypted form has procoagulant activity.
Thrombin generation via the TF pathway is normally rapidly controlled by TF pathway inhibitor (TFPI). TFPI is predominantly expressed by endothelial cells (up to 90% of the total intravascular pool) and in addition by platelets [6,11]. TFPI binds with high-affinity to TF-FVIIa and FXa present in plasma and on endothelial cells. TFPI contains three Kunitz (K) domains, each of which plays a specific role in inhibiting TF (Figure 1). The K2 domain initially binds FXa, and then the TF-FXa complex binds to TF-FVIIa via the K1 domain. The role of the K3 domain is not fully understood but it is thought to be involved in endothelial binding. Notably, in humans TFPI mRNA is alternatively spliced to produce TFPIα and TFPIβ [11]. TFPIβ, in which the K3 domain is absent, is the main regulator of TF activity, although this isoform accounts for only 20% of total TFPI.
Figure 1.

The anticoagulant action of TFPI. (a) The structure of TFPIα. Adapted with permission from Monroe and Key [6]. The anticoagulant action of TFPI is a two-stage process. (b) The second Kunitz (K2) domain binds first to a molecule of Xa and deactivates it. (c) The first Kunitz domain (K1) rapidly binds to an adjacent TF-VIIa complex, preventing further activation of X. The formation of this quaternary compound is necessary for the inhibitory action of TFPI on the TF-VIIa complex. TFPI can bind TF-VIIa, but this interaction is weak. The TFPI-Xa interaction is strong, whereas the interaction between TFPI-Xa and TF-VIIa is very strong and results in an essentially irreversible complex: TFPI-Xa-TF-VIIa. TF, tissue factor; TFPI, tissue factor pathway inhibitor; VIIa, activated factor VII; X, factor X; Xa, activated factor X.
Inhibition by TFPI of the TF pathway occurs by TFPI forming a stable complex, preventing factor X activation and conversion of prothrombin to thrombin. The complex is formed by binding
FXa followed by FXa-dependent binding of TF-VIIa. Under physiological conditions, TFPI is attached to the endothelium via proteoglycans (glycosaminoglycans bound to a core protein), which facilitates its TF-FVIIa-factor X inhibiting properties on the endothelial surface [12]. In sepsis pro-inflammatory cytokines reduce the synthesis of glycosaminoglycans on the endothelial surface, which probably impairs TFPI function. Although studies of TFPI activity in situations of TF-induced coagulation have yielded contradictory results, an increase in plasma TFPI activity in meningococcal sepsis has been associated with more severe coagulation and mortality, supporting the hypothesis that TFPI works less efficiently when it is not attached to the endothelium [13]. The role played by endogenous TFPI in anticoagulation in sepsis is illustrated by the fact that immunodepletion of TFPI-sensitized rabbits to LPS-induced DIC and the generalized Shwartzman reaction [14,15].
The pivotal role of TF in activation of coagulation during a systemic inflammatory response syndrome, such as that produced by endotoxemia or severe sepsis, has been established in numerous experiments. Generation of thrombin in humans intravenously injected with a low dose of LPS was preceeded by an increase in TF mRNA levels in circulating blood cells, enhanced expression of TF on circulating monocytes, and release of TF-containing microparticles [8,16]. Similarly, baboons infused with a lethal dose of Escherichia coli exhibited sustained activation of coagulation, which was associated with enhanced expression of TF on circulating monocytes, and patients with severe bacterial infection expressed TF activity on the surface of peripheral blood mononuclear cells [17]. More importantly, various strategies that prevent activation of the VIIa-TF pathway in endotoxemic humans and chimpanzees, and in bacteremic baboons abrogated the activation of the common pathway of coagulation [18-23]. Accordingly, mice with an almost complete absence of TF had reduced coagulation, inflammation, and mortality relative to control mice upon administration of high-dose LPS [24]. Similarly, deficient TF expression by hematopoietic cells reduced LPS-induced coagulation, inflammation, and mortality, which suggests that hematopoietic cells are the major pathologic site of TF expression during endotoxemia [24].
The potential of recombinant TFPI as a therapeutic entity in severe bacterial infection has been demonstrated by a double-blind, randomized, placebo-controlled study in which two groups of eight healthy male volunteers received LPS, administered on two separate occassions, followed by one of two doses of TFPI (0.0125 mg/kg or 0.05 mg/kg) or placebo [18]. Administration of LPS resulted in activation of thrombin generation, as reflected by significant increases in the plasma levels of thrombin-antithrombin complexes (TATc). At the higher TFPI dose, the LPS-induced increase in thrombin-antithrombin complexes was almost completely prevented, and low-dose TFPI tended to decrease thrombin-antithrombin complex formation. Experiments in baboons have demonstrated that TFPI prevents coagulation activation during severe E. coli sepsis [20,22]. In these animal models of lethal sepsis, inhibition of coagulation using recombinant TFPI not only prevented DIC and fibrinogen consumption, but it also protected against death.
