

Abstract
Twenty-two phages that infect Stenotrophomonas species were isolated through sewage enrichment and prophage induction. Of them, S1, S3, and S4 were selected due to their wide host ranges compared to those of the other phages. S1 and S4 are temperate siphoviruses, while S3 is a virulent myovirus. The genomes of S3 and S4, about 33 and 200 kb, were resistant to restriction digestion. The lytic cycles lasted 30 min for S3 and about 75 min for S1 and S4. The burst size for S3 was 100 virions/cell, while S1 and S4 produced about 75 virus particles/cell. The frequency of bacteriophage-insensitive host mutants, calculated by dividing the number of surviving colonies by the bacterial titer of a parallel, uninfected culture, ranged between 10−5 and 10−6 for S3 and 10−3 and 10−4 for S1 and S4. The 40,287-bp genome of S1 contains 48 open reading frames (ORFs) and 12-bp 5′ protruding cohesive ends. By using a combination of bioinformatics and experimental evidence, functions were ascribed to 21 ORFs. The morphogenetic and lysis modules are well-conserved, but no lysis-lysogeny switch or DNA replication gene clusters were recognized. Two major clusters of genes with respect to transcriptional orientation were observed. Interspersed among them were lysogenic conversion genes encoding phosphoadenosine phosphosulfate reductase and GspM, a protein involved in the general secretion system II. The attP site of S1 may be located within a gene that presents over 75% homology to a Stenotrophomonas chromosomal determinant.
Stenotrophomonas maltophilia, initially classified as Pseudomonas maltophilia (16) and later on as Xanthomonas maltophilia (34), is the main species of its genus and exists as aerobic, gram-negative rods with wide environmental distribution (8). It also behaves as an opportunistic human pathogen, causing nosocomial infections in immunocompromised patients; it has been isolated from the feces of 9.5% of oncology patients with associated diarrhea (1) and has also been involved in bacteremia and in respiratory, urinary, and soft-tissue infections (8, 25). The cases of pneumonia are especially worrying due to mortality rates of up to 50% (7). Most S. maltophilia strains are resistant to many broad-spectrum antimicrobial agents, including β-lactams such as amoxicillin-clavulanate, aztreonam and carbapenems, aminoglycosides, and ciprofloxacin. In fact, previous treatment with these antibiotics seems to be a factor predisposing toward Stenotrophomonas colonization (8, 19). Therefore, new approaches are needed for effective therapy of S. maltophilia infections. Among them, phage therapy may be envisaged as a possible alternative treatment.
The use of bacterial viruses as therapeutic agents was initiated in 1919, 3 years after their discovery, for the treatment of bacillary dysentery and continued up to the decade of the 1940s for the treatment of a variety of infectious diseases such as bubonic plague, cholera, and staphylococcal septicemia (9, 10, 23). These practices were interrupted by the advent of antibiotics, whose wider spectrum of action than that of phages opened the possibility to apply empirical treatments. However, the selection of antibiotic-resistant bacteria and the frequent occurrence of adverse effects associated with their use led to renewed interest in phages as therapeutic agents (5, 20, 27, 28, 33).
In addition, phages are very useful tools both for understanding the biology of their bacterial hosts, especially the regulation of gene expression, and for developing vectors that make their hosts amenable to recombinant DNA manipulation (18, 29).
Three phages active on S. maltophilia have been described previously in some detail. Two of them (φSMA5 and Smp14) are myoviruses with wide host ranges and genomes of about 250 and 160 kb that appear to be resistant to digestion by most restriction endonucleases (3, 4). The other one (φSMA9) has a 6.9-kb genome that harbors seven genes, the largest being an orthologue of the gene encoding the toxin Zot of the filamentous φLf bacteriophage of Vibrio cholerae (14).
In this communication, the isolation and characterization of several Stenotrophomonas phages are described. In addition, the sequencing of the genome of the first known temperate phage infecting S. maltophilia is reported.
MATERIALS AND METHODS
Microorganisms and culture conditions.
The 26 S. maltophilia strains used in this work as sources and as hosts of phages were obtained from the Spanish Type Culture Collection or from clinical samples (most of which were kindly provided by J. L. Martínez, Centro Nacional de Biotecnología, Spain) (Table 1).
TABLE 1.
Stenotrophomonas strains used in this work and host ranges of phages S1, S3, and S4
| Strain | Origin | Original species identification | Species classification after PCR genotyping (if different) | Other relevant feature(s) | Susceptibilitya to phage:
|
||
|---|---|---|---|---|---|---|---|
| S1 | S3 | S4 | |||||
| CECT 112 | Clinical sample | S. maltophilia | + | ||||
| CECT 113 | Clinical sample | S. maltophilia | |||||
| CECT 114 | Unknown | S. maltophilia | + | ||||
| CECT 115 | Oropharyngeal sample | S. maltophilia (type strain) | |||||
| CECT 757 | Sample from an infected monkey | Burkholderia pseudomallei | S. maltophilia | + | |||
| CECT 4793 | Water | S. maltophilia | Source of S1 | + | |||
| CECT 4091 | Forest soil | Burkholderia cepacia | S. maltophilia | ||||
| CECT 5308 | Vinegar | S. africana (type strain) | S. maltophilia | + | + | ||
| A1 | Sputum | S. maltophilia | Standard strain for propagation of S1 | + | + | ||
| 46021 | Sputum | S. maltophilia | + | + | |||
| C048 | Urine | S. maltophilia | + | ||||
| D388 | Urine | S. maltophilia | + | + | |||
| D457 | Bronchial aspirate | S. maltophilia | + | + | + | ||
| E301 | Urine | S. maltophilia | + | + | + | ||
| E539 | Pus from a wound | S. maltophilia | Source of S2; standard strain for propagation of S3 | + | + | ||
| E729 | Urine | S. maltophilia | |||||
| E759 | Sputum | S. maltophilia | |||||
| E923 | Sputum | S. maltophilia | + | + | |||
| E999 | Respiratory secretion | S. maltophilia | Host of S2 | + | + | ||
| F227 | Blood | S. maltophilia | Standard strain for propagation of S4 | + | + | ||
| F375 | Blood | S. maltophilia | + | + | |||
| F861 | Sputum | S. maltophilia | + | + | |||
| G51 | Blood | S. maltophilia | + | ||||
| C357 | Urine | S. maltophilia | |||||
| 5123-Z | Sputum | S. maltophilia | + | + | |||
| Ps6 | Burn specimen | Pseudomonas aeruginosa | S. maltophilia | ||||
+ indicates susceptibility.
