

Abstract
Listeria monocytogenes may persist for long periods in food processing environments. In some instances, this may be due to aggregation or biofilm formation. To investigate the mechanism controlling biofilm formation in the food-borne pathogen L. monocytogenes, we characterized LM-49, a mutant with enhanced ability of biofilm formation generated via transposon Tn917 mutagenesis of L. monocytogenes 4b G. In this mutant, a Tn917 insertion has disrupted the coding region of the gene encoding a putative ATP-binding cassette (ABC) transporter permease identical to Lmof2365_1771 (a putative ABC transporter permease) presented in the sequenced strain L. monocytogenes strain 4b F2365. This disrupted gene, denoted lm.G_1771, encoded a protein with 10 transmembrane helixes. The revertant, LM-49RE, was obtained by replacing lm.G_1771::Tn917 with lm.G_1771 via homologous recombination. We found that LM-49RE formed the same amount of biofilm biomass as the wild-type strain. Furthermore, transcription of the downstream lm.G_1770 gene was not influenced by the upstream Tn917 insertion, and the presence of Tn917 has no effect on biofilm formation. These results suggest that lm.G_1771 was solely responsible for the negative regulation of biofilm formation by L. monocytogenes 4b G. The immediate gene upstream of lm.G_1771 encoded an ATP-binding protein. Bioinformatics analysis suggested that these two genes were organized into an operon and that their proteins formed an export ABC transporter. Here, we report the characterization of the mutant and identification of a novel ABC transporter that functions in negative regulation of biofilm formation in L. monocytogenes.
The gram-positive bacterium Listeria monocytogenes is responsible for listeriosis outbreaks and causes infections in fetuses, newborns, and immunocompromised individuals, with a 28% mortality rate (19, 38). This bacterium is ubiquitous on food surfaces and in food processing facilities, where it is able to survive for several months or even years (14, 36). It has been suggested that adhesion or biofilm formation is the main reason for the persistence of L. monocytogenes on food contact surfaces since bacterial cells within biofilms are much more resistant to detergents, biocides, and antibiotics than their free-living (planktonic) counterparts (10, 20). Consequently, L. monocytogenes biofilms on food contact surfaces are difficult to completely eliminate and may cause secondary food contamination, imposing major challenges to the production of safe food in the food industries (37).
Biofilms are highly organized communities, and their formation includes three steps: initial attachment, microcolony formation, and community expansion (5). Therefore, bacterial cells within biofilms are metabolically different from their free-living forms. It is believed that extracellular signaling molecules (autoinducers of quorum sensing) are involved in regulating specific transcription factors, which then facilitate global controls in the bacterial genome expression, changing bacteria from a planktonic to a sessile lifestyle (15). Indeed, DNA microarray analysis has revealed that autoinducer 2 (AI-2) alters the expression of six transcription factors in Escherichia coli (7). Thus far, three quorum-sensing systems have been shown to function in bacterial cellular communication and to play roles in biofilm formation and maturation (1, 6). Firstly, acyl-homoserine lactones (AHLs) function as autoinducers of quorum sensing in the LuxI/LuxR signaling system found only in gram-negative bacteria (6, 13). Secondly, AI-2 signals of the LuxS communication system are used for quorum sensing in many distantly related bacteria such as the gram-negative Salmonella and the gram-positive Staphylococcus (34, 41). Thirdly, small peptides also function as quorum-sensing signals, exemplified by the agr system identified in Staphylococcus aureus and L. monocytogenes (15, 32, 41).
Recently, the biofilm-positive mutant LM-49 was isolated by screening a library constructed from L. monocytogenes 4b G using Tn917 mutagenesis. This mutant exhibited an enhanced ability to form biofilm compared to the wild-type strain (4). An elucidation of the function of the defective gene could lead to an improved understanding of biofilm formation of L. monocytogenes. Therefore, the purpose of this work was to characterize the functions of the mutant gene and to identify its mechanism for biofilm formation.
MATERIALS AND METHODS
Bacterial strains.
The L. monocytogenes strain 4b G was obtained from the Chinese Center for Disease Control (Hubei, China). Serotyping of this isolate using a Listeria antisera kit (Denka Seiken Co., Tokyo, Japan) revealed that it belongs to the serotype 4b group. The biofilm-positive mutant LM-49 was generated from L. monocytogenes 4b G by Tn917 mutagenesis (4). The LM-49RE is a revertant of the LM-49 mutant in which the mutant allele lm.G1771::Tn917 is replaced with the wild-type gene via homologous recombination (see below).
L. monocytogenes 4b G and its derivatives were cultured in either Trypticase soy broth (TSB) medium (Merck, Germany) or brain heart infusion (BHI) medium (Becton, Dickinson, and Co., Franklin Lakes, NJ). To select cells containing Tn917, TSB or BHI medium was supplemented with 5 μg/ml erythromycin (Erm), whereas the medium was supplemented with 10 μg/ml chloramphenicol (Cam) to select cells harboring the Listeria-E. coli shuttle vector pKSV7 or its derivatives. The incubation temperature was 37°C for all L. monocytogenes strains used in this work, unless otherwise indicated specifically. E. coli DH5α and TG1 were used as host strains for molecular cloning. All bacterial strains and plasmids used in this study are listed in Table 1.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| L. monocytogenes strains | ||
| 4b G | Wild-type L. monocytogenes, isolate strain | Chinese Center for Disease Control, Hubei, China |
| LM-49 | Enhanced biofilm-forming Tn917 mutant of L. monocytogenes | 4 |
| LM-49RE | Revertant L. monocytogenes strain from allelic recombination using pKSV7-AB | Sarah Vela |
| E. coli strains | ||
| DH5α | λ− φ80dlacZΔM15 Δ(lacZYA-argF)U196 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Laboratory stock |
| TG1 | supE hsdΔ5 thiΔ(lac-proAB) F′ [traD36 proAB+lacIqlacZΔM15] | Laboratory stock |
| Plasmids | ||
| pKSV7 | Temperature-sensitive shuttle vector | This study |
| pKSV7-AB | Allelic recombination vector | This study |
Southern blot analysis.