Tissue factor in the lungs during sepsis caused by extrapulmonary infection
A major role for TF in activating coagulation in the pulmonary compartment has been suggested [5,25]. In the lung, TF is expressed by alveolar macrophages and respiratory epithelial cells. E. coli sepsis in baboons resulted in a profound up-regulation of TF on alveolar epithelial cells, macrophages, and endothelium [26,27]. The notion that extrapulmonary infection may cause TF-dependent activation of coagulation in the lungs received support from a recent investigation conducted in patients with peritonitis [28]. Bronchoalveolar lavage (BAL) samples were obtained from these patients, who did not suffer from clinically detectable lung disease, on the day of surgery and – where possible – on days 2 and 3 thereafter. Remarkably, BAL fluid levels of TF and FVIIa were higher in patients with peritonitis than in control patients undergoing elective surgey or mechanical ventilation.
Inhibition of the TF pathway in experimentally induced sepsis in baboons was associated with markedly reduced lung injury, as reflected by histopathology, protein leak, wet/dry weight, and largely preserved lung function, as measured by gas exchange and lung compliance [29-31]. Accordingly, in rats challenged with high-dose LPS intravenously, recombinant TFPI not only attenuated activation of coagulation but also markedly diminished lung injury and pulmonary edema [32]. Moreover, in mice with abdominal sepsis caused by intra-peritoneal infection with E. coli, inhibition of the TF pathway by administration of the recombinant nematode anticoagulant protein c2 (rNAPc2) prevented not only systemic but also pulmonary coagulopathy [33]. The hypothesis that increased TF-dependent procoagulant activity in sepsis may be due, in part, to decreased expression or function of TFPI in the lung is supported by work recently conducted in septic baboons [27]. That study identified increased pulmonary levels of TF protein, mRNA, and activity, whereas TFPI protein, mRNA, and activity were concurrently reduced. Of interest, passive immunization against endogenous TFPI was associated with increased deposition of fibrin in the lung. These findings are consistent with a role for TF in pulmonary coagulation, whereas the relatively low levels of TFPI in septic lungs suggest an important role for TFPI in normal lung in preventing local coagulation.
The role of tissue factor in primary lung inflammation and infection
Although there are relatively little data on TF expression in acute respiratory distress syndrome (ARDS), one study has shown that in lungs TF is produced by alveolar macrophages and alveolar epithelial cells [34]. TF production by the alveolar compartment indicates that the alveolar epithelium expresses TF when it is exposed to inflammatory cytokines. Exposure of cultured alveolar epithelial cells (the cancer cell line A549) to human pulmonary edema fluid from patients with acute lung injury or ARDS induced TF expression in a dose-dependent and time-dependent manner; in contrast, little TF expression was seen in control lungs [34]. Although alveolar epithelial cells can produce TFPI, the amount of this endogenous protein is not sufficient to block increased TF procoagulant activity due to inflammation [35].
ARDS and pneumonia are associated with local activation of coagulation within the airways mediated by the TF pathway.
Patients with ARDS and/or pneumonia exhibited elevated TF and FVIIa activity in BAL fluid, together with decreased activity of the fibrinolytic system caused by enhanced release of plasminogen activator inhibitor-1 [36]. In accordance with this, patients who developed ventilator-associated pneumonia exhibited increased concentrations of thrombin-antithrombin complexes in lavage fluid collected from the airways, together with increases in soluble TF and FVIIa levels; these pro-coagulant changes were detected several days prior to clinical and microbiological diagnosis of pneumonia [37]. Of note, the procoagulant response to respiratory tract infection was found to occur in a compartmentalized manner; the changes in fibrin generation were restricted to the primary site of infection in patients with community-acquired pneumonia [38] or ventilator-associated pneumonia [39]. Additionally, a recent investigation provided evidence that mechanical ventilation per se can cause pulmonary activation of coagulation in healthy human lungs through activation of the TF pathway [40].
Experimental studies have revealed that bacterial antigens can directly activate the coagulation system in the lung. Although the host response to LPS, as a proinflammatory antigen of Gram-negative pathogens, has been well defined for many years, it was not until recently that progress was made in elucidating the pulmonary host response to infection with Gram-positive pathogens. Such studies have used exposure to lipoteichoic acid (LTA), a major constituent of the membrane of Gram-positive organism with many biologic properties in common with LPS [41,42]. In vitro and animal studies have shown that the cellular recognition and signaling receptors for LTA and LPS are Toll-like receptor 2 and 4, respectively [43]. Inhalation of nebulized LPS or bronchial instillation of LPS into a lung subsegment induced activation of coagulation in the bronchoalveolar space of healthy human volunteers [44-47]. This localized procoagulant response was accompanied by simultaneous downregulation of anticoagulant and fibrinolytic pathways, thereby further enhancing the procoagulant state in the lung [44-47]. The first data are now emerging regarding the human response to LTA in vivo [47]. In this study, segmental instillation was used to assess host responses in 24 healthy male volunteers. Using a flexible video bronchoscope, sterile saline was instilled into the right middle pulmonary lobe, followed by instillation of either LTA or LPS into the contralateral lung. This study provided the first evidence that LTA (like LPS) elicits an inflammatory response in the human lung together with local activation of coagulation. Indeed, instillation of LTA (or LPS) resulted in a marked dose-dependent increase in the number of cells recruited into BAL fluid, due to recruitment of neutrophils, whereas a concurrent rise in TATc and soluble TF levels reflected pulmonary coagulation activation [47,48].
