

Abstract
Gordonia westfalica Kb1 and Gordonia polyisoprenivorans VH2 induce the formation of an extracellular superoxide dismutase (SOD) during poly(cis-1,4-isoprene) degradation. To investigate the function of this enzyme in G. polyisoprenivorans VH2, the sodA gene was disrupted. The mutants exhibited reduced growth in liquid mineral salt media containing poly(cis-1,4-isoprene) as the sole carbon and energy source, and no SOD activity was detectable in the supernatants of the cultures. Growth experiments revealed that SodA activity is required for optimal growth on poly(cis-1,4-isoprene), whereas this enzyme has no effect on aerobic growth in the presence of water-soluble substrates like succinate, acetate, and propionate. This was detected by activity staining, and proof of expression was by antibody detection of SOD. When SodA from G. westfalica Kb1 was heterologously expressed in the sodA sodB double mutant Escherichia coli QC779, the recombinant mutant exhibited increased resistance to paraquat, thereby indicating the functionality of the G. westfalica Kb1 SodA and indirectly protection of G. westfalica cells by SodA from oxidative damage. Both sodA from G. polyisoprenivorans VH2 and sodA from G. westfalica Kb1 coded for polypeptides comprising 209 amino acids and having approximately 90% and 70% identical amino acids, respectively, to the SodA from Mycobacterium smegmatis strain MC2 155 and Micrococcus luteus NCTC 2665. As revealed by activity staining experiments with the wild type and the disruption mutant of G. polyisoprenivorans, this bacterium harbors only one active SOD belonging to the manganese family. The N-terminal sequences of the extracellular SodA proteins of both Gordonia species showed no evidence of leader peptides for the mature proteins, like the intracellular SodA protein of G. polyisoprenivorans VH2, which was purified under native conditions from the cells. In G. westfalica Kb1 and G. polyisoprenivorans VH2, SodA probably provides protection against reactive oxygen intermediates which occur during degradation of poly(cis-1,4-isoprene).
Reactive oxygen species (ROS) occur during aerobic metabolism and are known to cause damage to many biomacromolecules, with DNA appearing to be the most sensitive target of these agents (38). Therefore, living cells developed various mechanisms to protect cellular constituents against oxidative damage. The microbial oxidative stress response is a result of well-orchestrated reactions involving synthesis of many proteins and small molecules. These can be grouped into at least four categories. The first group includes enzymes like superoxide dismutase (SOD; EC 1.15.1.1) and catalase (EC 1.11.1.6) and small molecules for direct detoxification and/or protection against oxidative stress. Cellular components like DNA can also be protected by unspecific binding of proteins like Dps (DNA-binding protein from starved cells) (3, 38) and small antioxidant molecules like ascorbate and glutathione (42). The second category comprises enzymes responsible for repair of damaged cellular components like DNA (24) or protein by methionine sulfoxide reductase (EC 1.8.4.6) (1, 43). Proteins involved in signal transduction and regulation like SoxR and SoxS from Escherichia coli represent the third category (24), and the fourth group comprises proteins, like glucose-6-phosphate dehydrogenase, induced during the oxidative stress response (24).
SODs are a ubiquitous family of enzymes efficiently catalyzing the dismutation of superoxide anions to molecular oxygen and hydrogen peroxide according to the following equation (25, 26): 2 O2− + 2 H+ → O2 + H2O2. The primary function of SODs is the detoxification of cell-damaging superoxide anions, but they also play important roles during phytopathogenesis since oxidative stress is an important component of the plant defense response against microbial invasion (37). Furthermore, Mycobacterium tuberculosis expresses SOD during pathogenesis, leading to a T-cell response (22). For this bacterium, large amounts of SOD are found extracellularly. For a long time it was unclear whether the enzyme is actively secreted or whether the extracellular abundance of SOD is due to bacterial leakage or autolysis upon a high level of expression in combination with extracellular stability (54). The latest studies showed that SodA is actively secreted by M. tuberculosis, with involvement of SecA2 as an accessory secretion factor (16). Nocardia asteroides SOD has been implicated as a virulence factor, allowing the cells to survive intracellularly and to escape killing by phagocytic cells (2).
The tendency of natural and synthetic rubber to become autoxidized by atmospheric oxygen and ozone is a well-known phenomenon. This autoxidation process leads to the formation of activated oxygen species and is the main reason for the extensive use of antioxidants to protect rubber against aging and microbial attack (8). Degradation of poly(cis-1,4-isoprene) by species of the genus Gordonia and other bacteria has been previously described (for reviews, see references 5 and 44). Our previous studies led us to investigate the involvement and influence of extracellular SODs produced by strains of Gordonia westfalica and Gordonia polyisoprenivorans during degradation of rubber (36).
MATERIALS AND METHODS
Bacterial strains, plasmids, and oligonucleotides.
Strains of the genus Gordonia and of Escherichia coli and plasmids and primers used in this study are listed in Table 1. Cells of Gordonia spp. were cultivated in standard I (St-I) medium (Merck, Darmstadt, Germany) at 30°C on a rotary shaker at 180 rpm. For growth experiments with poly(cis-1,4-isoprene), 0.5% (vol/vol) natural latex concentrate (Neotex Latz; Weber & Schaer, Hamburg, Germany) or 0.3% (wt/vol) synthetic poly(cis-1,4-isoprene) with an average molecular mass of 800 kDa (182141; Aldrich, Steinheim, Germany) was added to liquid mineral salts medium (MSM), as described by Schlegel et al. (48), as the sole source of carbon and energy. Cells of E. coli strains were cultivated at 37°C in Luria-Bertani (LB) broth (46) on a rotary shaker at 180 rpm. Antibiotics were applied as described by Sambrook et al. (46) as indicated below. Other carbon sources were added to MSM as indicated below.