Genomic DNAs were prepared from L. monocytogenes 4b G and LM-49, and Southern blot analysis was done using standard procedures (30). In brief, the prepared genomic DNA was digested with EcoRI and run on a 1% agarose gel. The DNA fragments in the agarose gel were then transferred onto a Hybond-N+ nylon membrane (GE Health Care) via capillary transfer. The probe for the Southern analysis was generated by PCR from a plasmid containing Tn917 by using primers Tn4292 and Tn3768 (Table 2). The PCR product was purified, labeled with biotin using a Gene Images Random Priming DNA Labeling Kit (GE Health Care), and used as a probe for hybridization. Then, the hybridization signals on the membranes were detected using a Gene Image CDP-Star Detection kit (GE Health Care), following the instructions of the manufacturer, and recorded on Fuji X-ray film.
TABLE 2.
Oligonucleotides used as primers
| Primer name | Primer sequence | Location (bp) | Use |
|---|---|---|---|
| C427 | 5′-CTACATTACGCATTTGGA-3′ | 533-516a | iPCR |
| Tn917F | 5′-TGGTCTTAAGGCAGCCAGAG-3′ | 2180-2199a | iPCR |
| Tn4431 | 5′-CCATCAAGGGTATGAGTA-3′ | 4537-4520a | iPCR |
| Tn5054 | 5′-AAATAGATCCCGAAGTAAC-3′ | 5143-5161a | iPCR |
| Tn4292 | 5′-TTGAGCACGAGTCAGAGC-3′ | 4292-4275a | Southern blot probe |
| Tn3768 | 5′-AACAATATTCGATCAGGAGA-3′ | 3767-3786a | Southern blot probe |
| InsL | 5′-AGCCATCAAAGAAAGGAAGA-3′ | 1794567-1794586b | Amplification of insertion site |
| InsR | 5′-GTATCGGTGAAAGGCAACAG-3′ | 1795615-1795596b | Amplification of insertion site |
| LM DBR-F | 5′-CGCTCAAACCAATTTCTGCTA-3′ | 1794114-1794134b | RT-PCR |
| LM DBR-R | 5′-CTTCCTTTCTTTGATGGCTTT-3′ | 1794585-1794565b | RT-PCR |
| 16S rRNA-F | 5′-TTAGTGGCGGACGGGTGAGT-3′ | rrsA-rrsB | RT-PCR |
| 16S rRNA-R | 5′-CGGACAACGCTTGCCACCTA-3′ | rrsA-rrsB | RT-PCR |
| P1 | 5′-GTCCGCCTTAATAGAGCA-3′ | 1794392-1794409b | Identification of revertant |
| P2 | 5′-GTTCTACCGCTGGCTCTT-3′ | 1797225-1797208b | Identification of revertant |
| C1 | 5′-GGGCGTCTCAGCTCTAAAATCCG-3′ | 4385-4406a | Identification of allelic exchange |
| C2 | 5′-TAGGAACTATGAAAAAGGAACGGA-3′ | 1729-1706a | Identification of allelic exchange |
| M13fwd | 5′-GGTTTTCCCAGTCACGAC-3′ | Identification of allelic exchange | |
| M13rev | 5′-AGCGGATAACAATTTCACAC-3′ | Identification of allelic exchange |
Identification of the Tn917 insertion site by inverse PCR.
The DNA oligonucleotide primers used for PCRs were designed using the software Primer Premier, version 5 (33). To identify the target site of Tn917 in the mutant LM-49, two sets of primers were required, one for each flanking end (Fig. 1A). The primers C427 and Tn917F (Table 2) were used to clone the Tn917 5′ flanking sequence, while primers Tn4431 and Tn5054 (Table 2) were used to obtain its 3′ flanking sequence. An inverse PCR (iPCR) approach (22) was used since the primers in each set extend away from each other on a linear DNA template but face toward each other on a circular DNA template (Fig. 1A).
FIG. 1.

Identification of the Tn917 insertion site in the L. monocytogenes LM-49 mutant. (A) Flow chart for generating DNA templates for iPCR: HindIII digestion of total DNA, circularization of the linear fragments by ligation, and iPCR. Two sets of primers were required for iPCR, one for each end since there are two HindIII (H) restriction sites in Tn917. E, EcoRI restriction sites. (B) Southern analysis of Tn917 insertion in the LM-49 mutant. The location of the probe in Tn917 is indicated in panel A. (C) Agarose gel electrophoresis of iPCR products. M, 1-kb DNA Ladder Marker; lanes 1 and 3, iPCR with C427 and Tn917F primers; lanes 2 and 4: iPCR with primers Tn4431 and Tn5054. LM-49 denotes DNA templates prepared from the mutant, and wild type (WT) denotes templates from L. monocytogenes 4b G (negative control).
The genomic DNAs from the L. monocytogenes LM-49 and the wild-type L. monocytogenes 4b G (as negative control) strains were digested with HindIII at 37°C for 16 h in order to prepare the circular DNA used as a template for iPCR. The resulting DNA fragments were purified with a Nucleic Acid Purification kit (Qiagen) and ligated with T4 DNA ligase. The ligated circular DNA was then used as a template for iPCR and amplified using Ex-Tag DNA polymerase in the presence of 1 mM deoxynucleoside triphosphate mix. The amplification was done on a Peltier Thermal Cycler PTC225 (GMI, Ramsey, MN). The amplification conditions were as follows: an initial denaturation step at 95°C for 5 min; 34 cycles of 94°C for 30 s, followed by 50°C for 30 s, and 72°C for 2 min; and a final extension step at 72°C for 8 min. The PCR products were analyzed by agarose gel electrophoresis.
The purified PCR products were cloned into the PCR cloning vector pMD19-T (Takara Bio Inc.) following the procedure of the manufacturer, and the resulting plasmids were transformed into E. coli TG1 competent cells. The DNA sequences of both strands of the PCR products were then determined (Sangon Co. Ltd., Shanghai, China).
Construction of the plasmid pKSV7-AB for gene replacement and isolation of the revertant LM-49RE.