Animal studies on acute lung injury and pneumonia in which the TF pathway was blocked have indicated that TF indeed plays a key role in pulmonary coagulopathy in these conditions. In rats that received LPS intratracheally, intravenous infusion of site-inactivated FVIIa (which effectively blocks the TF pathway) decreased intraalveolar inflammation and fibrin deposition, which was accompanied by decreased pulmonary protein leakage and cytokine release [49]. In a mouse model of community-acquired pneumonia in which Streptococcus pneumoniae was instilled intranasally, enhanced TF expression was observed in infected mice compared with control mice; inhibition of TF-FVIIa by rNAPc2 attenuated the procoagulant response in the lung, highlighting the central role played by TF in this process [38]. Investigation of the effect of natural anticoagulants in development of S. pneumoniae pneumonia in the rat revealed that administration of TFPI, activated protein C, and antithrombin all significantly limited procoagulant changes [50]. Of interest, of these anticoagulants, only antithrombin reduced the bacterial load and the associated histopathologic changes in the lungs of infected rats [50].
The link between coagulation and inflammation
Protease-activated receptors (PARs) are emerging as important mediators of cellular responses to tissue injury, and are implicated in crosstalk between coagulation and inflammation [51]. The archetypal PAR is PAR1, which is a seven transmembrane domain, G-protein-coupled receptor. The amino terminus of PAR1 contains a protease cleavage site that, once cleaved by thrombin, results in a new amino terminus. The new amino-terminal sequence, SFLLRN, acts as a tethered ligand and binds intramolecularly to the hepta-helical body of the receptor to effect transmembrane signaling and G-protein activation within the cell [51]. In total, four PARs (1 to 4) have been identified, each of which can be activated by several proteases, resulting in exposure of the 'hidden' ligand and signal transduction (Figure 2). Thrombin is an essential player in PAR activation because this enzyme can activate PAR1, PAR3, and PAR4.
Figure 2.
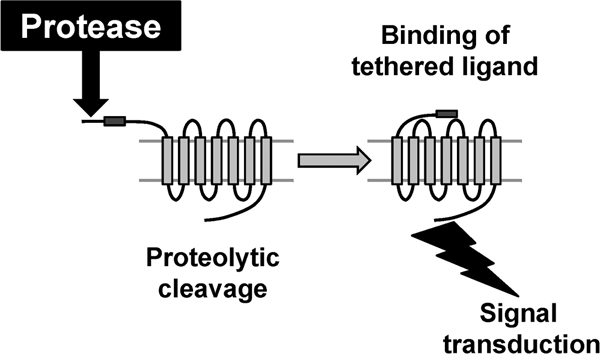
Mechanism of activation of PARs. Cleavage by a protease of the amino-terminal extracellular domain of PAR exposes the tethered ligand, which binds and activates the cleaved receptor. PAR, protease activated receptor.
The TF pathway can influence inflammation via interactions with PAR1 and PAR2 (Figure 3). The TF-FVIIa-FXa complex can signal through PAR1 and PAR2; in endothelial cell cultures stimulated with cytokines, signaling through PAR2 predominates [52]. Free FXa can also activate PAR1 and PAR2, although signaling of the ternary TF-FVIIa-FXa complex is at least fivefold more efficient [52]. Thus, although TF-FVIIa and FXa can activate PAR2 and PAR1 plus PAR2, respectively, relatively high concentrations are required, which has raised doubts regarding the biologic relevance of signaling by these upstream coagulation proteases. In contrast, FXa within the transient TF-FVIIa-FXa complex is a highly efficient activator of PAR1 and PAR2 at concentrations that are physiologically achievable [5]. Notably, because FXa exerts its procoagulant activity only after dissociation from TF-FVIIa, cell signaling by the TF-FVIIa-FXa complex precedes TF-dependent coagulation. As such, cell signaling via PAR1 and PAR2 is directly coupled to the mechanism by which TF initiates coagulation [5].
Figure 3.

Activation of PAR1 and PAR2 by the TF pathway. In the transient TF-VIIa-Xa complex, Xa is an efficient activator of PAR1 and PAR2. Downstream TF-dependent coagulation occurs after dissociation of Xa from this ternary complex. This essential procoagulant event results in less efficient PAR signaling because either TF-VIIa or free Xa is less capable of PAR activation than the TF-VIIa-Xa complex. Adapted with permission from Ruf and Riewald [5]. PAR, protease activated receptor; TF, tissue factor; VIIa, activated factor VII; Xa, activated factor X.