TABLE 1.
Bacterial strains, plasmids and primers used in this study
| Strain, plasmid, or primer | Relevant characteristicsa | Reference, source, or alternate strain designation |
|---|---|---|
| Strains | ||
| E. coli XL1-Blue | recA1 endA1 gyrA96 thi1 hsdR17 (rK− mK+) supE44 relA1 λ−lac [F′ proAB lacIqlacZΔM15 Tn10(Tcr)] | 19 |
| E. coli QC779 | As GC4468 except (sodA::MudPR13)25 Φ(sodB-kan)1-Δ2 | 20 |
| E. coli Rosetta-gami B (DE3)pLys | Δ(ara-leu)7697 ΔlacX74 ΔphoA PvuII phoR araD139 ahpC galE galK rpsL (DE3) F′ [lac+lacIqpro] gor-522::Tn10 trxB pLysSRARE (Camr Strr Tetr) | Novagen |
| M. smegmatis mc2 155 | ept-1 | 50 |
| G. polyisoprenivorans VH2 | Wild type, rubber degrading | DSM44266 |
| G. westfalica Kb1 | Wild type, rubber degrading | DSM44215T |
| Plasmids | ||
| pBluescript SK(−) | E. coli cloning vector; Apr | Stratagene |
| pGEM-T Easy | E. coli T cloning vector; Apr | Promega |
| pET23a | E. coli expression vector; Apr | Novagen |
| pNC9501 | E. coli/Rhodococcus shuttle vector; Kmr; thiostreptonr | H. Saleki, Japan Energy Corporation |
| Primers | ||
| P.SOD11 | GA(CT)CTGGA(CT)TACGACTA(CT)GC | |
| P.SOD22 | CAGGTAGAA(AC)GC(AG)TG(CT)TCCCA | |
| P.IPCRCS3 | TTGGGGGACAGGTTCTTC | |
| P.IPCRCS4 | TCCAGGGTTCGGGCTGGG | |
| P.SPB | AAAAGGATCCGTGGCTGAATACACGCTTGCCG | |
| P.SPE | AAAAGAATTCTCAGGCAGGCTCGATGAGGCC | |
| P.SODVH2F1 | CACCACA(GC)CAAGCACCACGC(AGCT)(AG)C(GC)TA | |
| P.SODVH2B1 | GCGTG(CT)TCCCACATGTC(GC)TC(GC)A(GC)(GC) | |
| P.SODVH2up1bio | TGTCGTTGGCGCCCTTGACGTAC | |
| P.SODVH2down1bio | TCATCCCGGTCGTCATGCTCG | |
| P.walker1 | CTAATACGACTCACTATAGGGNNNNATGC | |
| P.walker2 | CTAATACGACTCACTATAGGGNNNNGATC | |
| P.walker3 | CTAATACGACTCACTATAGGGNNNNTAGC | |
| P.walker4 | CTAATACGACTCACTATAGGGNNNNCTAG | |
| P.nested | CTAATACGACTCACTATAGGG | |
| P.SODVH2up1 | GACGTACGTAGCGTGGTGCTTG | |
| P.SODVH2down1 | GCTCGACGACATGTGGGAACAC | |
| P.SODVH2-647 | AGCTGCTGTTTTCCGGAGCGG | |
| P.SODVH2+9 | GGAACTCAGGCGGGGACGATG | |
| P.pETsod_Nde | CATATGGCTGAATACACCTTGCCGGATCTC | |
| P.pETsod_Xho_his | CTCGAGGGCGGGGACGATGAGGCCCTTG |
In primer sequences, recognition sites for restriction enzymes are underlined.
Paraquat assay.
For the paraquat growth assay, the sodA sodB double mutant E. coli QC779 and recombinant strains derived from E. coli QC779 harboring plasmid pSKsod or only the vector pBluescript SK− as a control were cultivated in LB medium at 37°C in the presence of an appropriate antibiotic to an optical density at 600 nm (OD600) of 1.0. Afterwards, the cultures were rapidly chilled in a water-ice mixture and were kept cold until they were used for inoculation of prewarmed LB medium with an initial OD600 of 0.02. The cultures were then grown at 37°C, and 0.05 mM of the herbicide paraquat (methyl viologen) was added as a redox cycling agent, which promotes formation of the superoxide radical anion inside cells (27); cell growth was monitored photometrically at 600 nm.
Isolation, analysis, and manipulation of DNA.
Plasmid DNA was prepared from crude lysates of E. coli cells by the alkaline extraction method (12). Total DNA of Gordonia sp. cells was prepared as described by Ausubel et al. (6) with the following modification. Cells of 50-ml cultures were harvested by centrifugation and suspended in 8.5 ml TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0), and 1 ml lysozyme solution (10 mg/ml TE) was added. After incubation at 37°C for 2 h, 500 μl of a sodium dodecyl sulfate (SDS) solution (100 g/liter) and 50 μl of a proteinase K solution (20 g/liter TE) were added and mixed gently. After additional incubation at 37°C for 1 h, 5 ml 5 M NaCl and 1.5 ml of a cetyltrimethylammonium bromide solution (100 g per liter of 0.7 M NaCl) were added, and the solution was incubated at 65°C for 20 min. DNA was transferred to G. polyisoprenivorans by electroporation as described by Arenskötter et al. (4). All other genetic procedures and manipulations were conducted as described by Sambrook et al. (46).
Directional genome walking.