A 1,049-bp DNA fragment that flanked the Tn917 insertion site was amplified from the wild-type L. monocytogenes 4b G genome by PCR, using primers InsL and InsR (Table 2). This PCR product was sequenced and cloned into pMD19-T, resulting in the plasmid pMD19-AB. The SalI-EcoRI fragment containing the PCR product was then cloned into the Listeria-E. coli shuttle plasmid pKSV7, yielding the plasmid pKSV7-AB for gene replacement.
pKSV7-AB was transformed into LM-49 competent cells via electroporation (25), and transformants harboring the plasmid were selected at 30°C in BHI medium containing 10 μg/ml Cam. Cells containing an integrated pKSV7-AB (pop-in recombinants) were selected by growth at 41°C (nonpermissive temperature for replication of this plasmid) for 40 generations. One pop-in recombinant was chosen and cultured in nonselective medium at 30°C for 30 generations. Under these conditions, all five types of recombinants could grow up, i.e., the strain containing the integrated pKSV7-AB after the first crossover and the four possible outcomes after the second allelic exchange, as illustrated in Fig. 3A. The pop-out recombination types I and II containing plasmids pKSV7-Tn917 and pKSV7-AB, respectively, could grow on Cam plates at 30°C, while type III (revertant) and type IV (LM-49) with no plasmid could not grow on Cam plates. Several resulting strains were characterized by PCR, and a confirmed revertant strain, LM-49RE, was used for further experiments.
FIG. 3.

Construction of the LM-49 revertant via homologous recombination. (A) Flow chart of LM-49 revertant construction (1). In the LM-49 strain harboring the gene replacement plasmid pKSV7-AB, the plasmid may integrate into the host chromosome at either site a or site b (Pop in) (2). Four possible outcomes of pop-out recombination are shown. The PCR primer sets P1+P2, M13fwd+C1, and M13rev+C2 were used for distinguishing between these recombinations by PCR. (B) Identification of pKSV7-AB integration by colony PCR. A total of 30 colonies were purified and tested. The results from eight of them are shown. M, 1-kb DNA ladder marker; lanes 1 to 8, purified transformants PCR checked with M13fwd+C1 or M13rev+C2 as primers. (C) Analysis of plasmid and Tn917 junctions for checking type I and II colonies by colony PCR. (D) PCR identification of the revertant LM-49RE.
RNA preparation and reverse transcription-PCR (RT-PCR) analyses.
L. monocytogenes 4b G and LM-49 cells were grown in BHI medium to an optical density at 600 nm (OD600) of 0.7 for exponential phase samples and to 1.3 for stationary phase samples. Sessile cells from biofilms of both strains grown on glass slides were scraped off with cotton sticks, resuspended in 1 ml of 0.85% NaCl, and pelleted by centrifugation. The cells were resuspended in a 10 mg/ml solution of lysozyme and incubated at 37°C for 15 min to make the cell wall more permeable. Total RNA was extracted with Trizol reagent, following the manufacturer's procedure (Invitrogen). Six units of Turbo DNase I (Ambion) was used to treat 10 μg of RNA at 37°C for 2 h to ensure complete removal of residual DNA. DNase I was then extracted with phenol (pH 4.5), the RNA was precipitated with ethanol, and the RNA pellet was resuspended in 20 μl of H2O pretreated with 0.1% diethyl pyrocarbonate.
cDNA was synthesized by RT-PCR in 10-μl reaction mixtures containing 100 ng of RNA, 2 pmol of RT-PCR primer (Table 2), 10 U of RNase inhibitor, and 25 U of Moloney murine leukemia virus reverse transcriptase (Ambion), and the mixture was incubated at 42°C for 30 min. One unit of HotStart Taq DNA Polymerase (Qiagen, Germany) was used for RT-PCRs in the presence of 1 mM deoxynucleoside triphosphate mix in a 25-μl reaction system.
For analysis of lm.G_1770 expression, three samples of each cDNA preparation were amplified for 32, 35, and 38 cycles with conditions of 94°C for 30 s, 55°C for 30 s, and 72°C for 40 s. The PCRs for 16S rRNA expression were 22, 25, and 28 cycles with conditions of 94°C for 30 s, 63°C for 30 s, and 72°C for 1 min. The synthesized DNA was analyzed by agarose gel electrophoresis.
Biofilm formation assay.
Biofilm formation by L. monocytogenes was monitored using the microplate assay described by Djordjevic et al. (8). In brief, L. monocytogenes cells were inoculated in 96-well microplates and cultured at 37°C for 48 h. Free-moving (planktonic) cells in the liquid medium were removed by gentle pipetting, while sessile cells in the biofilms remained attached to the wells of the plate. To estimate the cell mass of the biofilms, 1% crystal violet was added to each well to stain the cells. After the unbound dye was removed and plates were rinsed with water, the crystal violet bound to the sessile cells in the biofilms was recovered by ethanol extraction, and the amount was determined by measuring the absorbance at OD595 with a DU-800 spectrophotometer (Beckman Instruments). The biomass of a biofilm was expressed as the amount of violet crystal bound to the plates. To determine the number of attached cells in the biofilms, the sessile cells were scraped off from the wells of microplates using sterile cotton sticks and resuspended. Serial decimal dilutions were then made with 0.85% NaCl solution, and the diluted cells were spread on BHI plates. This procedure was repeated three times for each sample to assess the reproducibility of the data.
Fluorescence microscopy of biofilm.
Samples for fluorescence microscopy were prepared on glass slides using a method described by Marsh et al. (17) with the modification that fluorescein isothiocyanate (FITC) (21) was used instead of acridine orange. Briefly, petri dishes containing 20 ml of TSB medium and a sterilized glass slide were inoculated with Listeria cells and incubated at 37°C for 72 h, during which time Listeria cells formed biofilms on the glass slide. The slide was then rinsed three times with sterile water to remove all planktonic cells, air dried, and immersed in 0.1% FITC (Sigma-Aldrich) for 15 to 20 min at room temperature in the dark to stain the sessile cells in the biofilms. FITC was poured off, excess dye was removed by rinsing the slide three times with water, and the biofilms formed were examined with the computer-assisted Olympus fluorescence microscope BX51 (Olympus company, Japan) and photographed.
Nucleotide sequence accession number.