Strain propagation was performed in 2× TY broth (22) under agitation (250 rpm) at 30°C. Solid medium contained 1.5% (wt/vol) agar (Roko). Phage enumeration was performed by the double-layer technique (22) using soft 2× TY (0.75% agar) to which 10 mM CaCl and 10 mM MgSO4 were added in the upper layer. Bacterial stocks were stored at −70°C in 2× TY supplemented with 20% (final concentration) glycerol.
Bacteriophage enrichment and isolation from sewage.
Six samples of water, obtained from the aeration and decantation tanks of local sewage plants, were incorporated into preparations of 2× TY liquid medium, which were used (unsterilized) to set up two successive enrichment cultures by inoculation with a mix of the 26 Stenotrophomonas strains (1% of an overnight culture of each strain). After overnight incubation at 30°C with shaking, the cultures were centrifuged and the supernatants were filtered through 0.22-μm-pore-diameter filters and used as a source of phages in a new enrichment step with the same mix of strains (in sterile medium). Supernatants of these second cultures were filtered and tested for the presence of inhibition halos by placing drops onto lawns of each of the 26 bacterial strains. Phages from the lysis zones were suspended in SM buffer (22) and diluted to get single plaques. The procedure was repeated twice, and the purified phages were stored at −70°C in SM buffer with 50% glycerol.
Phage isolation from lysogenic strains.
Exponential cultures of each Stenotrophomonas strain were treated with mitomycin C or with ciprofloxacin at the MIC for the strain and half the MIC (the MICs of mitomycin C ranged from 4 to 16 μg/ml, and those of ciprofloxacin ranged from 4 to 8 μg/ml). Supernatant aliquots of these cultures (5 μl) were placed onto lawns of each of the 26 bacterial strains, and after incubation, the lawns were inspected for the appearance of inhibition halos. Phages were isolated from the lysis zones by following the procedure described above.
Host range, phage purification, and functional studies.
Phage host ranges were determined by placing 5-μl drops of each viral suspension onto lawns of each of the 26 Stenotrophomonas strains. Large-scale phage propagation and purification through CsCl gradients were performed as described in reference 30.
Curves from one-step analyses of growth in 2× TY at a multiplicity of infection of 0.03 were generated as described in reference 15.
The frequency of emergence of bacteriophage-insensitive mutants (BIMs) was determined after the infection of exponential cultures at a multiplicity of infection of 10. The BIM frequency was calculated by dividing the number of surviving colonies by the bacterial titer of a parallel, uninfected culture.
The experiments described above were performed at least in triplicate.
Electron microscopy.
Phage gradient drops were placed onto 0.022-μm-pore-diameter filters floating on SM buffer at room temperature for 30 min (with two changes of buffer) to eliminate CsCl. Aliquots were negatively stained with 2% (wt/vol) uranyl acetate on copper grids covered with a film of carbon and ionized. Grids were observed with a JEOL 1200 EXII microscope stabilized at 100 KV. Images were obtained on Scientia photographic plates (AGFA).
DNA and protein techniques.
Phage DNA extraction from CsCl-purified virion suspensions dialyzed against SM buffer was performed as described in reference 12.
Pulsed-field gel electrophoresis was used to determine the sizes of phage genomes. Phage DNA was loaded into a 1% agarose gel in 0.5× Tris-borate-EDTA buffer (pH 8) and resolved using a CHEF-DR III pulsed-field system (Bio-Rad Laboratories, Hercules, CA) at 6 V/cm for 20 h and 14°C. Linear ramped pulse times of 3 to 63.8 s were employed.
Enzymatic treatments of phage DNA were carried out by following the instructions provided by the suppliers of the enzymes. DNA was visualized after separation in 0.7% agarose gels in Tris-borate-EDTA buffer and staining with ethidium bromide under UV light.
Genotypic identification of the 26 bacterial strains used in this work was performed by multiplex PCR using primers specific for S. maltophilia, the Burkholderia cepacia complex, and Pseudomonas aeruginosa, plus a pair targeting a universal 16S RNA stretch as a positive control (6). The reactions were set up with 25-μl aliquots using 2.5-μl samples of exponential cultures without any processing as DNA sources.
Phage structural proteins were extracted and purified as described in reference 12 and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with a Mini-PROTEAN III system (Bio-Rad). Proteins were stained with Coomassie R-250 blue. The bands of interest were excised manually and digested with porcine trypsin (Promega), and the resulting peptides were analyzed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry, essentially as described previously (13).
Phage genome sequencing and analysis.
Approximately 10 μg of S1 phage DNA was sheared by sonication, and segments were selected on the basis of size (2 to 3 kbp) and cloned into pUC18. Individual clones were sequenced and assembled. Trace assembly was done with the phredPhrap package (11). Generally, the assembled phage genome sequences had at least fivefold coverage in both orientations. Primer walking was sometimes used to close gaps in the sequence.
The cos sites were localized through analysis of the restriction patterns of ligated and unligated S1 DNA samples. To obtain the sequence of the single-stranded cos extensions, the ends of S1 DNA were sequenced directly using primers that pointed outwards. The nucleotide sequences obtained were aligned with that of the circular S1 genome. They overlapped by 12 bases that were considered to constitute the cosN site.
Open reading frames (ORFs) were predicted by the Clone Manager 7 and the ORF Finder (http://ncbi.nlm.nih.gov/gorf.html) software and by visual inspection. The primary nucleotide sequence in all reading frames was scanned for the start codons AUG and GUG, with a threshold of 50 codons. BLASTX and BLASTP (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) were used to search for proteins homologous to the ORF products. Structural predictions and motif searches were performed with InterProScan (http://www.ebi.ac.uk/InterProScan/), Pfam (http://pfam.sanger.ac.uk/search?tab=searchSequenceBlock), YASPIN (http://zeus.cs.vu.nl/programs/yaspinwww/), and TMHMM Server version 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/). Likely σ70 target promoter sequences were identified using BPROM (http://www.softberry.com). Putative terminator sequences were detected with the Terminator function of GCG (version 10.2). The search for putative tRNA-encoding genes was performed with tRNAscan-SE 1.21 (http://selab.janelia.org/tRNAscan-SE/). The prediction of +1 and −1 frameshifting was carried out with FSFinder 2 (http://wilab.inha.ac.kr/fsfinder2/).