An internal 400-bp DNA fragment of sodA was obtained from G. polyisoprenivorans by applying wobble primers derived from highly conserved regions of the sodA sequences of G. westfalica and several mycobacterial strains. The up- and downstream regions of this internal sodA fragment of G. polyisoprenivorans were explored by a directional genome walking method using PCR (39). For each direction, one biotinylated oligonucleotide, which binds closely to the 5′ end of the respective DNA strand, was designed. Four degenerate walker primers described by Mishra et al. (39), expected to anneal in the flanking regions up- and downstream of the known internal sodA sequence, were obtained. The first set of eight PCRs was carried out using genomic DNA of G. polyisoprenivorans as the template and each biotinylated primer in combination with each of the four walking primers under PCR conditions described previously to amplify the flanking regions. Streptavidin-coupled magnetic beads (Roche, Switzerland) were then applied according to the manufacturer's protocol to isolate biotinylated PCR products, which were then used as the template for a second set of PCRs. For this, for each direction a specific nested primer binding even more closely to the 5′ end of the known sequence than the corresponding biotinylated oligonucleotide was designed. Each specific nested primer was then used together with a general nested primer, also described by Mishra et al. (39), for PCR, and the obtained amplification products were then cloned into pGEM-T Easy (Promega, Madison, WI) and sequenced.
DNA sequence analysis.
DNA sequences were determined with IRD800-labeled primers by using the SequiTherm Excel II Long-Read L-C kit and a model 4200 sequencer (LI-COR Biosciences, Lincoln, NE).
Preparation of extracellular protein fractions of Gordonia spp.
For preparative purposes, cells of Gordonia spp. were harvested from the aqueous phase by 30 min of ultracentrifugation at 100,000 × g and 4°C, and the clear supernatant was subsequently concentrated 200- to 1,000-fold by ultrafiltration by applying a Difco Amicon chamber equipped with a PM 10 membrane under nitrogen at 4°C. The concentrated extracellular proteins were equilibrated with 10 mM sodium phosphate buffer (pH 7) by a second filtration process. For analytical purposes small amounts of extracellular proteins were also concentrated from cell-free supernatants by applying Vivaspin 500 centrifugal filter units with a polyethersulfone membrane (Sartorius, Göttingen, Germany) having a cutoff of 10 kDa.
Preparation of soluble protein from E. coli.
E. coli cells were washed twice with 100 mM potassium phosphate buffer (pH 7.0) and were subsequently resuspended in 2 volumes of 10 mM potassium phosphate buffer (pH 7.0). During disruption by sonication employing a Sonifier 250 (Branson Sonic Power Company) with an amplitude of 16 μm (3 min/ml; 50% output control), the cells were cooled with an ice-salt mixture. Soluble membrane-free protein fractions were then prepared by 60 min of ultracentrifugation of the crude extract at 100,000 × g and 4°C.
SOD activity assay.
In supernatants of Gordonia sp. cultures grown in the presence of synthetic poly(cis-1,4-isoprene), SOD enzyme activity was directly measured by applying a SOD activity kit (Sigma-Aldrich, Munich, Germany) according to the manufacturer's manual. In contrast, in the soluble protein fractions of recombinant E. coli strains, SOD activity was measured by applying the xanthine oxidase-nitroblue tetrazolium (NBT) assay modified as described by Beauchamp and Fridovich (11). One unit of SOD is defined as the amount required to inhibit the reduction rate of NBT by 50%. The amount of soluble protein was determined as described by Bradford (15).
Cloning and expression of six-His-tagged SodA.
The coding region of sodA from G. polyisoprenivorans VH2 was amplified by PCR by applying the primers P.pETsod_Nde and P.pETsod_Xho_his. The amplified PCR product lacking the stop codon of sodA was then cloned into pGEM-T Easy, excised by restriction with NdeI and XhoI, and ligated to NdeI-XhoI-linearized plasmid pET23a DNA. The resulting plasmid, pET23a:sodAhis, was subsequently transferred to E. coli Rosetta-gami B(DE3)pLys for expression. This strain was cultivated in LB medium at 37°C to an OD600 of 0.5, and expression was then induced by addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 1 mM for 3 h. The cells were harvested and washed with sodium phosphate buffer (50 mM, pH 7.4) before six-His-tagged SodA was isolated to homogeneity by applying Ni-nitrilotriacetic acid (NTA) columns (see below).
Isolation of six-His-tagged SodA and generation of anti-SodA antibodies.
C-terminally six-His-tagged SodA was isolated under denaturing conditions from recombinant E. coli strains expressing sodA from G. polyisoprenivorans VH2 under the control of the T7 promoter by applying Ni-NTA spin columns (Qiagen, Hilden, Germany). Cells were lysed in buffer A (6 M guanidine hydrochloride, 0.1 M NaH2PO4, 0.01 M Tris-Cl; pH 8.0) to achieve effective denaturation of the enzyme. All subsequent steps were carried out according to the manufacturer's protocol. The purified protein was separated by SDS-polyacrylamide gel electrophoresis (PAGE), excised from the gel, and analyzed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry before it was used for generation of polyclonal antibodies in rabbits. This was done by Eurogentec (Belgium). The immunoglobulin G fractions from the blood sera were obtained by chromatography on protein A-Sepharose (29).
SDS-polyacrylamide gel electrophoresis, Western blot analysis, and N-terminal amino acid sequence analysis.