The entire nucleotide sequence of lm.G_1771 was deposited in the GenBank database under accession number FJ216726.
RESULTS
Tn917 transposition inactivated the gene encoding a putative ABC transporter permease in LM-49.
Previously, an L. monocytogenes mutant, LM-49, exhibiting enhanced biofilm formation (biofilm-positive mutant) was isolated from a mutant library generated by Tn917 mutagenesis (4). We chose to investigate the mechanism of the biofilm formation in Listeria using this mutant. To ascertain that LM-49 was generated from a single Tn917 transposition event, a Tn917-specific sequence (Fig. 1A) was amplified by PCR, labeled with biotin, and used as a probe in Southern blot analysis of the mutant and wild-type DNA (Materials and Methods). As shown in Fig. 1B, the Tn917 probe specifically recognized only one hybridized fragment from the LM-49 genome and no hybridized signal from the L. monocytogenes 4b G wild type. These results indicated that the LM-49 mutant carried only one copy of Tn917.
Next, the LM-49 gene harboring the Tn917 insertion was identified by iPCR since the Tn917 sequence is known. As illustrated in Fig. 1A, oppositely oriented Tn917 primers point toward each other on the circularized DNA, and this enabled the Tn917-flanking sequence of the host chromosome, including the target site of the Tn917 transposition, to be cloned. The key step in iPCR is to generate suitable circular DNA molecules. Hence, the HindIII restriction enzyme (that cuts twice in Tn917) was used to cleave the LM-49 genomic DNA, yielding linear DNA fragments. These fragments were ligated with T4 DNA ligase, resulting in circular DNA substrates for iPCR (Fig. 1A). Two specific PCR products of 1.5 and 2.0 kb were obtained, one for each primer set (Fig. 1C). The 1.5-kb fragment comprised the Tn917 upstream-flanking fragment, while the 2.0-kb fragment contained the Tn917 downstream-flanking sequence.
The two PCR fragments were cloned into the pMD19-T vector and sequenced. Analyses of the sequences obtained indicated the following: (i) the target gene encoded a putative ABC transporter permease of 659 amino acids, which was 100% identical to the permease encoded by Lmof2365_1771; (ii) the insertion point was at 1,715 bp after the ATG start codon of the gene; (iii) the transposon was oriented in the same transcriptional direction as the target gene; and (iv) the 5-bp target sequence (5′-AAACT-3′) was duplicated (Fig. 2A). We named this gene lm.G_1771, referring to its homology with Lmof2365_1771 present in L. monocytogenes strain 4b F2365, and we named the mutant allele in LM-49 lm.G_1771::Tn917.
FIG. 2.

(A) Genetic map of the lm.G_1771::Tn917 mutant allele. Putative promoter elements identified upstream of lm.G_1772 and lm.G_1770 are indicated. A typical 5-bp target sequence duplicated by Tn917 transposition is indicated in boldface letters and underlined. (B) Effect of Tn917 insertion on the expression of downstream gene. a, exponential growth phase; b, stationary growth phase; c, sessile growth phase (biofilms). WT, the wild-type strain; LM-49, the biofilm-positive mutant. PCR with the purified RNA as a template did not yield any product (data not shown). This experiment was repeated three times with similar results.
Tn917 insertion did not change transcription of a downstream gene and the phenotype in biofilm formation.
In addition to mutagenesis by transposition into target genes, Tn917 mutagenesis may produce polar effects on the expression of downstream genes in the mutant by introducing transcriptional termination and/or changing transcriptional readthrough (9, 16). To study this possible polar effect, we used semiquantitative RT-PCR to analyze the expression of lm.G_1770 immediately downstream of the lm.G_1771 gene in L. monocytogenes 4b G and in LM-49 (Fig. 2A). As shown in Fig. 2B, the expression of lm.G_1770 in the wild-type and mutant strains remained the same for cells from exponential, stationary, and sessile growth phases. Thus, inactivation of the ABC transporter permease gene lm.G_1771 by Tn917 transposition did not have a polar effect on the downstream gene. Furthermore, sequence analysis of the promoter region of lm.G_1770 revealed a putative promoter 33 bp upstream of the ATG start codon (Fig. 2A). Thus, the expression of this gene was very likely only from this promoter.
To exclude the possibility that the Tn917 insertion element influences biofilm formation, we also constructed a deletion mutant. This mutant had the same phenotype for biofilm formation as LM-49 (data not shown).
Construction of a revertant of LM-49 by replacing lm.G_1771::Tn917 with the wild-type gene lm.G_1771.
To demonstrate that the Tn917 insertion in lm.G_1771 (lm.G_1771::Tn917) was solely responsible for the enhanced biofilm formation of the mutant, a revertant was constructed by replacing lm.G_1771::Tn917 with the wild-type gene lm.G_1771 via homologous recombination (Fig. 3A).
The temperature-sensitive plasmid pKSV7-AB, which contains the lm.G_1771 fragment, was transformed into LM-49. The transformation mixtures were spread onto Cam-containing plates to select for Camr transformants at a permissible temperature (30°C). A transformant was chosen and grown at the nonpermissible temperature of 41°C, thereby forcing integration of the plasmid into the chromosome to yield the pKSV7-AB pop-in recombinants. About 30 colonies were randomly picked and analyzed by PCR to identify Tn917 and pKSV7 junctions that were characteristic for the two possible types of pKSV7-AB pop-in (Fig. 3A, panel 1). A PCR product of the expected size was obtained from amplification with primers M13fwd and C1 but not with M13rev and C2 (Fig. 3B), suggesting that site “a” was preferred to site “b” in the pop-in recombination.
To obtain revertants, one purified pop-in recombinant (LM-49::pKSV7-AB) was grown for 30 generations at 30°C. Approximately 700 colonies were replica plated onto plates containing either Erm or Cam and incubated at 30°C. Colonies with type I and type II recombinations grew on both antibiotic-containing plates since they contained plasmids pKSV7-Tn917 and pKSV7-AB, respectively. Colonies with type IV recombination (LM-49) grew on Erm plates but not on Cam plates. Finally, pop-out revertants (type III) grew on neither Erm nor Cam plates. To further confirm the pop-out event, PCR amplification of plasmid and Tn917 junctions was performed to distinguish between type I and II colonies (Fig. 3A, panel 2). As expected, both M13fwd+C1 and M13rev+C2 primers yielded a PCR product for type I recombination and none for type II (Fig. 3C). Furthermore, similar PCR fragments of lm.G_1771 were obtained from a revertant (LM-49RE) and from the L. monocytogenes 4b G wild type using the primers P1 and P2 (Fig. 3D). The same results were obtained for the 18 revertants found (data not shown).