The roles played by the two PARs that are known to be influenced by the TF pathway in endotoxemia and sepsis, namely PAR1 and PAR2, have been the topic of several investigations. In an early study, mice deficient in either PAR1 or PAR2 administered high-dose LPS exhibited unaltered inflammation and survival [24]. However, a combination of thrombin inhibition by hirudin and PAR2 deficiency reduced inflammation and mortality [24]. In another investigation conducted in mice with LPS-induced shock, mice deficient for PAR1, PAR2, PAR4, or PAR1 plus PAR2 did not exhibit improved survival or decreased cytokine responses compared with wild-type mice [53]. Together these studies suggest that multiple PARs mediate crosstalk between coagulation and inflammation during endotoxemia.
Kaneider and coworkers [54] utilized cell penetrating peptides (so-called pepducins) to delineate the roles played by PAR1 and PAR2 in LPS shock and sepsis induced by cecal ligation and puncture. Using either agonistic or antagonistic pepducins targeted at PARs, they provided evidence that activation of PAR1 is harmful during the early phases of endotoxemia and sepsis, facilitating pulmonary leak and DIC, but that it becomes beneficial at later stages in a PAR2-dependent way by a mechanism that involves a time-dependent switch in the inflammatory functions of endothelial PAR1, dependent on the ability of PAR1 to transactivate PAR2. Specifically, LPS first induced barrier-disrupting effects on the vascular endothelium and activation of coagulation. However, subsequently – by a negative feedback loop – stimulation of the endothelium with LPS induced relocalization of PAR1-PAR2 complexes to the plasma membrane, whereupon PAR1 switched its signaling by transactivation of PAR2 protective pathways, resulting in barrier repair and inhibition of DIC.
This intriguing time-dependent switch of PAR1 from an exacerbating receptor to a protective one is consistent with studies showing that PAR1 deficiency conferred no net survival benefit in models of endotoxemia or sepsis [24,53,54]. Remarkably, PAR1 deficiency was reported to protect mice against LPS-induced lethality in a LD80 (80% lethal dose) model of endotoxemia [55]. Although PAR1-deficient mice initially developed levels of inflammatory markers indistinguishable from those in wild-type mice, they exhibited reduced late-stage inflammation, as indicated by a reduction in interleukin-6 and interleukin-1 levels 12 hours after LPS injection. When wild-type mice were treated with the thrombin inhibitor hirudin or a PAR1-specific antagonist 10 hours after LPS challenge, both interventions protected against lethality. Reconstitution of inflammation in PAR1-deficient mice could be achieved by adoptive transfer of wild-type bone marrow or of purified dendritic cells.
In a series of elegant experiments, support was provided for the hypothesis that a severe infection starts with a local inflammation resulting in systemic release of inflammatory mediators, which may cause vascular dysfunction [55]. This early response also involves a procoagulant response, which is TF mediated. In this early phase, coagulation and inflammation do not interact, but later there appears to be a tightly regulated interaction between coagulation and inflammation, driven by the interaction between thrombin and PAR1, and specifically PAR1 expressed by dendritic cells. These findings suggest a mechanism by which dendritic cells play a key role in lymphatic dissemination of coagulation and inflammation in deregulated innate immune responses during ongoing sepsis and endotoxemia [55]. Clearly, the studies on the roles played by PAR1 and PAR2 in endotoxic shock and sepsis discussed here [24,53-55] are not entirely consistent, although differences can be explained – at least in part – by differences in the severity of the insults.
With respect to the functions of PAR1 and PAR2 in the lungs, these receptors have been implicated in inflammatory and fibroproliferative processes in the pulmonary compartment [56]. PAR1 plays an important role in the acute inflammation and chronic fibrotic phase of bleomycin-induced lung injury; PAR1 deficiency was associated with less inflammatory cell recruitment, collagen accumulation, and expression of profibrotic factors in this model [57]. TF may be involved in this mechanism, considering that intratracheal gene transfer of TFPI not only attenuated pulmonary coagulation in the same model but also the associated fibrotic response [58]. Data on the role of PAR2 in lung inflammation are somewhat contradictory. Protective anti-inflammatory effects of PAR2 activation in mice were documented in rodent models of LPS-induced lung injury [59,60]. In contrast, intrapulmonary delivery of a PAR2-activating peptide resulted in enhanced inflammation and lung leakage in another study [61], and PAR2 was found to exert a similar proinflammatory role in models of asthma [62,63]. Among mice with Pseudomonas aeruginosa pneumonia, those with PAR2 deficiency exhibited diminished clearance of bacteria from the respiratory tract caused by reduced capacity of PAR2-deficient neutrophils to phagocytose and kill Pseudomonas [64].
It should be noted that TF inhibition did not result in attenuation of inflammation in all models examined. For example, during human endotoxemia, recombinant human TFPI completely prevented activation of coagulation while not influencing the release of proinflammatory cytokines into the circulation [18]. Conceivably, the crosstalk between TF-driven coagulation and PAR-driven inflammation is dependent – at least in part – on the severity of the primary insult and the organ that is affected.
Altogether, there is ample evidence for significant roles of PAR1 and PAR2 in sepsis and lung injury. Further studies are warranted to define more precisely the involvement of PAR1 and PAR2 signaling in these conditions and the contribution that the TF pathway makes in this setting.