Cumulative GC skew analysis with a system from In Silico Biology, Inc. (Yokohama, Japan) was used to locate the origin of replication.
Nucleotide sequence accession number.
The sequence of the phage S1 genome has been deposited in GenBank under accession number EU849489.
RESULTS
Phage isolation and host range determination.
The 26 Stenotrophomonas strains used in this work as sources and as hosts of phages were confirmed to belong to the genus by PCR genotyping (6). This genotyping allowed the reclassification of three isolates that were identified previously as Burkholderia cepacia, Burkholderia pseudomallei, and Pseudomonas aeruginosa by phenotypic methods.
Two procedures were undertaken for phage isolation: (i) the induction of resident prophages and (ii) the enrichment of sewage samples. In the first procedure, presumptive prophages were induced with mitomycin C or with ciprofloxacin and allowed to form plaques on lawns of each of the 26 bacterial strains. As a result, two phages, named S1 and S2, were isolated from S. maltophilia CECT 4793 and E539, respectively.
Complementarily, water samples taken from a sewage plant were used as potential sources of phages. By this procedure, two successive rounds of enrichment with a mix of all 26 Stenotrophomonas strains were applied prior to the placement of drops of the resulting cultures onto lawns of each strain. This procedure yielded 20 phages, which were named S3 to S22.
Of the two temperate phages isolated after the induction of lysogenic cultures, S1 inhibited the growth of four strains while S2 could be propagated only on its original indicator strain. A narrow host range was also the rule among the phages obtained from sewage, with two exceptions: S3 inhibited the growth of 12 strains, while S4 acted on 18 strains. The host ranges of these phages appeared to be confined to Stenotrophomonas, in that none were able to affect any of a collection of 14 Pseudomonas aeruginosa and 5 Burkholderia cepacia strains isolated from clinical samples (data not shown). The phages S1, S3, and S4 were chosen for further study because they had the widest host ranges within their categories.
Structure of the virions.
S1, S3, and S4 were propagated and purified through two successive CsCl gradients and observed under the electron microscope. All showed an icosahedral capsid and a tail, which allows their inclusion in the order Caudovirales (Fig. 1). S1 and S4 belong to the family Siphoviridae (they present a noncontractile tail), while S3 is a member of the family Myoviridae due to its contractile tail. The diameters of the capsids (means ± standard deviations) of S1, S3, and S4 were, respectively, 61.4 ± 1.35, 82.92 ± 1.25, and 87.5 ± 1.5 nm. The tail of S1, with a length of 129.2 ± 1.3 nm and a width of 9.93 ± 0.66 nm, is remarkable for its extreme flexibility, compared with the tail of S4 (201.87 ± 1.22 nm long and 10.7 ± 0.24 nm wide). The tail of S3 is 136.9 ± 0.59 nm by 21.43 ± 0.0 nm and has a basal plate with protruding spikes.
FIG. 1.

Transmission electron micrographs of the bacteriophage S1, S3, and S4 virions, negatively stained with 2% uranyl acetate.
The structural protein patterns were characteristic of each phage, as determined by SDS-PAGE. S1 showed two major bands with apparent sizes of 35 and 28 kDa (Fig. 2), S3 rendered two major polypeptides of 35 and 50 kDa and two less abundant species of 67 and 80 kDa, and S4 showed three proteins of 29, 32, and 36 kDa.
FIG. 2.
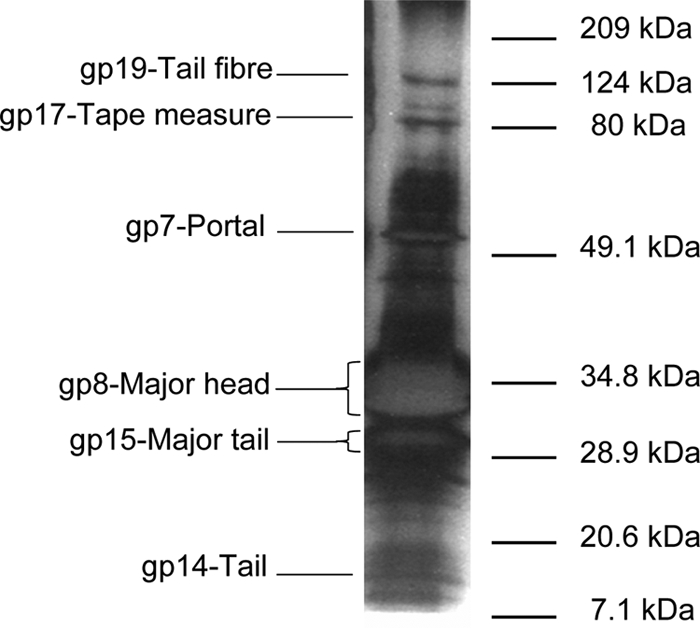
SDS-PAGE analysis of the S1 virion proteins. Those identified by mass spectrometry are indicated on the left. The unlabeled bands contained mixes of two or more structural proteins. The migration of the molecular mass markers is indicated on the right. The gel track was heavily overloaded to make evident the relationship between the tape measure and tail fiber proteins.
The genomic DNA of S1 was susceptible to restriction with BamHI, BglII, EcoRI, EcoRV, and HindIII, among other enzymes, allowing the determination of its length to be about 38.5 kb. The DNA molecules of S3 and S4 were not susceptible to restriction with any of the following enzymes: BamHI, BglII, DraI, EcoRI, EcoRV, HindIII, KpnI, NdeI, PacI, PstI, SacI, SalI, Sau3AI, SphI, and XbaI. The DNA of these phages remained unrestricted even when the phages had been propagated on alternative hosts to eliminate the possibility of host-induced DNA modification, suggesting that these phages either contain atypical bases or encode DNA-modifying enzymes that may protect them against restriction-modification systems. The sizes of the genomes of S3 and S4 were determined to be about 33 and 200 kb, respectively, by pulsed-field gel electrophoresis.
Functional properties of the phages.
Phage S3 showed a much faster development cycle than S1 and S4 (Fig. 3). While its eclipse phase lasted about 30 min, those of the other two phages took 75 min. Its rise period was also shorter, 30 min versus 45 min for S1 and 60 min for S4. The burst sizes were calculated to be about 75 virions/cell for S1, 100 virions/cell for S3, and 80 virions/cell for S4.
FIG. 3.