Conservation of the Lm.G_1771 ABC transporter permease and related gene products among gram-positive bacteria.
A search for sequence homologues of Lm.G_1771 permease in the GenBank database revealed that the Lm.G_1771 permease is highly conserved in Listeria species; its homologues were found in all sequenced Listeria genomes showing more than 95% sequence identity. Structural analysis of the Lm.G_1771 protein sequence further revealed that it contained 10 transmembrane fragments (http://www.cbs.dtu.dk/services/TMHMM/). Moreover, genes lm.G_1771 and lm.G_1772 (which encodes a putative ATP-binding protein) were clustered, and they most likely form an operon (Fig. 2A). These results suggest that the Lm.G_1771 permease and Lm.G_1772 ATP-binding protein constitute an uncharacterized ABC transporter.
Furthermore, a similar ABC transporter was found to be encoded by other bacterial genomes as well. By searching the public databases with the protein sequences of Lm.G_1771 and Lm.G_1772, several open reading frames with significant similarity to the components of the putative transporter were found, one of which is a known drug efflux protein that exports bacitracin (24). They all comprise two components, the permease and the ATP-binding protein, and it appears that the genes encoding them are also organized into operons (Fig. 4). The sequence similarity between the identified Listeria ABC transporter and the Bacillus bacitracin drug efflux protein was the highest among these drug efflux proteins: 26% sequence identity between their permease components (Lm.G_1771 versus BecB) and 54% identity for the ATP-binding components (Lm.G_1772 versus BecA). Thus, Lm.G_1771 and Lm.G_1772 are likely to encode an export ABC transporter. Preliminary data, however, do not indicate that bacitracin susceptibility has changed in the LM-49 mutant (data not shown). In the following, we call this putative ABC transporter the Lm.G_1771 transporter.
FIG. 4.

Organization of the putative Lm.G_1771 ABC transporter and its homologous systems identified in bacterial genomes. The filled boxes indicate the homologues of the putative Lm.G_1771 permease, and the dotted boxes show the open reading frames encoding an ATP-binding protein. The percentages under the gene symbols indicate the sequence identities between Lm.G_1771, Lm.G_1772, and their homologues. The homologues were identified by BlastP searches (27) and aligned using the Clustal W program (12). L.m 4b F2365, L. monocytogenes strain 4b F2365; L.m EGD-e, L. monocytogenes EGD-e; B.t israelensis, Bacillus thuringiensis serovar israelensis; B.s str.168, Bacillus subtilis subsp. subtilis strain 168; S. aureus MW2, S. aureus subsp. aureus MW2.
Lm.G_1771 ABC transporter permease as a negative regulator of biofilm formation by Listeria.
Biofilm formation by L. monocytogenes 4b G and its biofilm-positive mutant LM-49 and corresponding revertant LM-49RE was studied using microplate assay and fluorescence microscopy. As seen in Fig. 5A, the LM-49RE revertant and the wild-type strain produced similar amounts of biofilm, while the LM-49 mutant formed a thicker biofilm layer. Quantification of the biomass using crystal violet staining also showed that the LM-49 mutant formed more biofilm than either the revertant or parental strain (Fig. 5A). Furthermore, fluorescence microscopy showed that the biofilms formed by these strains exhibited distinct morphologies; the wild-type strain and the revertant LM-49RE showed the same tread-like pattern, whereas large cell aggregates were built up along the treads in the LM-49 biofilm (Fig. 5B). These results suggest that the Lm.G_1771 transporter could be an integral part of signal transduction participating in the negative regulation of biofilm formation in Listeria.
FIG. 5.

Characterization of the biofilms produced by different L. monocytogenes strains. (A) Biomass quantification using crystal violet staining. Pictures of biofilm formed in microplates are shown above each column. (B) Fluorescence micrograph. Pictures in panels I and IV represent the biofilm formed by the LM-49 mutant; panels II and V represent the wild type (WT), and panels III and VI represent the revertant LM-49RE. Upon incubation on glass slides for 72 h, L. monocytogenes formed biofilms.
The Lm.G_1771 transporter may export a signal inhibiting biofilm formation.
The enhanced biofilm-forming ability of LM-49 would be expected if the mutant cells were defective in exporting a signal inhibiting biofilm formation. Therefore, if wild-type and mutant cells were to be mixed, their ability to form biofilm should now be similar, since all cells would be exposed to the signal from the wild-type cells. On the other hand, if the mutant cells were not defective in exporting a signaling molecule, there would be no signal in the extracellular environment to inhibit biofilm formation in the mixed culture, and then the ratio of LM-49 to wild-type cells would be higher in the biofilms since the mutant itself has higher ability of biofilm formation than the wild type.
To test this hypothesis, we mixed wild-type and mutant cells at a 1:1 ratio in triplicate and incubated the mixture in a microplate at 37°C for 48 h, allowing biofilms to form. Pure cultures of the wild-type and mutant strains were incubated similarly.
The biofilm biomass in all cultures and the number of wild-type and mutant cells in the biofilms from mixed culture were determined by crystal violet quantification and counting CFU on plates (the mutant is Ermr), respectively. The mutant LM-49 produced approximately twofold more biofilm mass than the wild type in pure culture (Fig. 6), and the ratio of wild-type to mutant cells in the mixed culture basically remained unchanged (52% wild-type cells versus 48% mutant cells). The growth rate and the maximum OD in stationary phase were also essentially the same for both the wild-type and mutant strains (data not shown). The results of this experiment are in agreement with our notion that, in contrast to the wild-type strain, the mutant LM-49 strain is unable to export a signal inhibiting biofilm formation.
FIG. 6.