Samples were resuspended in gel loading buffer (0.6%, wt/vol, SDS; 1.25%, wt/vol, β-mercaptoethanol; 0.25 mM EDTA; 10%, vol/vol, glycerol; 0.001%, wt/vol, bromophenol blue; and 12.5 mM Tris-HCl, pH 6.8). Proteins were denatured by 5 min of incubation at 100°C and separated in 11.5% (wt/vol) SDS-polyacrylamide gels as described by Laemmli (34). Proteins were stained with Coomassie brilliant blue R-250 (56). Proteins blotted from SDS-polyacrylamide gels onto nitrocellulose BA83 membranes (pore size, 0.2 mm; Schleicher & Schuell, Dassel, Germany) using a Semidry Fast Blot B33 apparatus (Biometra, Göttingen, Germany) were analyzed immunologically as described by Hein et al. (28). To determine the N-terminal amino acid sequence, the proteins were blotted from an SDS-polyacrylamide gel onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA) according to the method of Towbin et al. (52) by use of a Semidry Fast Blot B33 apparatus and were analyzed by automated Edman degradation.
Native PAGE and SOD activity staining.
Cells were harvested by 20 min of centrifugation at 4,000 × g and 4°C, washed twice with 100 mM sodium phosphate buffer (pH 7.0), and resuspended in the same buffer. Cell disruption was done twice for 10 min in a type MM301 bead mill (Retsch, Haan, Germany). The cell tube was cooled down with fluid nitrogen before and after the first cell disruption. Native polyacrylamide gels were prepared as described by Laemmli (34). Protein samples were loaded with 5× loading buffer containing 20% (wt/vol) sucrose and 0.1% (wt/vol) bromine phenol blue. The native polyacrylamide gel was handled as described by Juhnke et al. (32), and SOD activity was detected by the method of Beauchamp and Fridovich (11). Bands of SOD activity appeared as transparent zones on a purple background.
Purification of SodA from G. polyisoprenivorans VH2.
Purification of SodA was carried out as described by Beaman et al. (10) with some modifications. G. polyisoprenivorans VH2 cells were cultivated in 30 liters MSM containing 0.2% (wt/vol) sodium propionate. This carbon source was used to compare the intracellular active SOD with the extracellular active SOD expressed by G. polyisoprenivorans VH2 cultivated in the presence of poly(cis-1,4-isoprene) as the sole carbon source. Cell disruption was done twice in a bead mill, and debris and unbroken cells were removed by 1 h of centrifugation at 100,000 × g. Anion-exchange chromatography was performed as the second purification step, and the cleared lysate was applied at a flow rate of 0.5 ml min−1 to a Q-Sepharose column, which had been washed earlier with 0.01 M Tris-HCl and 0.1 mM EDTA, pH 7.8 (buffer A), at a flow rate of 5 ml min−1. After the column had been washed with buffer A, proteins were eluted with a linear gradient of 500 ml buffer A containing 0 to 1,000 mM NaCl at a flow rate of 5 ml min−1. Fractions exhibiting SodA activity were pooled, concentrated to 5 ml, and dialyzed against buffer A overnight at 4°C. The combined sample was purified by anion-exchange chromatography again. The Q-Sepharose column was washed with buffer A and equilibrated with buffer A containing 0.2 M NaCl at a flow rate of 5 ml min−1. The dialyzed fraction was applied to the column at a flow rate of 0.5 ml min−1. After the column was washed with buffer A containing 0.2 M NaCl, proteins were eluted by applying a linear gradient with NaCl concentrations increasing from 0.2 to 0.45 M (250 ml) at a flow rate of 5 ml min−1. Fractions with SodA activity were combined and concentrated by ultrafiltration.
Nucleotide sequence accession numbers.
The DNA sequences of G. westfalica sodA and the downstream flanking region and G. polyisoprenivorans sodA and the upstream flanking region are available under GenBank accession numbers AJ312188 and EF178278, respectively.
RESULTS
Proteins secreted by G. westfalica during rubber degradation.
Cleavage of poly(cis-1,4-isoprene) as the initial step of degradation occurs extracellularly or at the cell surface. Enzymes responsible for rubber cleavage by Streptomyces sp. strain K30 and Xanthomonas sp. strain Y35 were found to be secreted into the extracellular environment (31, 45, 58). Therefore, it is supposed that other accessory proteins essential for the initial step of rubber degradation are also localized outside the cells. Cell-free culture supernatants of G. westfalica and G. polyisoprenivorans grown in the presence of poly(cis-1,4-isoprene) were therefore derived from various growth phases, concentrated, and analyzed by SDS-PAGE to identify such proteins. On comparison of the resulting extracellular protein pattern to that of cultures grown in the presence of succinate, it was found that one predominant protein band with an apparent molecular mass of 25 kDa was specifically present in cultures degrading the polymer, as shown in Fig. 1 (lane WT) for supernatants obtained from a G. polyisoprenivorans VH2 culture. To determine the amino-terminal sequences of these proteins, the extracellular proteins from poly(cis-1,4-isoprene) cultures of G. westfalica cells and also from G. polyisoprenivorans VH2 cells were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes and the corresponding protein bands were excised and subjected to Edman degradation. When the obtained amino-terminal sequences (EYALPVLDYDYAAL and AEYTLPDLPY, respectively) were compared to sequences of proteins in databases, it was found that they exhibited about 78% and 90% identical amino acids to the amino-terminal sequences of a manganese SOD of Nocardia asteroides GUH-2 (accession no. P53651) and a SOD of Kineococcus radiotolerans SRS30216 (NC009664.1), respectively. The sequences followed the methionine encoded by the translational start codon, and no hints of a leader sequence were obtained.
FIG. 1.