One-step growth curves for phages S1, S3, and S4 on exponential cultures of different Stenotrophomonas strains (Table 1) incubated in 2× TY at 30°C under agitation. The curves are representative of the data obtained from several experimental replicas.
The efficiencies of plating on different hosts differed significantly among the phages; the efficiency for S1, when plated onto any other than its original host, dropped to 10−4 PFU/ml, while S4 showed a range between no effect on efficiency and an efficiency of 10−3. Conversely, S3 did not show notable differences in its plating efficiency among the hosts tried.
Finally, the appearance of BIMs was tested. The frequency was the lowest for S3, with levels that ranged between 10−5 and 10−6 depending on the host, while in the case of S1, these values fell to levels on the order of 10−3 to 10−4. S4 BIMs emerged at a frequency of approximately 10−4.
The long one-step curve for S4, together with the relatively high frequency with which insensitive mutants were generated, suggested that S4 might be a temperate phage. To test this possibility, colonies of presumptively insensitive mutants were isolated, washed with a chelating agent (10 mM sodium pyrophosphate at 37°C for 30 min) to eliminate free virions (30), and plated onto lawns of a strain susceptible to S4. Colonies that generated an inhibition zone were then subjected to phage induction with mitomycin C, thus confirming the temperate nature of S4. It is then assumed that the high frequency of BIMs observed after exposure to S1 and S4 was due to host lysogenization.
Analysis of the bacteriophage S1 genome.
The genome of S1 was determined to be located in a double-stranded DNA molecule 40,287 bp long (Fig. 4). It has 12-bp single-stranded 5′-end extensions with the sequence 5′-GGTATTCGGACG-3′. The G+C content is 63.7%, slightly lower than the 66% of the two sequenced S. maltophilia genomes (GenBank accession numbers NC011071 and AM743169).
FIG. 4.

Physical and genetic map of the bacteriophage S1. The 48 orf genes are correlatively numbered and are indicated by arrows proportional to their length and pointing in the direction of transcription. The functional modules detected are indicated above the scheme, and the products of several putatively or experimentally identified genes are shown below. PAPS, phosphoadenosine phosphosulfate.
In the genome, 48 putative ORFs with more than 50 codons were recognized (Table 2). The majority of them had an AUG start codon, while five (orf5, orf13, orf20, orf26, and orf47) initiated at GUG. The genome is highly compressed: only 3% is noncoding, the start codons of 15 ORFs overlap with the stop codons of the previous ORFs, and in four cases, the end of one gene is located inside the following one. The genome was annotated after comparison to sequences in current databases (Fig. 2). Significant matches to 35 ORFs were obtained, and biological functions were assigned to 25 of these ORFs. No tRNA genes were found.
TABLE 2.
Characteristics of bacteriophage S1 ORFs and their deduced gene productsa
| ORF | Nucleotide start position | Nucleotide end position | Length (nt) | Product length (aa) | Product mass (kDa) (pI) | Predicted identity(ies) or function(s) of product | Strain with closest hit (E value) | % Amino acid identity (% similarity) | Accession no. | Predicted domain (E value) |
|---|---|---|---|---|---|---|---|---|---|---|
| orf1 | 40 | 2454 | 2,415 | 804 | 89.6 (7.35) | Topoisomerase-primase/VirE | Azoarcus sp. strain EbN1 (0.0) | 46 (63) | ref YP_161152.1 | NACHT domain (PF05729) (0.2) |
| orf2 | 2454 | 2804 | 351 | 116 | 12.7 (11.23) | Unknown | ||||
| orf3 | 2797 | 3441 | 645 | 214 | 22.7 (4.66) | Terminase small subunit | Azoarcus sp. strain EbN1 (5e−13) | 34 (46) | ref YP_161150.1 | Phage_Nu1 (PF07471) (0.71) |
| orf4 | 3438 | 5363 | 1,926 | 641 | 72.1 (8.89) | Terminase large subunit | Azoarcus sp. strain EbN1 (0.0) | 59 (73) | ref YP_161149.1 | Terminase_GpA (PF05876) (9.1e−124) |
| orf5 | 5357 | 5677 | 321 | 106 | 12.0 (6.59) | Unknown | ||||
| orf6 | 5757 | 5972 | 219 | 71 | 8.03 (11.22) | Peptidase | Synechococcus elongatus PCC 6301 (9e−05) | 41 (68) | ref YP_171509.1 | |
| orf7 | 5974 | 7464 | 1,491 | 496 | 53.7 (6.14) | Portal protein | Delftia acidovorans SPH-1 (2e−105) | 44 (60) | ref YP_001565698.1 | Phage portal_2 (PF05136) (4e−09) |
| orf8 | 7461 | 9443 | 1,983 | 660 | 71.7 (5.26) | Peptidase/scaffold/major head | Sphingomonas wittichii RW1 (3e−98) | 47 (59) | ref YP_001262687.1 | Peptidase_ U35 (PF04586) (2.2e−05) |
| orf9 | 9466 | 9831 | 366 | 121 | 11.7 (4.49) | Putative RecA/RadA recombinase | Polaromonas naphthalenivorans CJ2 (2e−17) | 45 (59) | ref YP 982481.1 | |