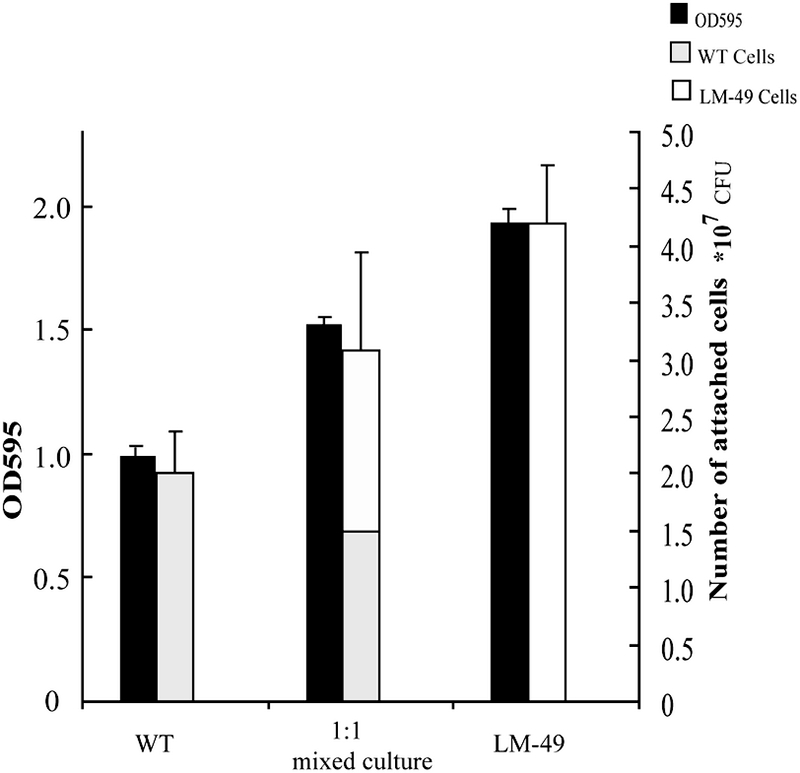
Characterization of signal transport in the LM-49 mutant. Biofilms formed by pure cultures of the wild type (WT) and LM-49 and by a culture containing a 1:1 mixture thereof. The biofilm biomasses were estimated using crystal violet staining and viable cell determination.
DISCUSSION
In this study, we have identified a new transporter that is involved in negative regulation of biofilm formation by L. monocytogenes. When one of the components of the ABC transporter was inactivated by a mutation, the resulting L. monocytogenes LM-49 mutant exhibited a stronger capacity for biofilm formation. Since biofilm formation is a complex process causing major physiological changes, the process could be regulated by global transcriptional factors that, in turn, respond to a signaling molecule such as a quorum-sensing signal. Therefore, the Lm.G_1771 ABC transporter may be an efflux protein exporting a signaling molecule that activates a pattern of genome expression characteristic of planktonic growth of L. monocytogenes.
A typical ABC transporter consists of a few subunits and contains one of the following structural domains: a nucleotide-binding domain, a transmembrane domain, or a solute-binding domain (SBD) (35). All ABC transporters are either exporters or importers. Specifically, importers harbor at least one SBD, whereas exporters have no SBD (28). Although lm.G_1771 encodes a permease with 10 transmembrane fragments and lm.G 1772 encodes an ATP-binding protein, none of the neighboring genes encodes an SBD (Fig. 4), which suggests that the encoded ABC transporter is an exporter. Furthermore, it is very likely that this ABC transporter operon consists of only two genes, lm.G_1772 and lm.G_1771, since they share a common promoter (Fig. 2A). The immediate downstream gene, lm.G_1770, encoding a putative DNA-binding response regulator, does not belong to the putative operon since it has its own promoter (Fig. 2A). In fact, as seen from semiquantitative RT-PCR analyses, its expression levels did not change in the wild-type and LM-49 strains (Fig. 2B).
Lm.G_1771 may represent an unknown transporter in Listeria because all known Listeria genomes encode such a transporter with high sequence similarity (>95%) while Lm.G_1771 shows only 26% sequence identity to BceB (YtsD) in Bacillus subtilis, the closest homologue of the permease among those identified in non-Listeria bacterial species. The LM-49 and its parent wild-type strain exhibited the same level of resistance to bacitracin (data not shown), which further indicates that the permease function differs from that of the Bacillus bacitracin drug efflux protein (23).
However, it has been established that several transporters are responsible for transporting signaling molecules that regulate bacterial biofilm formation. These chemical signals include AHL, the furanone derivative AI-2, and signal peptides. Although it has been suggested that small signal molecules can diffuse freely across the cellular membrane, active transport is also involved in exporting small signals. For example, while the AHL signals N-3-oxo-hexanoyl-homoserine lactone (HSL) and N-butyryl-HSL are freely diffusible, N-3-oxo-dodecanoyl-HSL has to be exported by the MexAB-OprM active efflux pump (26). Furthermore, for bacteria using AI-2 signals for quorum sensing to regulate their bioluminescence and biofilm formation (2, 18), the AI-2 molecules diffuse freely across the cell membrane in some bacteria and are detected by a two-component regulatory system as in Vibrio harveyi (31), while in other bacteria the AI-2 signals are imported into the cell via an lsr ABC transporter as in Salmonella enterica serovar Typhimurium and E. coli (40). With large signaling molecules such as signal peptides, active transport is always responsible for both exporting and importing, as in the case of the agr signaling system of S. aureus where the autoinducer peptide is exported by the AgrB transporter (39). These transporters are integral parts of their corresponding signaling systems. Therefore, inactivating any of these signal transporters should abolish the corresponding signal transmission and, consequently, terminate the programmed regulation of genome expression. As for the LM-49 mutant, mutation in the Lm.G_1771 transporter has resulted in the derepression of biofilm formation, leading to the observed biofilm-positive phenotype. However, the Lm.G_1771 transporter does not exhibit sequence similarity to any known transporters of signaling molecules. It could, therefore, represent a novel exporter for chemical signals in bacteria.