Extracellular protein fractions of G. polyisoprenivorans VH2 and mutants grown in the presence of poly(cis-1,4-isoprene) for 30 days at 30°C. Culture supernatants were concentrated approximately 200-fold, and the proteins were subsequently separated in an SDS-polyacrylamide gel (12.5%, wt/vol, acrylamide). M, low-molecular-weight calibration kit (Pharmacia, Uppsala, Sweden); WT, G. polyisoprenivorans VH2 wild type; I3 and I5, “irregular” mutants; B, C, and D, sodA disruption mutants. The position of SodA in the gel is indicated by an arrow; *, protein occurring in “irregular” mutants and in G. polyisoprenivorans VH2 sodAΔKm.
Cloning of the complete sodA gene of G. westfalica.
G. westfalica was chosen for further studies, because it was genetically much more accessible than G. polyisoprenivorans. To amplify the central region of the G. westfalica sodA gene, primer P.SOD11 and primer P.SOD22 were used. An approximately 450-bp PCR product was obtained, cloned into pGEM-T Easy yielding pGEM-TSOD1122, and sequenced. The amino acid sequence obtained shared 66% identity with that of SodA from N. asteroides GUH-2. A digoxigenin-labeled pGEM-TSOD1122 DNA identified one single fragment of about 6,000 bp in SacI-digested total DNA of G. westfalica by DNA-DNA hybridization. Therefore, a partial genomic library of SacI-digested genomic DNA was prepared in pBluescript SK− DNA in E. coli XL1-Blue. DNA-DNA hybridization with digoxigenin-labeled pGEM-TSOD1122 DNA and analysis of the nucleotide sequence, which was obtained by applying the primer hopping strategy, identified the incomplete sodA on the smaller fragment. To obtain also the sequence of the DNA region beyond the SacI restriction site encoding the first 25 amino acids of SodA, inverse PCR was applied to religated BamHI-digested genomic G. westfalica DNA by employing primers P.IPCRCS3 and P.IPCRCS4. The obtained product was cloned into plasmid pGEM-T Easy and sequenced.
A contiguous DNA sequence of 4,439 bp comprising the entire sodA gene (630 bp) plus 10 and 3,799 bp of the upstream and downstream regions, respectively, was obtained (Fig. 2). The sequenced gene contained the coding region for the N-terminal sequence of SodA that had been determined by Edman degradation. The translational product of sodA exhibited significant homologies to various SODs, with the highest homology (70% identical amino acids) to the putative SOD of Micrococcus luteus NCTC 2665 (NCBI nr accession no. ZP_02944066.1), as shown in the phylogenetic tree (Fig. 3). Since only 10 bp upstream of the start codon of sodA was obtained, no complete Shine-Dalgarno sequence could be identified, and genes in sequences upstream of sodA remained unknown. Three genes encoding a putative ABC transporter (a probable transmembrane protein, an ATP-binding protein, and a protein with homologies to hypothetical proteins of unknown function) are located downstream of sodA.
FIG. 2.

Relationship between SODs from different organisms. The dendrogram was constructed using the CLUSTAL_X program as described by Thompson et al. (51). Relatedness is represented by the branch length. Bar, 0.1 amino acid substitution. GenBank accession numbers for the organisms represented are as follows: Gordonia sp. strain Kb2, CAC85367; Mycobacterium smegmatis MC2 155, YP_890640; Micrococcus luteus NCTC 2665, ZP_02944066; Brevibacterium linens BL2, ZP_00380098; Kineococcus radiotolerans SRS30216, YP_001363305; Nocardia farcinica IFM 10152, YP_116327; Mycobacterium abscessus, YP_001700872; Nocardia brasiliensis ATCC 700358 SodA, ABD64088; Rhodococcus sp. strain RHA1 SodA, YP_703964; Nocardia asteroides GUH2, P53651; Mycobacterium fortuitum, Q59519; Mycobacterium sp. strain MCS, YP_642204; Mycobacterium avium subsp. paratuberculosis K10, NP_959121; Mycobacterium avium 104, YP_879475; Kocuria rhizophila DC2201, YP_001853893; Mycobacterium lepraemurium, O86165; Mycobacterium smegmatis MC2 155 SodA, YP_890845; Mycobacterium avium subsp. paratuberculosis, ABZ81482; Renibacterium salmoninarum ATCC 33209, YP_001625417; Mycobacterium avium subsp. avium, ABZ81479.1; Arthrobacter sp. strain FB24, YP_831566.1; Arthrobacter pascens DMDC12, ABG76965.1; Mycobacterium avium subsp. paratuberculosis, ABZ81484.1; Mycobacterium avium subsp. hominissuis, ABZ81485.1; Mycobacterium leprae TN, NP_301180.1; Mycobacterium tuberculosis H37Rv, NP_218363.1; Arthrobacter aurescens TC1, YP_947831.1; Arthrobacter nitroguajacolicus, YP_001210461; Mycobacterium bovis AF 2122/97, NP_857513.1.
FIG. 3.

Screening for sodA disruption mutants of G. polyisoprenivorans VH2 by colony PCR. Single colonies of putative sodA disruption mutants were suspended in 50 μl TE buffer, and the suspension was then boiled for 15 min. After centrifugation, 0.5 μl was applied as a template for a 10-μl PCR mixture, and the products were separated in a 1% (wt/vol) agarose gel. M, λ DNA digested with PstI; WT, G. polyisoprenivorans VH2 wild type; I3, “irregular” mutant; B, C, and D, sodAΔKm disruption mutants of G. polyisoprenivorans VH2.
Functional expression of G. westfalica SodA in an E. coli sodA sodB double mutant.