| orf10 | 9912 | 10343 | 432 | 143 | 15.3 (10.34) | Structural protein | Acidovorax avenae subsp. citrulli AAC00-1 (3e−32) | 50 (65) | ref YP_970706.1 | BAR domain (PFAM PF03114) (0.32) |
| orf11 | 10340 | 10678 | 339 | 112 | 12.4 (5.79) | Holin | Signal-peptide, Transmembrane_regions | |||
| orf12 | 10675 | 11166 | 492 | 163 | 17.07 (8.94) | Rz | Pseudomonas phage M6 (4e−06) | 31 (48) | ref YP_001294529.1 | Signal-peptide, phage_ lysis (PF03245) (0.41) |
| orf12.1 | 10799 | 11119 | 321 | 106 | 11.8 (12.00) | Rz1 | Rz1 motif (PF06085) (0.11) | |||
| orf13 | 11163 | 11486 | 324 | 107 | 11.6 (4.65) | Lysin | ||||
| orf14 | 11542 | 11961 | 450 | 139 | 14.7 (4.20) | Minor tail protein | ||||
| orf15 | 11975 | 12775 | 801 | 266 | 28.3 (4.63) | Major tail protein | Shewanella oneidensis MR-1 (1e−08) | 26 (42) | ref NP_716312.1 | |
| orf16 | 12829 | 13497 | 669 | 222 | 24.8 (11.92) | Unknown | ||||
| orf17 | 13490 | 17236 | 3,747 | 1248 | 131.7 (5.32) | Tape measure | Bordetella bronchiseptica RB50 (1e−38) | 25 (40) | ref NP_888251.1 | |
| orf18 | 17236 | 17652 | 417 | 138 | 15.2 (5.72) | Transmembrane protein | Pseudomonas aeruginosa 2192 (3e−10) | 37 (53) | ref ZP_00970870.1 | |
| orf19 | 17656 | 21261 | 3,606 | 1201 | 126.5 (4.84) | Tail fiber | Ralstonia solanacearum GMI1000 (0.0) | 36 (53) | ref NP_518996.1 | Phage short tail fiber protein gp12, middle domain (PF09089) (0.73) |
| orf20 | 21258 | 22292 | 1,035 | 344 | 36.1 (5.74) | Unknown | Ralstonia solanacearum GMI1000 (3e−13) | 25 (41) | ref NP_518997.1 | |
| orf21 | 22237 | 24411 | 2,175 | 724 | 77.8 (8.81) | Tail protein | Ralstonia solanacearum GMI1000 (8e−62) | 29 (42) | ref NP_519000.1 | |
| orf22 | 24401 | 24634 | 234 | 77 | 8.5 (4.80) | Unknown | Shewanella baltica OS223 (0.001) | 37 (63) | ref ZP_01843215.1 | |
| orf23 | 24782 | 25573 | 792 | 263 | 30.2 (6.19) | DNA methylase | Xylella fastidiosa Ann-1 (2e−103) | 69 (80) | ref ZP_00684054.1 | N6 Mtase (PF02384) (1.4e−13) |
| orf24 | 26242 | 25577 | 666 | 221 | 24.1 (5.89) | Unknown | Bacillus sp. NRRL B-14911 (1e−33) | 40 (53) | ref ZP_01170832.1 | DUF159 (PF02586) (1.1e−58) |
| orf25 | 27391 | 26420 | 972 | 323 | 36.4 (7.77) | Unknown | ||||
| orf26 | 27820 | 27395 | 426 | 141 | 15.9 (9.82) | Unknown | Sphingomonas sp. strain SKA58 (0.093) | 31 (54) | ref ZP_01304913.1 | |
| orf27 | 28014 | 28526 | 513 | 170 | 19.3 (11.40) | Unknown | S. maltophilia R551-3 (3e−45) | 66 (77) | ref ZP_01642485.1 | |
| orf28 | 29795 | 28530 | 1,266 | 421 | 46.9 (10.31) | Integrase | Syntrophus aciditrophicus SB (3e−135) | 57 (74) | ref YP_461774.1 | Phage_integrase (PF00589) (2.5e−15) |
| orf29 | 30390 | 30001 | 390 | 129 | 14.02 (9.51) | AlpA-like transcription regulator | Chromobacterium violaceum ATCC 12472 (1e−13) | 46 (71) | ref NP_900318.1 | Phage_AlpA (PF05930) (2e−22) |
| orf30 | 31618 | 30623 | 996 | 331 | 36.8 (8.48) | PAPS reductase | Xanthomonas axonopodis pv. Citri strain 306 (3e−132) | 68 (81) | ref NP_642602.1 | PAPS_reduct (PF01507) (4.3e−16) |
| orf31 | 31646 | 32467 | 822 | 273 | 30.2 (6.51) | GspM-like protein | S. maltophilia R551-3 (7e−13) | 58 (66) | ref ZP_01642616.1 | GspM motif (PF04612) (0.82) |
| orf32 | 33302 | 32469 | 834 | 277 | 30.5 (4.75) | DNA methylase | Ralstonia eutropha H16 (0.72) | 30 (51) | ref YP_728546.1 | N6 Mtase (PF02384) (0.27) |
| orf33 | 33568 | 33299 | 270 | 89 | 9.3 (6.09) | Unknown | ||||
| orf34 | 33804 | 33565 | 240 | 79 | 9.1 (10.80) | Unknown | ||||
| orf35 | 34517 | 33804 | 714 | 237 | 25.9 (4.49) | Unknown | ||||
| orf36 | 34906 | 34514 | 393 | 130 | 14.3 (9.29) | Unknown | Pseudomonas aeruginosa 2192 (1e−04) | 40 (56) | ref ZP_00976133.1 | |
| orf37 | 35402 | 34914 | 489 | 162 | 17.5 (10.78) | Unknown | Xanthomonas oryzae pv. Oryzae KACC10331 (0.038) | 58 (63) | ref YP_199271.1 | |
| orf38 | 35869 | 35399 | 471 | 156 | 17.6 (11.58) | Unknown | ||||
| orf39 | 35911 | 36504 | 594 | 197 | 20.5 (12.37) | Unknown | ||||
| orf40 | 36510 | 36854 | 345 | 114 | 12.3 (12.40) | Unknown | ||||
| orf41 | 36851 | 37312 | 462 | 153 | 15.8 (12.31) | Unknown | ||||
| orf42 | 37433 | 37984 | 552 | 183 | 19.5 (8.72) | Unknown | “Dechloromonas aromatica” RCB (0.041) | 44 (55) | ref YP_285023.1 | |
| orf43 | 38395 | 38024 | 372 | 123 | 13.9 (6.13) | Unknown | S. maltophilia R551-3 (3e−12) | 39 (56) | ref ZP_01642516.1 | |
| orf44 | 38969 | 38310 | 660 | 219 | 23.7 (6.38) | Repressor | Xylella fastidiosa Ann-1 (1e−05) | 32 (57) | ref ZP_00681945.1 | HTH 3 (PF01381) (0.0049) |
| orf45 | 39183 | 39392 | 210 | 69 | 7.08 (5.61) | Unknown | ||||
| orf46 | 39389 | 39919 | 531 | 176 | 19.3 (5.55) | Unknown | S. maltophilia R551-3 (3e−40) | 48 (69) | ref ZP_01642514.1 | |
| orf47 | 39921 | 40151 | 231 | 76 | 8.5 (10.34) | Unknown |
nt, nucleotides; aa, amino acids; PAPS, phosphoadenosine phosphosulfate.