The LM-49 mutant and the wild-type strain were cocultured at a 1:1 ratio for a biofilm formation assay using microplates. The biofilms of the mixed culture as well as the composition of the cells in the biofilms were analyzed. Interestingly, we found in the biofilms formed by the mixed culture that the ratio of mutant to wild-type cells did not change, suggesting that the wild-type cells produced a signal molecule and were able to inhibit biofilm formation in the LM-49 mutant cells. Therefore, we postulate that the L. monocytogenes LM-49 mutant may be defective in exporting a signal that leads to the inhibition of biofilm formation in L. monocytogenes. Consequently, the Lm.G_1771 transporter could be an exporter of this signal.
It remains to be determined which signal is exported by the Lm.G_1771 transporter. Currently, two quorum-sensing signaling systems are known in L. monocytogenes: the agr peptide and the AI-2 signals (2, 23, 26). Clearly, the Lm.G_1771 transporter cannot be responsible for transporting an agr signal since the complete pathway of the agr signal transduction has been identified in this bacterium (29). However, it is not clear whether this transporter transports an AI-2 signal. Genetic analyses of luxS, the gene encoding the enzyme for AI-2 biosynthesis, have indicated that AI-2 functions as an inhibitor of biofilm formation in L. monocytogenes EGD-e (3, 32), and it has been reported that defects in AI-2 biosynthesis has resulted in more than a 17-fold increase in biofilm formation (3). The Lm.G_1771 transporter could constitute an integral part of the AI-2 signaling system since some AI-2 molecules rely on an exporter for export into the extracellular environments (11). If so, LM-49 could be important in further elucidation of the AI-2 signaling system. Alternatively, the Lm.G_1771 transporter may export a novel signal.
In conclusion, we have shown that the lm.G_1771 gene encoded a putative ABC transporter permease. When lm.G_1771 was inactivated, it caused enhanced biofilm-formation of L. monocytogenes. The upstream gene, lm.G_1772, encodes an ATP binding protein, and the products of these two genes may form a putative ABC transporter. We also found that the sequence of this ABC transporter was highly conserved in Listeria species. Finally, the ratio of wild-type to mutant cells in the biofilm formed by a mixed culture was more or less constant, suggesting that this ABC transporter most likely exported a signal inhibiting biofilm formation of L. monocytogenes. Further investigation is required to elucidate precisely how the ABC transporter controls L. monocytogenes biofilm formation.
Acknowledgments
This work was jointly supported by grants 2006BAK02A14 from the Ministry of Science and Technology of China and 071422011 and 07dz19508 from the Science and Technology Commission of Shanghai Municipality to X. Shi, Danish Free Research Councils grants 274-07-0116 (Forskningsrådet for Teknologi og Produktion [FTP]) and 272-05-400 (Forskningsrådet for Natur og Univers) to Q. She, and grant 274-05-0073 (FTP) to S. Knøchel.
We thank Sarah Vela (University of California at Berkeley, CA) for providing plasmid pKSV7, Frank Aarestrup (Technical University of Denmark) for help with antibiotic resistance determinations, and Bjarne Albrechtsen (University of Copenhagen) for critical reading of the manuscript.
Footnotes
Published ahead of print on 3 October 2008.
REFERENCES
- 1.Abraham, W. R. 2006. Controlling biofilms of gram-positive pathogenic bacteria. Curr. Med. Chem. 13:1509-1524. [DOI] [PubMed] [Google Scholar]
- 2.Bassler, B. L., M. Wright, and M. R. Silverman. 1994. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: sequence and function of genes encoding a second sensory pathway. Mol. Microbiol. 13:273-286. [DOI] [PubMed] [Google Scholar]
- 3.Challan Belval, S., L. Gal, S. Margiewes, D. Garmyn, P. Piveteau, and J. Guzzo. 2006. Assessment of the roles of LuxS, S-ribosyl homocysteine, and autoinducer 2 in cell attachment during biofilm formation by Listeria monocytogenes EGD-e. Appl. Environ. Microbiol. 72:2644-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, Y. H., and X. M. Shi. 2005. Mutagenesis on biofilm formation of Listeria monocytogenes by Tn917 transposon insertion. Wei Sheng Wu Xue Bao 45:952-954. [PubMed] [Google Scholar]
- 5.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318-1322. [DOI] [PubMed] [Google Scholar]
- 6.Davies, D. G., M. R. Parsek, J. P. Pearson, B. H. Iglewski, J. W. Costerton, and E. P. Greenberg. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295-298. [DOI] [PubMed] [Google Scholar]
- 7.DeLisa, M. P., C. F. Wu, L. Wang, J. J. Valdes, and W. E. Bentley. 2001. DNA microarray-based identification of genes controlled by autoinducer 2-stimulated quorum sensing in Escherichia coli. J. Bacteriol. 183:5239-5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Djordjevic, D., M. Wiedmann, and L. A. McLandsborough. 2002. Microtiter plate assay for assessment of Listeria monocytogenes biofilm formation. Appl. Environ. Microbiol. 68:2950-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dobinsky, S., K. Bartscht, and D. Mack. 2002. Influence of Tn917 insertion on transcription of the icaADBC operon in six biofilm-negative transposon mutants of Staphylococcus epidermidis. Plasmid 47:10-17. [DOI] [PubMed] [Google Scholar]
- 10.Frank, J. F., and R. A. Koffi. 1990. Surface-adherent growth of Listeria monocytogenes is associated with increased resistance to surfactant sanitizers and heat. J. Food Prot. 53:550-554. [DOI] [PubMed] [Google Scholar]
- 11.Herzberg, M., I. K. Kaye, W. Peti, and T. K. Wood. 2006. YdgG (TqsA) controls biofilm formation in Escherichia coli K-12 through autoinducer 2 transport. J. Bacteriol. 188:587-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higgins, D. G., J. D. Thompson, and T. J. Gibson. 1996. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 266:383-402. [DOI] [PubMed] [Google Scholar]