The PCR product obtained with primers P.SPB and P.SPE and DNA of G. westfalica as the template was restricted with BamHI plus EcoRI and ligated with BamHI-EcoRI-linearized pBluescript SK−, yielding plasmid pSKsod. By this means, sodA was cloned in colinear orientation to the lacZ promoter and in frame with the region of lacZ responsible for α-complementation. To investigate the fusion of the N-terminal LacZ region with the entire SodA from G. westfalica with regard to complementation of the SOD-negative phenotype, pSKsod was transformed into the sodA sodB double mutant QC779 of E. coli. Soluble protein fractions from cells harboring pSKsod and cultivated for 12 h at 37°C in LB medium containing an appropriate antibiotic and 1 mM IPTG for expression of SodA exhibited a specific SOD activity of 62.6 U/mg protein, whereas in the control harboring pBluescript SK− only 6.8 U/mg protein was measured. This clearly indicated functional expression of the G. westfalica SodA in E. coli. This was also confirmed by the paraquat assay. After 42 h of cultivation in the presence of paraquat, E. coli QC779 harboring pSKsod reached about a threefold-higher OD600 (2.1) than E. coli QC779 harboring pBluescript SK− (0.8). Consequently, the G. westfalica SodA protected E. coli QC779 from oxidative damage by superoxide radicals and compensated for the absence of the E. coli SODs, as indicated by an increased paraquat resistance.
Sodium azide (NaN3) as a Mn SOD inhibitor affects rubber degradation by G. westfalica.
Different compounds inhibit the activity of SODs depending on the metal ion present at the active site of the enzyme. Cu/Zn SODs are inhibited by CN− ions, whereas Fe and Mn SODs are effectively inhibited by N3− ions (40). Therefore, we studied the effect of NaN3 on growth of G. westfalica in MSM with poly(cis-1,4-isoprene) or nonrelated carbon sources like gluconate as the sole carbon source and in complex media like St-I. With only 0.5 mM NaN3 in the medium, growth of cells with poly(cis-1,4-isoprene) was completely inhibited, whereas it was only slightly reduced on gluconate and not at all in St-I medium. Growth on any substrate was completely inhibited by 7 mM NaN3.
Cloning of sodA from G. polyisoprenivorans.
Several attempts to inactivate SodA by gene disruption in G. westfalica to obtain final proof of its involvement in rubber degradation failed due to the absence of an efficient gene transfer system for G. westfalica (4). Since G. polyisoprenivorans VH2 also secreted noticeable amounts of SodA protein (Fig. 1) in the presence of poly(cis-1,4-isoprene) and since this strain is readily accessible for DNA uptake by electroporation (4, 7), we continued the studies with this closely related bacterium.
Primers P.SODVH2F1 and P.SODVH2B1 were deduced from nucleotide sequences highly conserved in many sodA genes of actinomycetes and yielded by PCR a central region of the sodA gene comprising 423 bp from total genomic DNA of strain VH2. The up- and downstream regions of this central sodA region were obtained by directional PCR-based genome walking (39) employing one of the biotinylated primers P.SODVH2up1bio and P.SODVH2down1bio in combination with each of the walker primers P.walker1 to P.walker4 in the first round. After purification of biotinylated amplification products, a more specific PCR product was then amplified in the second round using primers P.SODVH2up1 and P.SODVH2down1 individually, in combination with primer P.nested. The obtained PCR products were cloned into pGEM-T Easy, and the sequences were assembled, yielding a 1,285-bp fragment. Putative start (GTG) and stop (TGA) codons were identified at positions 647 and 1277, respectively. Therefore, the G. polyisoprenivorans sodA gene comprises 630 bp coding for a protein of 209 amino acids. Highest homologies of 90% and 69% identical amino acids were obtained with SodA from G. westfalica and with SodA from M. smegmatis MC2 155 (accession no. YP_890640.1) and M. luteus NCTC 2665 (accession no. ZP_02944066.1), respectively (Fig. 2). SOD homologues of Gordonia strains are closely related to other sodA-encoded proteins from representative species of gram-positive bacteria.
Construction of sodA disruption mutants of G. polyisoprenivorans.
The 1,285-bp sequence comprising the entire sodA coding region (positions 647 to 1279) exhibited a unique restriction site for ClaI at position 810, which was 163 nucleotides downstream of the putative start codon. For this reason a 1,284-bp fragment was amplified by PCR using primers P.SODVH2-647 and P.SODVH2+9 and cloned into pGEM-T Easy. This vector was used because of the missing cleavage site for ClaI in the vector. Since the resulting plasmid, pGEM-T-1.3sod, isolated from E. coli TOP10 could not be digested with ClaI, indicating methylation at its recognition site, it was transformed into E. coli ET12567 lacking DNA-methylating enzymes. It could then be linearized with ClaI. The 3′ protruding ends were blunted with T4 DNA polymerase, and an approximately 1,000-bp SmaI-SmaI kanamycin resistance (Km) cassette was inserted at position 163 of sodA.
The 2.3-kbp sodAΔKm DNA fragment was amplified by PCR using again P.SODVH2-647 and P.SODVH2+9, and the resulting linear DNA fragment was purified, dialyzed, and transferred to G. polyisoprenivorans VH2 by electroporation. Recombinant clones were selected for chromosomal integration of the 2.3-kbp sodAΔKm fragment on St-I medium agar plates containing kanamycin (50 μg/ml). Two individual transformation reactions yielded in total more than 400 kanamycin-resistant colonies. Colony PCR using again P.SODVH2-647 and P.SODVH2+9 gave, surprisingly, only 18 clones that did not possess the wild-type 1,284-bp PCR product, only the 2.3-kbp sodAΔKm knockout PCR product (Fig. 3), as expected for knockout mutants. All other clones (more than 382) exhibited both the wild-type 128-bp fragment and the 2.3-kbp sodAΔKm fragment, indicating unspecific integration of the 2.3-kbp sodAΔKm DNA fragment somewhere else in the chromosome.