Two major blocks of gene orientation were observed. One starts at orf39 and goes through the cohesive ends up to orf23; only orf43 and orf44 may be transcribed in the alternative direction. The other block extends from orf38 to orf24 and harbors two putative genes (orf27 and orf31) that may be read in the opposite direction. In phage genomes, the genes tend to be clustered into functional modules. In the case of S1, the structural group is the most conspicuous and can be divided into four submodules, in the following order: packaging, head morphogenesis, lysis, and tail morphogenesis. The packaging region comprises orf3 and orf4, which would encode the small and large terminase subunits, respectively. The product of the latter gene has a motif typical of large terminases that generate 5′ protruding cohesive ends (2) as expected from the cosN experimental data. The head morphogenesis module comprises the region from orf5 to orf10. gp7 and gp8 were shown directly to be virion components (Fig. 2). The first shares motifs with phage portal proteins, while gp8 may be the major capsid protein. However, orf8 has a coding capacity for a protein of 660 amino acids and 71.7 kDa, while the product detected in the virions has an apparent mass of 35 kDa as determined by SDS-PAGE. Interestingly, gp8 matches three types of proteins in the databases: its amino-terminal third (up to residue 192) matches proteases; its middle part, which in addition is predicted to be structured almost exclusively as a row of α-helices, matches scaffold proteins; and its carboxyl terminus matches major head proteins. MALDI-TOF analysis of the most abundant virion protein yielded peptides that matched gp8 from its 360th residue onwards, possibly indicating that native gp8 became autoprocessed by its protease domain, giving rise to the scaffold and the major capsid polypeptides. The latter would have about 300 amino acids and a mass of about 33 to 35 kDa, in good agreement with the apparent size observed by SDS-PAGE. Unfortunately, the NH2-terminal end of this protein was blocked, so we are at present unable to precisely determine where processing takes place.
The lysis cassette would comprise orf11 to orf13, apparently corresponding to the holin, the tandem Rz-Rz1, and the endolysin. The putative holin was unusual since only one transmembrane domain was detected in it. However, its location and size suggest that it may behave as a bona fide holin. The next orf encodes both Rz and Rz1. The Rz protein has a typical signal peptide at its amino terminus, while Rz1 is translated in the +1 frame with respect to Rz and gives rise to a polypeptide rich in proline (accounting for 17 of 106 residues). Finally, gp13 has an endolysin domain and a weak sequence relationship to peptidoglycan binding proteins, which together with its location next to a holin, allows the endolysin function to be ascribed to it.
The tail module appears to extend from orf14 to orf21. gp14 is a minor component of the virion, as are gp15 (major tail protein), gp17 (tape measure), and gp19 (tail fiber) (Fig. 2). As expected for tail measure proteins, the secondary structure of gp17 is almost exclusively α-helix. The protein may be processed during particle formation, since it migrates as a 90-kDa polypeptide, whereas the gene sequence indicates a size of 131.7 kDa. In fact, MALDI-TOF analysis provided only peptides that corresponded to the first 924 amino acids of the protein (orf17 has a coding capacity for a polypeptide of 1,248 residues), suggesting that processing occurs at its C-terminal end. Between the major tail and the tape measure genes lies orf16, which does not have the translational frameshift site frequently encountered in this position in other double-stranded DNA phages (37).
The other two modules that are commonly present in temperate siphoviruses, i.e., the lysis-lysogeny switch and the replication cluster, are not easily recognizable in S1. The only protein showing homology with phage transcription regulation proteins is gp29, whose determinant, in addition, is followed by the putative phage integrase gene. However, gp29 has a motif homologous to the positive transcriptional regulator AlpA (35) rather than to the phage repressor CI, it does not have a clear helix-turn-helix motif, and no cro-like repressor gene is found adjacent to orf29. The product of orf28 may be the phage integrase because it presents the C-terminal catalytic domain of DNA breaking-rejoining enzymes and, more particularly, of Cre integrases.
S1 does not have a replication module, but some genes that may be involved in DNA metabolism are scattered along the genome. To this category belong orf1 and orf23, as well as the putative origin of replication. This cis-acting element was identified between orf44 and orf45, in a 214-bp noncoding region that presents direct and inverted repeats typical of replication origins, through cumulative GC skew analysis. gp1 is predicted to be a topoisomerase-primase that has also been found in phages of Vibrio and Burkholderia species (24, 31). Finally, orf23 encodes a putative N6-adenine methyltransferase.
DISCUSSION
Most of the 22 phages specific for Stenotrophomonas that were either induced from lysogens or isolated from sewage had a narrow host range, in most cases restricted to just 1 of the 26 strains used. The exceptions to the rule were S3, S4, and to a lesser extent, S1.
The genomic DNA of S3 and S4 is resistant to digestion by restriction endonucleases. This property may contribute to the wide host ranges of both phages. In fact, the other two Stenotrophomonas phages of the order Caudovirales isolated to date are extremely resistant to DNA restriction as well and infect a wide range of susceptible strains: 42.5 and 70% (of 87 strains) for Smp14 and φSMA5, respectively (3, 4). In contrast, S1, whose DNA is readily digested, is able to infect only 4 of 26 strains (15%). Smp14 DNA presents, in addition to the normal four deoxynucleotides, small proportions of N4-deoxymethylcytidine and two other unusual bases, suggested to be deoxythymidine and deoxyguanosine analogues, which are claimed to be responsible for its resistance to digestion (4). This may be true for S3 and S4 as well. In any case, the genetic determinants that render the S3 and S4 DNA refractory to restriction are probably located in the phages' own genomes rather than being a trait conferred by particular hosts, because the phages remained insensitive to digestion after development on strains whose DNA was a substrate for restriction enzymes.
S3 is proposed as a model phage to be used in therapeutically aimed experiments, not only because of this resistance to restriction, but also because it seems to be virulent (although this property has to be confirmed by genome sequencing), which prevents the risk of lysogen generation; it attacks a wide range of clinical strains; it has a fast developmental cycle; its plaquing frequency is independent of the strain used as a host; and resistant mutants (BIMs) are infrequent.
S1, S2, and S4 are the only temperate phages isolated from Stenotrophomonas to date. Of the two that infected more than one strain, S1 is more amenable to study because its genomic DNA is relatively short compared to that of S4 (40 versus 200 kb) and is susceptible to restriction endonucleases: S1 was therefore chosen as a representative of its group, and its genome sequence was determined. The modular organization observed in most double-stranded DNA bacteriophages occurs only in the left half of the S1 genome, which encodes the morphogenetic functions (DNA packaging, head morphogenesis, cell lysis, and tail morphogenesis). However, no lysis-lysogeny switch or DNA replication modules were obvious, so only informed guesses on the possible implication of a few genes in these functions can be formulated, the tasks of the remaining ORFs being unknown due to the lack of homology of their products to sequences from the databases.