Conclusion
Activation of coagulation is an important part of the initial host response to tissue injury and/or upon the first encounter with a pathogen. TF is the key mediator of coagulation activation. In addition, the TF pathway can influence inflammatory cell signaling via PAR1 and PAR2, in particular through the transient ternary TF-FVIIa-FXa complex. All components that contribute to the procoagulant and proinflammatory effects of the TF pathway are expressed in the lungs, which probably explains – at least in part – the lung-protective effects of TF inhibition in models of sepsis or lung injury. In the years to come it will be a challenge to unravel further the precise molecular mechanisms that contribute to the effects of activation of the TF pathway in the lungs and other organs.
Abbreviations
ARDS: acute respiratory distress syndrome; BAL: bronchoalveolar lavage; DIC: disseminated intravascular coagulation; FVIIa: activated factor VII; FXa: activated factor X; LPS: lipopolysaccharide; LTA: lipoteichoic acid; PAR: protease-activated receptor; rNAPc2: recombinant nematode anticoagulant protein c2; TF: tissue factor; TFPI: tissue factor pathway inhibitor.
Competing interests
The author has received research grant money from Novartis and Eli Lilly, the manufacturers of recombinant human tissue factor pathway inhibitor and recombinant human activated protein C, respectively. He is a consultant to Novartis and LeoPharma (manufacturer of recombinant human antithrombin).
Acknowledgments
Acknowledgements
This article is based on a presentation made at a satellite symposium, 'Severe community-acquired pneumonia update: mortality, mechanisms and medical intervention', held on 21 April 2008 in Barcelona, Spain as part of the 18th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID). It is published as part of Critical Care Volume 12 Supplement 6, 2008. The full contents of the supplement are available online at http://ccforum.com/supplements/12/S6
Publication of the supplement has been sponsored by Novartis.
References
- Opal SM, Garber GE, LaRosa SP, Maki DG, Freebairn RC, Kinasewitz GT, Dhainaut JF, Yan SB, Williams MD, Graham DE, Nelson DR, Levy H, Bernard GR. Systemic host responses in severe sepsis analyzed by causative microorganism and treatment effects of drotrecogin alfa (activated) Clin Infect Dis. 2003;37:50–58. doi: 10.1086/375593. [DOI] [PubMed] [Google Scholar]
- Dhainaut JF, Yan SB, Joyce DE, Pettilä V, Basson B, Brandt JT, Sundin DP, Levi M. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost. 2004;2:1924–1933. doi: 10.1111/j.1538-7836.2004.00955.x. [DOI] [PubMed] [Google Scholar]
- Levi M, Poll T van der. Two-way interactions between inflammation and coagulation. Trends Cardiovasc Med. 2005;15:254–259. doi: 10.1016/j.tcm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Schouten M, Wiersinga WJ, Levi M, Poll T van der. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol. 2008;83:536–545. doi: 10.1189/jlb.0607373. [DOI] [PubMed] [Google Scholar]
- Ruf W, Riewald M. Tissue factor-dependent coagulation protease signaling in acute lung injury. Crit Care Med. 2003;31(suppl):S231–S237. doi: 10.1097/01.CCM.0000057848.27456.04. [DOI] [PubMed] [Google Scholar]
- Monroe DM, Key NS. The tissue factor-factor VIIa complex: procoagulant activity, regulation, and multitasking. J Thromb Haemost. 2007;5:1097–1105. doi: 10.1111/j.1538-7836.2007.02435.x. [DOI] [PubMed] [Google Scholar]
- Giesen PL, Rauch U, Bohrmann B, Kling D, Roqué M, Fallon JT, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci USA. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aras O, Shet A, Bach RR, Hysjulien JL, Slungaard A, Hebbel RP, Escolar G, Jilma B, Key NS. Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood. 2004;103:4545–4553. doi: 10.1182/blood-2003-03-0713. [DOI] [PubMed] [Google Scholar]
- Nieuwland R, Berckmans RJ, McGregor S, Böing AN, Romijn FP, Westendorp RG, Hack CE, Sturk A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95:930–935. [PubMed] [Google Scholar]
- Szotowski B, Antoniak S, Rauch U. Alternatively spliced tissue factor: a previously unknown piece in the puzzle of hemostasis. Trends Cardiovasc Med. 2006;16:177–182. doi: 10.1016/j.tcm.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Crawley JT, Lane DA. The haemostatic role of tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2008;28:233–242. doi: 10.1161/ATVBAHA.107.141606. [DOI] [PubMed] [Google Scholar]