- 13.Juhas, M., L. Eberl, and B. Tummler. 2005. Quorum sensing: the power of cooperation in the world of Pseudomonas. Environ. Microbiol. 7:459-471. [DOI] [PubMed] [Google Scholar]
- 14.Kathariou, S. 2002. Listeria monocytogenes virulence and pathogenicity, a food safety perspective. J. Food Prot. 65:1811-1829. [DOI] [PubMed] [Google Scholar]
- 15.Kong, K. F., C. Vuong, and M. Otto. 2006. Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol. 296:133-139. [DOI] [PubMed] [Google Scholar]
- 16.Le Coq, D., S. Aymerich, and M. Steinmetz. 1991. Dual effect of a Tn917 insertion into the Bacillus subtilis sacX gene. J. Gen. Microbiol. 137:101-106. [DOI] [PubMed] [Google Scholar]
- 17.Marsh, E. J., H. Luo, and H. Wang. 2003. A three-tiered approach to differentiate Listeria monocytogenes biofilm-forming abilities. FEMS Microbiol. Lett. 228:203-210. [DOI] [PubMed] [Google Scholar]
- 18.McNab, R., S. K. Ford, A. El-Sabaeny, B. Barbieri, G. S. Cook, and R. J. Lamont. 2003. LuxS-based signaling in Streptococcus gordonii: autoinducer 2 controls carbohydrate metabolism and biofilm formation with Porphyromonas gingivalis. J. Bacteriol. 185:274-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mead, P. S., L. Slutsker, V. Dietz, L. F. McCaig, J. S. Bresee, C. Shapiro, P. M. Griffin, and R. V. Tauxe. 1999. Food-related illness and death in the United States. Emerg. Infect. Dis. 5:607-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mustapha, A., and M. B. Liewen. 1989. Destruction of Listeria monocytogenes by sodium hypochlorite and quaternary ammonium sanitizers. J. Food Prot. 52:306-311. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen, A. T., T. Tolker-Nielsen, K. B. Barken, and S. Molin. 2000. Role of commensal relationships on the spatial structure of a surface-attached microbial consortium. Environ. Microbiol. 2:59-68. [DOI] [PubMed] [Google Scholar]
- 22.Ochman, H., J. W. Ajioka, D. Garza, and D. L. Hartl. 1990. Inverse polymerase chain reaction. Nat. Biotechnol. 8:759-760. [DOI] [PubMed] [Google Scholar]
- 23.Ohki, R., Giyanto, K. Tateno, W. Masuyama, S. Moriya, K. Kobayashi, and N. Ogasawara. 2003. The BceRS two-component regulatory system induces expression of the bacitracin transporter, BceAB, in Bacillus subtilis. Mol. Microbiol. 49:1135-1144. [DOI] [PubMed] [Google Scholar]
- 24.Ohki, R., K. Tateno, Y. Okada, H. Okajima, K. Asai, Y. Sadaie, M. Murata, and T. Aiso. 2003. A bacitracin-resistant Bacillus subtilis gene encodes a homologue of the membrane-spanning subunit of the Bacillus licheniformis ABC transporter. J. Bacteriol. 185:51-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park, S. F., and G. S. Stewart. 1990. High-efficiency transformation of Listeria monocytogenes by electroporation of penicillin-treated cells. Gene 94:129-132. [DOI] [PubMed] [Google Scholar]
- 26.Pearson, J. P., C. Van Delden, and B. H. Iglewski. 1999. Active efflux and diffusion are involved in transport of Pseudomonas aeruginosa cell-to-cell signals. J. Bacteriol. 181:1203-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearson, W. R. 1995. Comparison of methods for searching protein sequence databases. Protein Sci. 4:1145-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quentin, Y., G. Fichant, and F. Denizot. 1999. Inventory, assembly and analysis of Bacillus subtilis ABC transport systems. J. Mol. Biol. 287:467-484. [DOI] [PubMed] [Google Scholar]
- 29.Saenz, H. L., V. Augsburger, C. Vuong, R. W. Jack, F. Gotz, and M. Otto. 2000. Inducible expression and cellular location of AgrB, a protein involved in the maturation of the staphylococcal quorum-sensing pheromone. Arch. Microbiol. 174:452-455. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 31.Schauder, S., and B. L. Bassler. 2001. The languages of bacteria. Genes Dev. 15:1468-1480. [DOI] [PubMed] [Google Scholar]
- 32.Sela, S., S. Frank, E. Belausov, and R. Pinto. 2006. A mutation in the luxS gene influences Listeria monocytogenes biofilm formation. Appl. Environ. Microbiol. 72:5653-5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh, V. K., A. K. Mangalam, S. Dwivedi, and S. Naik. 1998. Primer premier: program for design of degenerate primers from a protein sequence. BioTechniques 24:318-319. [DOI] [PubMed] [Google Scholar]
- 34.Taga, M. E., J. L. Semmelhack, and B. L. Bassler. 2001. The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol. Microbiol. 42:777-793. [DOI] [PubMed] [Google Scholar]
- 35.Tam, R., and M. H. Saier, Jr. 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 57:320-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tompkin, R. B. 2002. Control of Listeria monocytogenes in the food-processing environment. J. Food Prot. 65:709-725. [DOI] [PubMed] [Google Scholar]
- 37.Trachoo, N. 2003. Biofilms and the food industry. Songklanakarin J. Sci. Technol. 25:807-815. [Google Scholar]
- 38.Vazquez-Boland, J. A., M. Kuhn, P. Berche, T. Chakraborty, G. Dominguez-Bernal, W. Goebel, B. Gonzalez-Zorn, J. Wehland, and J. Kreft. 2001. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 14:584-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vuong, C., H. L. Saenz, F. Gotz, and M. Otto. 2000. Impact of the agr quorum-sensing system on adherence to polystyrene in Staphylococcus aureus. J. Infect. Dis. 182:1688-1693. [DOI] [PubMed] [Google Scholar]
- 40.Walters, M., and V. Sperandio. 2006. Quorum sensing in Escherichia coli and Salmonella. Int. J. Med. Microbiol. 296:125-131. [DOI] [PubMed] [Google Scholar]
- 41.Xu, L., H. Li, C. Vuong, V. Vadyvaloo, J. Wang, Y. Yao, M. Otto, and Q. Gao. 2006. Role of the luxS quorum-sensing system in biofilm formation and virulence of Staphylococcus epidermidis. Infect. Immun. 74:488-496. [DOI] [PMC free article] [PubMed] [Google Scholar]