Noticeably, mutants A to O, which did not harbor the 128-bp wild-type fragment exhibited delayed growth on St-I medium after electroporation and a colony morphology slightly different from those of the wild type, with colonies being rougher. The phenotype of only two mutants (mutants I3 and I5) did not differ from that of the wild type. DNA-DNA hybridization experiments employing sodA as a digoxigenin-labeled probe also confirmed disruption of sodA in each of the rough mutants (mutants A to O) (Fig. 4). In comparison to those of the wild type, the hybridizing ApaI fragments of these mutants were approximately 1 kb larger, corresponding to the size of the inserted kanamycin resistance cassette. However, the ApaI DNA fragments of mutants I3 and I5 were approximately 2.5 to 3.0 kb larger than the corresponding fragments of the wild type hybridizing to the sodA probe, suggesting an irregular integration of the 2.3-kbp sodAΔKm DNA fragment.
FIG. 4.

DNA-DNA hybridization experiments. Total DNA of G. polyisoprenivorans VH2 and of putative sodA mutants was digested with ApaI and separated in a 1% (wt/vol) agarose gel. sodA DNA labeled with digoxigenin by PCR was used as a probe. M, λ DNA digested with PstI; WT, G. polyisoprenivorans VH2 wild type; I3 and I5, “irregular” mutants; B, C, D, and H, sodAΔKm disruption mutants of G. polyisoprenivorans VH2.
Effect of sodA disruption on growth.
If SodA has some function for poly(cis-1,4-isoprene) degradation in Gordonia spp., its absence should have a deleterious effect on the utilization of this polymer. The effect of sodA inactivation on growth of mutants B, C, D, and H of G. polyisoprenivorans VH2 and of the “irregular” mutants, I3 and I5, of G. polyisoprenivorans VH2 in the presence of poly(cis-1,4-isoprene) was abundantly clear (Fig. 5, right). After 45 days of incubation, MSM cultures of the wild type with 0.2% (wt/vol) synthetic poly(cis-1,4-isoprene) as the sole carbon source reached an OD600 of 3.32, whereas the G. polyisoprenivorans VH2sodAΔKm insertion mutants (mutants B, C, D, and H) grew much slower and to an OD600 of only 1.32. Both irregular mutants (I3 and I5) also differed from the wild type, but to a lesser extent: they grew to an OD600 of 2.45.
FIG. 5.

Growth behavior of sodA mutants. Cells were cultivated in duplicate; graphs show the arithmetic means with standard deviations of two experiments. (Right) Effect of sodA disruption on growth in the presence of poly(cis-1,4-isoprene) or other carbon sources. ▪, wild type of G. polyisoprenivorans VH2; ▴ sodAΔKm disruption mutants B, C, D, and H of G. polyisoprenivorans VH2; • “irregular” mutants I3 and I5 of G. polyisoprenivorans VH2. Cells were grown in the presence of synthetic poly(cis-1,4-isoprene) as the sole source for carbon and energy, and growth was recorded for an incubation period of 45 days by measuring the optical density. (Left) Growth of the wild type and of the sodAΔKm mutants B, C, D, and H of G. polyisoprenivorans VH2 in the presence of 1% (wt/vol) sodium acetate or 1% (wt/vol) sodium succinate: ▪, wild type of G. polyisoprenivorans VH2 grown in the presence of acetate (cells were cultivated in duplicate. •, sodAΔKm disruption mutants B and C of G. polyisoprenivorans grown on acetate; ▴, wild type of G. polyisoprenivorans VH2 grown on succinate (cells were cultivated in duplicate); ▾, sodAΔKm disruption mutants B, C, D and H of G. polyisoprenivorans grown on succinate.
SODs are of general importance for detoxification of bacterial cells during aerobic growth. Therefore, the effect of the inactivated sodA gene not only on growth in the presence of poly(cis-1,4-isoprene) but also on growth in the presence of other carbon sources like acetate and succinate was investigated. MSM cultures containing either 1% (wt/vol) sodium acetate or succinate were inoculated with St-I precultures of the wild-type G. polyisoprenivorans VH2 or the mutant G. polyisoprenivorans VH2 sodAΔKm grown for 48 h, and growth was monitored by measuring OD600 (Fig. 5, left). All four sodAΔKm disruption mutants mentioned above exhibited growth almost identical to that of the wild type, indicating that SodA did not affect growth on substrates not related to isoprenoids. The effect of a lack of SodA on growth on carbon sources other than poly(cis-1,4-isoprene) was also confirmed by comparing levels of colony growth of the wild type and of the mutants on solid MSM containing various carbon sources. In these cases absolutely no effect could be observed, whereas the effect of SodA on utilization of the polymer on solidified latex MSM was clearly visible.
Effect of sodA disruption on the presence of extracellular SodA and enzyme activity.
At the end of the cultivation experiments described above, the extracellular protein fractions were analyzed by SDS-PAGE (Fig. 1). As expected, SodA protein was present in the supernatants of cultures of G. polyisoprenivorans and also of both irregular mutants, I3 and I5, but was completely absent in cultures of G. polyisoprenivorans VH2 sodAΔKm mutants B, C, D, and H. In the culture supernatants of all regular and irregular mutants, there was an additional protein with an apparent molecular mass of 30 kDa which was absent in the supernatant of the wild type. Since it was present if the wild-type allele had been replaced (G. polyisoprenivorans VH2 sodAΔKm mutants A to O) or if it was integrated irregularly, as in mutants I3 and I5, this protein probably is the kanamycin phosphotransferase.