The transcription pattern of S1 presents two groups of ORFs running divergently and only two of them in each row going in the opposite direction. Among them, orf31 may represent a lysogenic conversion gene (moron) because its product resembles GspM, a protein involved in the general secretion system II, frequently used by virulence factors to reach the surrounding medium (21). However, no clear −10 and −35 sequences are found in front of it. Adjacent to orf31 lies the determinant for a homologue of phosphoadenosine phosphosulfate reductase, which has been found previously in other phages that infect gram-negative bacteria, such as φE125 and BcepB1A, which infect Burkholderia (32, 36), and coliphage 186 (26). This protein may confer an advantage on the host in the assimilation of inorganic phosphate through its reduction to organic thiols (31).
The opposite direction of transcription of orf43 and orf44 with respect to that of the surrounding genes may also be considered to be an indication of a moron; however, no function could be ascribed to their products apart from a hypothetical interaction with DNA, suggested by the helix-turn-helix motif of orf44.
The case of orf27 is peculiar; it is located immediately behind the integrase gene, with a gap of only 4 nucleotides. This position, together with the considerable degree of secondary structure implied in its sequence (Fig. 5), suggests that the attP sequence may be included in this gene. On the other hand, the direction of transcription of orf27 is opposite that of the surrounding genes, and overall, orf27 has 75% identity to a hypothetical-protein-encoding orf located in the chromosome of S. maltophilia (Fig. 5). This finding may suggest that the integrity of the cellular gene would be restored after the integration of the phage genome into the bacterial chromosome, thus keeping whatever function was played by its gene product.
FIG. 5.

Comparison of the nucleotide sequences of orf27 and a particular Stenotrophomonas chromosomal orf. The zones of more than 60 nucleotides showing a degree of identity of 85% or higher are shaded. Zones of putative secondary structure are indicated by arrows above the sequences (IR, inverted repeat; DR, direct repeat). Numbers at the ends of the sequences are nucleotide positions.
Another peculiarity of the S1 genome is that its left extremity is not occupied by the small terminase subunit gene, as in many bacteriophages, but by the determinant of a protein that is considered to be implicated in DNA replication due to the presence of a topoisomerase-primase motif typical of DnaG-related primases. However, this protein is also related to VirE, which was first discovered in virulent but not in harmless strains of Dichelobacter nodosus, the etiological agent of ovine foot root (17). For this reason, it was considered to be a virulence factor when found in temperate phages resident in pathogenic bacteria (24), a function that cannot be excluded for S1 in lysogenic Stenotrophomonas strains.
The genome of S1 is a new example of the extraordinary biodiversity shown by bacteriophages: we cannot even guess what may be the roles played by the products of almost half of its genes, even though we know that they must include the determinants for such a widespread and widely studied process as the lysis-lysogeny decision.
Acknowledgments
This work was supported by CICYT grants SAF2004-0033 and BFU2007-65781 from the Ministry of Science and Technology (Spain) and the FEDER Plan. N.S. and R.M. are holders, respectively, of a fellowship associated with the first grant and a scholarship from FICYT (Principado de Asturias). P.G. is a fellow of the Spanish Ministry of Education Ramón y Cajal Research Programme.
The provision of some of the Stenotrophomonas strains used in this work by Jose L. Martinez is gratefully acknowledged. We also thank K. F. Chater for critical reading of the manuscript.
Footnotes
Published ahead of print on 24 October 2008.
REFERENCES
- 1.Apisarnthanarak, A., V. J. Fraser, W. M. Dunne, J. R. Little, J. Hoppe-Bauer, J. L. Mayfield, and L. B. Polish. 2003. Stenotrophomonas maltophilia intestinal colonization in hospitalized oncology patients with diarrhea. Clin. Infect. Dis. 37:1131-1135. [DOI] [PubMed] [Google Scholar]
- 2.Casjens, S. R., E. B. Gilcrease, D. A. Winn-Stapley, P. Schicklmaier, H. Schmieger, M. L. Pedulla, M. E. Ford, J. M. Houtz, G. F. Hatfull, and R. W. Hendrix. 2005. The generalized transducing Salmonella bacteriophage ES18: complete genome sequence and DNA packaging strategy. J. Bacteriol. 187:1091-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang, H., C. R. Chen, J. W. Lin, G. H. Shen, K. M. Chang, Y. H. Tseng, and S. F. Weng. 2005. Isolation and characterization of novel giant Stenotrophomonas maltophilia phage φSMA5. Appl. Environ. Microbiol. 71:1387-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, C. R., C. H. Lin, J. W. Lin, C. I. Chang, Y. H. Tseng, and S. F. Weng. 2007. Characterization of a novel T4-type Stenotrophomonas maltophilia virulent phage Smp14. Arch. Microbiol. 188:191-197. [DOI] [PubMed] [Google Scholar]
- 5.Clewley, J. P. 2003. The day of the phage. Commun. Dis. Public Health 6:260-262. [PubMed] [Google Scholar]
- 6.Da Silva Filho, L., A. F. Tateno, L. F. Velloso, J. E. Levi, S. Fernandes, C. N. Bento, J. C. Rodrigues, and S. R. Ramos. 2004. Identification of Pseudomonas aeruginosa, Burkholderia cepacia complex and Stenotrophomonas maltophilia in respiratory samples from cystic fibrosis patients using multiplex PCR. Pediatr. Pulmonol. 37:537-547. [DOI] [PubMed] [Google Scholar]
- 7.del Toro, M. D., J. Rodríguez, L. Martínez, A. Pascual, R. Pérez, E. J. Perea, and M. A. Muniain. 2006. Epidemiology, clinical features and prognosis of infections due to Stenotrophomonas maltophilia. Enferm. Infecc. Microbiol. Clin. 24:4-9. [DOI] [PubMed] [Google Scholar]