- Ott I, Miyagi Y, Miyazaki K, Heeb MJ, Mueller BM, Rao LV, Ruf W. Reversible regulation of tissue factor-induced coagulation by glycosyl phosphatidylinositol-anchored tissue factor pathway inhibitor. Arterioscler Thromb Vasc Biol. 2000;20:874–882. doi: 10.1161/01.atv.20.3.874. [DOI] [PubMed] [Google Scholar]
- Brandtzaeg P, Sandset PM, Joo GB, Ovstebo R, Abildgaard U, Kierulf P. The quantitative association of plasma endotoxin, antithrombin, protein C, extrinsic pathway inhibitor and fibrinopeptide A in systemic meningococcal disease. Thromb Res. 1989;55:459–470. doi: 10.1016/0049-3848(89)90054-6. [DOI] [PubMed] [Google Scholar]
- Sandset PM, Warn-Cramer BJ, Maki SL, Rapaport SI. Immunodepletion of extrinsic pathway inhibitor sensitizes rabbits to endotoxin-induced intravascular coagulation and the generalized Shwartzman reaction. Blood. 1991;78:1496–1502. [PubMed] [Google Scholar]
- Sandset PM, Warn-Cramer BJ, Rao LV, Maki SL, Rapaport SI. Depletion of extrinsic pathway inhibitor (EPI) sensitizes rabbits to disseminated intravascular coagulation induced with tissue factor: evidence supporting a physiologic role for EPI as a natural anticoagulant. Proc Natl Acad Sci USA. 1991;88:708–712. doi: 10.1073/pnas.88.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco RF, de Jonge E, Dekkers PE, Timmerman JJ, Spek CA, van Deventer SJ, van Deursen P, van Kerkhoff L, van Gemen B, ten Cate H, Poll T van der, Reitsma PH. The in vivo kinetics of tissue factor messenger RNA expression during human endotoxemia: relationship with activation of coagulation. Blood. 2000;96:554–559. [PubMed] [Google Scholar]
- Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. 1983;49:5–7. [PubMed] [Google Scholar]
- de Jonge E, Dekkers PE, Creasey AA, Hack CE, Paulson SK, Karim A, Kesecioglu J, Levi M, van Deventer SJ, Poll T van Der. Tissue factor pathway inhibitor dose-dependently inhibits coagulation activation without influencing the fibrinolytic and cytokine response during human endotoxemia. Blood. 2000;95:1124–1129. [PubMed] [Google Scholar]
- Taylor FB, Jr, Chang A, Ruf W, Morrissey JH, Hinshaw L, Catlett R, Blick K, Edgington TS. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ Shock. 1991;33:127–134. [PubMed] [Google Scholar]
- Creasey AA, Chang AC, Feigen L, Wun TC, Taylor FB, Jr, Hinshaw LB. Tissue factor pathway inhibitor reduces mortality from Escherichia coli septic shock. J Clin Invest. 1993;91:2850–2856. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M, ten Cate H, Bauer KA, Poll T van der, Edgington TS, Büller HR, van Deventer SJ, Hack CE, ten Cate JW, Rosenberg RD. Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J Clin Invest. 1994;93:114–120. doi: 10.1172/JCI116934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr C, Bild GS, Chang AC, Peer GT, Palmier MO, Frazier RB, et al. Recombinant E. coli-derived tissue factor pathway inhibitor reduces coagulopathic and lethal effects in the baboon gram-negative model of septic shock. Circ Shock. 1994;44:126–137. [PubMed] [Google Scholar]
- Taylor FB, Chang AC, Peer G, Li A, Ezban M, Hedner U. Active site inhibited factor VIIa (DEGR VIIa) attenuates the coagulant and interleukin-6 and -8, but not tumor necrosis factor, responses of the baboon to LD100 Escherichia coli. Blood. 1998;91:1609–1615. [PubMed] [Google Scholar]
- Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, Andrade-Gordon P, Frank RD, Mackman N. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood. 2004;103:1342–1347. doi: 10.1182/blood-2003-09-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M, Schultz MJ, Rijneveld AW, Poll T van der. Bronchoalveolar coagulation and fibrinolysis in endotoxemia and pneumonia. Crit Care Med. 2003;31(suppl):S238–S242. doi: 10.1097/01.CCM.0000057849.53689.65. [DOI] [PubMed] [Google Scholar]
- Drake TA, Cheng J, Chang A, Taylor FB., Jr Expression of tissue factor, thrombomodulin, and E-selectin in baboons with lethal Escherichia coli sepsis. Am J Pathol. 1993;142:1458–1470. [PMC free article] [PubMed] [Google Scholar]
- Tang H, Ivanciu L, Popescu N, Peer G, Hack E, Lupu C, Taylor FB, Jr, Lupu F. Sepsis-induced coagulation in the baboon lung is associated with decreased tissue factor pathway inhibitor. Am J Pathol. 2007;171:1066–1077. doi: 10.2353/ajpath.2007.070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Till JW, Levi M, Bresser P, Schultz MJ, Gouma DJ, Boermeester MA. Early procoagulant shift in the bronchoalveolar compartment of patients with secondary peritonitis. J Infect Dis. 2006;194:1331–1339. doi: 10.1086/508290. [DOI] [PubMed] [Google Scholar]
- Welty-Wolf KE, Carraway MS, Miller DL, Ortel TL, Ezban M, Ghio AJ, Idell S, Piantadosi CA. Coagulation blockade prevents sepsis-induced respiratory and renal failure in baboons. Am J Respir Crit Care Med. 2001;164:1988–1996. doi: 10.1164/ajrccm.164.10.2105027. [DOI] [PubMed] [Google Scholar]
- Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, Petersen LC, Piantadosi CA. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med. 2003;167:1200–1209. doi: 10.1164/rccm.200204-287OC. [DOI] [PubMed] [Google Scholar]
- Welty-Wolf KE, Carraway MS, Ortel TL, Ghio AJ, Idell S, Egan J, Zhu X, Jiao JA, Wong HC, Piantadosi CA. Blockade of tissue factor-factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol. 2006;290:L21–L31. doi: 10.1152/ajplung.00155.2005. [DOI] [PubMed] [Google Scholar]
- Enkhbaatar P, Okajima K, Murakami K, Uchiba M, Okabe H, Okabe K, Yamaguchi Y. Recombinant tissue factor pathway inhibitor reduces lipopolysaccharide-induced pulmonary vascular injury by inhibiting leukocyte activation. Am J Respir Crit Care Med. 2000;162:1752–1759. doi: 10.1164/ajrccm.162.5.9911018. [DOI] [PubMed] [Google Scholar]
- Weijer S, Schoenmakers SH, Florquin S, Levi M, Vlasuk GP, Rote WE, Reitsma PH, Spek CA, Poll T van der. Inhibition of the tissue factor/factor VIIa pathway does not influence the inflammatory or antibacterial response to abdominal sepsis induced by Escherichia coli in mice. J Infect Dis. 2004;189:2308–2317. doi: 10.1086/421031. [DOI] [PubMed] [Google Scholar]
- Bastarache JA, Wang L, Geiser T, Wang Z, Albertine KH, Matthay MA, Ware LB. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62:608–616. doi: 10.1136/thx.2006.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastarache JA, Wang L, Wang Z, Albertine KH, Matthay MA, Ware LB. Intra-alveolar tissue factor pathway inhibitor is not sufficient to block tissue factor procoagulant activity. Am J Physiol Lung Cell Mol Physiol. 2008;294:L874–L881. doi: 10.1152/ajplung.00372.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther A, Mosavi P, Heinemann S, Ruppert C, Muth H, Markart P, Grimminger F, Walmrath D, Temmesfeld-Wollbrück B, Seeger W. Alveolar fibrin formation caused by enhanced procoagulant and depressed fibrinolytic capacities in severe pneumonia. Comparison with the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2000;161:454–462. doi: 10.1164/ajrccm.161.2.9712038. [DOI] [PubMed] [Google Scholar]
- Schultz MJ, Millo J, Levi M, Hack CE, Weverling GJ, Garrard CS, Poll T van der. Local activation of coagulation and inhibition of fibrinolysis in the lung during ventilator associated pneumonia. Thorax. 2004;59:130–135. doi: 10.1136/thorax.2003.013888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijneveld AW, Weijer S, Bresser P, Florquin S, Vlasuk GP, Rote WE, Spek CA, Reitsma PH, Zee JS van der, Levi M, Poll T van der. Local activation of the tissue factor-factor VIIa pathway in patients with pneumonia and the effect of inhibition of this pathway in murine pneumococcal pneumonia. Crit Care Med. 2006;34:1725–1730. doi: 10.1097/01.CCM.0000218807.20570.C2. [DOI] [PubMed] [Google Scholar]
- Choi G, Schultz MJ, van Till JWO, Bresser P, Zee JS van der, Boermeester MA, Levi M, Poll T van der. Disturbed alveolar fibrin turnover during pneumonia is restricted to the site of infection. Eur Respir J. 2004;24:786–789. doi: 10.1183/09031936.04.00140703. [DOI] [PubMed] [Google Scholar]
- Choi G, Wolthuis EK, Bresser P, Levi M, Poll T van der, Dzoljic M, Vroom MB, Schultz MJ. Mechanical ventilation with lower tidal volumes and positive end-expiratory pressure prevents alveolar coagulation in patients without lung injury. Anesthesiology. 2006;105:689–695. doi: 10.1097/00000542-200610000-00013. [DOI] [PubMed] [Google Scholar]
- Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol. 2008;6:276–287. doi: 10.1038/nrmicro1861. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Maris NA, de Vos AF, Bresser P, Zee JS van der, Meijers JC, Lijnen HR, Levi M, Jansen HM, Poll T van der. Activation of coagulation and inhibition of fibrinolysis in the lung after inhalation of lipopolysaccharide by healthy volunteers. Thromb Haemost. 2005;93:1036–1040. doi: 10.1160/TH04-08-0492. [DOI] [PubMed] [Google Scholar]
- Maris NA, de Vos AF, Bresser P, Zee JS van der, Jansen HM, Levi M, Poll T van der. Salmeterol enhances pulmonary fibrinolysis in healthy volunteers. Crit Care Med. 2007;35:57–63. doi: 10.1097/01.CCM.0000249827.29387.4E. [DOI] [PubMed] [Google Scholar]