- 8.Denton, M., and K. G. Kerr. 1998. Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia. Clin. Microbiol. Rev. 11:57-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'Hérelle, F. 1925. Essai de traîtment de la peste bubonique par le bactériophage. Presse Méd. 33:1393-1394. [Google Scholar]
- 10.D'Hérelle, F. 1936. Traitement et prophylaxie par le bactériophage de maladies épidémiques de nature bactérienne. Medecine. 17:23-32. [Google Scholar]
- 11.Ewing, B., and P. Green. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186-194. [PubMed] [Google Scholar]
- 12.Garcia, P., V. Ladero, and J. E. Suarez. 2003. Analysis of the morphogenetic cluster and genome of the temperate Lactobacillus casei bacteriophage A2. Arch. Virol. 148:1051-1070. [DOI] [PubMed] [Google Scholar]
- 13.Garcia, P., I. Rodriguez, and J. E. Suarez. 2004. A −1 ribosomal frameshift in the transcript that encodes the major head protein of bacteriophage A2 mediates biosynthesis of a second essential component of the capsid. J. Bacteriol. 186:1714-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagemann, M., D. Hasse, and G. Berg. 2006. Detection of a phage genome carrying a zonula occludens like toxin gene (zot) in clinical isolates of Stenotrophomonas maltophilia. Arch. Microbiol. 185:449-458. [DOI] [PubMed] [Google Scholar]
- 15.Herrero, M., C. G. de los Reyes-Gavilán, J. L. Caso, and J. E. Suárez. 1994. Characterization of φ393-A2, a bacteriophage that infects Lactobacillus casei. Microbiology 140:2585-2590. [Google Scholar]
- 16.Hugh, R. 1981. Pseudomonas maltophilia sp. nov. nom. rev. Int. J. Syst. Bacteriol. 31:195. [Google Scholar]
- 17.Katz, M. E., P. M. Howarth, W. K. Yong, G. G. Riffkin, L. J. Depiazzi, and J. I. Rood. 1991. Identification of three gene regions associated with virulence in Dichelobacter nodosus, the causative agent of ovine footrot. J. Gen. Microbiol. 137:2117-2124. [DOI] [PubMed] [Google Scholar]
- 18.Murray, N. 2006. Phage-based expression systems, p. 674-685. In R. Calendar (ed.), The bacteriophages, 2nd ed. Plenum Publishing Corp., New York, NY.
- 19.Nicodemo, A. C., and J. I. Paez. 2007. Antimicrobial therapy for Stenotrophomonas maltophilia infections. Eur. J. Microbiol. Infect. Dis. 26:229-237. [DOI] [PubMed] [Google Scholar]
- 20.Parisien, A., B. Allain, J. Zhang, R. Mandeville, and C. Q. Lan. 2008. Novel alternatives to antibiotics: bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides. J. Appl. Microbiol. 104:1-13. [DOI] [PubMed] [Google Scholar]
- 21.Py, B., L. Loiseau, and F. Barras. 2001. An inner membrane platform in the type II secretion machinery of Gram-negative bacteria. EMBO Rep. 2:244-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 23.Schless, R. A. 1932. Staphylococcus aureus meningitis: treatment with specific bacteriophage. Am. J. Dis. Child. 44:813-822. [Google Scholar]
- 24.Seguritan, V., I. W. Feng, F. Rohwer, M. Swift, and A. M. Segall. 2003. Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J. Bacteriol. 185:6434-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senol, E. 2004. Stenotrophomonas maltophilia: the significance and role as a nosocomial pathogen. J. Hosp. Infect. 57:1-7. [DOI] [PubMed] [Google Scholar]
- 26.Sivaprasad, A. V., R. Jarvinen, A. Puspurs, and J. B. Egan. 1990. DNA replication studies with coliphage 186. III. A single phage gene is required for phage 186 replication. J. Mol. Biol. 213:449-463. [DOI] [PubMed] [Google Scholar]
- 27.Smith, H. W., and M. B. Huggins. 1982. Successful treatment of experimental Escherichia coli infections in mice using phage: its general superiority over antibiotics. J. Gen. Microbiol. 128:307-318. [DOI] [PubMed] [Google Scholar]
- 28.Smith, H. W., M. B. Huggins, and K. M. Shaw. 1987. The control of experimental Escherichia coli diarrhoea in calves by means of bacteriophage. J. Gen. Microbiol. 133:1121-1126. [DOI] [PubMed] [Google Scholar]
- 29.Suarez, J. E., and K. F. Chater. 1980. Cloning in Streptomyces: a bifunctional replicon comprising pBR 322 cloned into a Streptomyces phage. Nature 286:527-529. [DOI] [PubMed] [Google Scholar]
- 30.Suárez, J. E., and K. F. Chater. 1981. Development of a DNA cloning system in Streptomyces using phage vectors. Cienc. Biol. 6:99-110. [Google Scholar]
- 31.Summer, E., J. J. Gill, C. Upton, C. F. Gonzalez, and R. Young. 2007. Role of phages in the pathogenesis of Burkholderia, or “Where are the toxin genes in Burkholderia phages?” Curr. Opin. Microbiol. 10:410-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Summer, E. J., C. F. Gonzalez, M. Bomer, T. Carlile, A. Embry, A. M. Kucherka, J. Lee, L. Mebane, W. C. Morrison, L. Mark, M. D. King, J. J. LiPuma, A. K. Vidaver, and R. Young. 2006. Divergence and mosaicism among virulent soil phages of the Burkholderia cepacia complex. J. Bacteriol. 188:255-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Summers, W. C. 2001. Bacteriophage therapy. Annu. Rev. Microbiol. 55:437-451. [DOI] [PubMed] [Google Scholar]
- 34.Swings, J., P. De Vos, M. Van den Mooter, and J. De Ley. 1983. Transfer of Pseudomonas maltophilia (Hugh 1981) to the genus Xanthomonas as Xanthomonas maltophilia (Hugh 1981) comb. nov. Int. J. Syst. Bacteriol. 33:409-413. [Google Scholar]
- 35.Trempy, J. E., J. E. Kirby, and S. Gottesman. 1993. Alp suppression of Lon: dependence on the slpA gene. J. Bacteriol. 176:2061-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woods, D. E., J. A. Jeddeloh, D. L. Fritz, and D. DeShaezer. 2002. Burkholderia thailandensis E125 harbors a temperate bacteriophage specific for Burkholderia mallei. J. Bacteriol. 184:4003-4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu, J., R. W. Hendrix, and R. L. Duda. 2004. Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol. Cell 16:11-21. [DOI] [PubMed] [Google Scholar]