- Poll T van der, Levi M, Nick JA, Abraham E. Activated protein C inhibits local coagulation after intrapulmonary delivery of endotoxin in humans. Am J Respir Crit Care Med. 2005;171:1125–1128. doi: 10.1164/rccm.200411-1483OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogerwerf JJ, de Vos AF, Bresser P, Zee JS van der, Pater JM, de Boer A, Tanck M, Lundell DL, Her-Jenh C, Draing C, von Aulock S, Poll T van der. Lung inflammation induced by lipoteichoic acid or lipopolysaccharide in humans. Am J Respir Crit Care Med. 2008;178:34–41. doi: 10.1164/rccm.200708-1261OC. [DOI] [PubMed] [Google Scholar]
- Hoogerwerf JJ, de Vos AF, Levi M, Bresser P, Zee JS van der, Draing C, von Aulock S, Poll T van der. Activation of coagulation and inhibition of fibrinolysis in the human lung upon bronchial instillation of lipoteichoic acid and lipopolysaccharide. Crit Care Med. 2008. [DOI] [PubMed]
- Miller DL, Welty-Wolf K, Carraway MS, Ezban M, Ghio A, Suliman H, Piantadosi CA. Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intra-tracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 2002;26:650–658. doi: 10.1165/ajrcmb.26.6.4688. [DOI] [PubMed] [Google Scholar]
- Choi G, Hofstra JJ, Roelofs JJ, Rijneveld AW, Bresser P, Zee JS van der, Florquin S, Poll T van der, Levi M, Schultz MJ. Antithrombin inhibits bronchoalveolar activation of coagulation and limits lung injury during Streptococcus pneumoniae pneumonia in rats. Crit Care Med. 2008;36:204–210. doi: 10.1097/CCM.0b013e318168cdc3. [DOI] [PubMed] [Google Scholar]
- Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25–27. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA. 2001;98:7742–7747. doi: 10.1073/pnas.141126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerer E, Cornelissen I, Kataoka H, Duong DN, Zheng YW, Coughlin SR. Roles of protease-activated receptors in a mouse model of endotoxemia. Blood. 2006;107:3912–3921. doi: 10.1182/blood-2005-08-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. 'Role reversal' for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;452:654–658. doi: 10.1038/nature06663. [DOI] [PubMed] [Google Scholar]
- Sokolova E, Reiser G. A novel therapeutic target in various lung diseases: airway proteases and protease-activated receptors. Pharmacol Ther. 2007;115:70–83. doi: 10.1016/j.pharmthera.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Howell DC, Johns RH, Lasky JA, Shan B, Scotton CJ, Laurent GJ, Chambers RC. Absence of proteinase-activated receptor-1 signaling affords protection from bleomycin-induced lung inflammation and fibrosis. Am J Pathol. 2005;166:1353–1365. doi: 10.1016/S0002-9440(10)62354-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijiyama N, Ueno H, Sugimoto I, Sasaguri Y, Yatera K, Kido M, Gabazza EC, Suzuki K, Hashimoto E, Takeya H. Intratracheal gene transfer of tissue factor pathway inhibitor attenuates pulmonary fibrosis. Biochem Biophys Res Commun. 2006;339:1113–1119. doi: 10.1016/j.bbrc.2005.11.127. [DOI] [PubMed] [Google Scholar]
- Moffatt JD, Jeffrey KL, Cocks TM. Protease-activated receptor-2 activating peptide SLIGRL inhibits bacterial lipopolysaccharide-induced recruitment of polymorphonuclear leukocytes into the airways of mice. Am J Respir Cell Mol Biol. 2002;26:680–684. doi: 10.1165/ajrcmb.26.6.4693. [DOI] [PubMed] [Google Scholar]
- Morello S, Vellecco V, Roviezzo F, Maffia P, Cuzzocrea S, Cirino G, Cicala C. A protective role for proteinase activated receptor 2 in airways of lipopolysaccharide-treated rats. Biochem Pharmacol. 2005;71:223–230. doi: 10.1016/j.bcp.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Su X, Camerer E, Hamilton JR, Coughlin SR, Matthay MA. Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol. 2005;175:2598–2605. doi: 10.4049/jimmunol.175.4.2598. [DOI] [PubMed] [Google Scholar]
- Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, Gater PR, Geppetti P, Bertrand C, Stevens ME. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol. 2002;169:5315–5321. doi: 10.4049/jimmunol.169.9.5315. [DOI] [PubMed] [Google Scholar]
- Ebeling C, Forsythe P, Ng J, Gordon JR, Hollenberg M, Vliagoftis H. Proteinase-activated receptor 2 activation in the airways enhances antigen-mediated airway inflammation and airway hyperresponsiveness through different pathways. J Allergy Clin Immunol. 2005;115:623–630. doi: 10.1016/j.jaci.2004.11.042. [DOI] [PubMed] [Google Scholar]
- Moraes TJ, Martin R, Plumb JD, Vachon E, Cameron CM, Danesh A, Kelvin DJ, Ruf W, Downey GP. Role of PAR2 in murine pulmonary pseudomonal infection. Am J Physiol Lung Cell Mol Physiol. 2008;294:L368–L377. doi: 10.1152/ajplung.00036.2007. [DOI] [PubMed] [Google Scholar]