Analysis of SOD enzyme activity in the supernatants confirmed the observations described above. Whereas SOD activity in supernatants of the wild type was approximately 1.8 U/ml, it was only 1.0 U/ml in the supernatants of the irregular mutants, I3 and I5, and below 0.01 U/ml in the supernatants of the disruption mutants (G. polyisoprenivorans VH2 sodAΔKm mutants A to O).
Additionally, analysis of the intracellular and extracellular protein patterns of the regular disruption strains by native PAGE in combination with activity staining demonstrated total absence of SOD activity in comparison to that associated with protein patterns of the irregular disrupted mutants (Fig. 6).
FIG. 6.

Detection of SOD activity in regular and irregular G. polyisoprenivorans VH2 disruption mutants during growth in the presence of poly(cis-1,4-isoprene) or sodium propionate by native PAGE. Cells were incubated at 30°C in MSM containing 0.2% (wt/vol) sodium propionate (lanes 1 to 7) or 0.2% (wt/vol) poly(cis-1,4-isoprene) (lanes 8 to 14), and crude extracts were analyzed. Lanes: 1 and 8, G. polyisoprenivorans mutant B; 2 and 9, G. polyisoprenivorans mutant C; 3 and 10, G. polyisoprenivorans mutant D; 4 and 11, G. polyisoprenivorans mutant F; 5 and 12, G. polyisoprenivorans mutant H; 6 and 13, G. polyisoprenivorans “irregular” mutant I3; 7 and 14, G. polyisoprenivorans “irregular” mutant I5.
Complementation of the VH2 sodAΔKm mutant.
The E. coli/Rhodococcus shuttle vector pNC9501 was used to restore SOD activity in disruption mutants of G. polyisoprenivorans VH2. Therefore, the vector pGEM-T-1.3sod was restricted with EcoRI, yielding a linearized vector and a 1.3-kbp DNA fragment comprising sodA with its putative promoter region. The 1.3-kbp EcoRI sodA DNA fragment was ligated with the EcoRI-linearized E. coli/Rhodococcus shuttle vector pNC9501, yielding pNC9501::sodA, and transformed into G. polyisoprenivorans VH2 sodAΔKm sodA disruption mutants B, C, D, H, I3, and I5. All randomly chosen transformants, which were selected on St-I agar plates containing thiostrepton (25 μg/ml), harbored pNC9501::sodA. Cultivation experiments in liquid MSM with poly(cis-1,4-isoprene) as the sole carbon source revealed that growth of G. polyisoprenivorans VH2 sodAΔKm was restored by pNC9501::sodA to a level comparable to that of the wild type. Moreover, SOD enzyme activity staining in nondenaturing polyacrylamide gels demonstrated expression of active SOD in cells of all complemented mutants like that in wild-type cells and unlike that in the disruption mutants (Fig. 7).
FIG. 7.
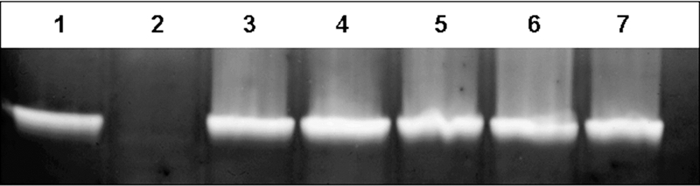
Detection of SOD activity in wild-type G. polyisoprenivorans VH2, G. polyisoprenivorans VH2 sodAΔKm mutant C, and the SodA-complemented G. polyisoprenivorans VH2 sodAΔKm(pNC9501::sodA) mutant. Cells were incubated in MSM containing 0.2% (wt/vol) poly(cis-1,4-isoprene) at 30°C for 14 days. Lanes contained soluble protein fractions. Lanes: 1, wild-type G. polyisoprenivorans VH2; 2, G. polyisoprenivorans VH2 sodAΔKm mutant C; 3 to 7, different transformants of G. polyisoprenivorans VH2 sodAΔKm harboring plasmid pNC9501::sodA for genetic complementation.
Purification of six-His-tagged SodA.
The coding region of sodA from G. polyisoprenivorans VH2 was cloned into the E. coli expression vector pET23a under the control of the T7 promoter, yielding plasmid pET23a:sodAhis. Recombinant strains of E. coli Rosetta-gami B(DE3)pLys harboring this plasmid were employed for heterologous expression as described in Materials and Methods. Six-His-tagged SodA was purified to homogeneity with Ni-NTA columns (Qiagen, Hilden, Germany) by applying buffer A for denaturation (6 M guanidine hydrochloride, 0.1 M NaH2PO4, 0.01 M Tris-Cl; pH 8.0) since purification after lysis with buffer B (8 M urea, 0.1 M NaH2PO4, 0.01 M Tris-Cl; pH 8.0) was not successful. The purified protein was separated by SDS-PAGE, excised from the gel, and analyzed by MALDI-TOF mass spectrometry (data not shown) before it was used for generation of antibodies.
Native PAGE and SOD activity staining.
To compare supernatants of cells grown with sodium succinate, intracellular and extracellular proteins from cultures cultivated with water-soluble substrates (glucose or sodium salts of propionate, acetate, or gluconate), latex, and synthetic rubber were analyzed by native PAGE with activity staining. The SOD activity was visualized with NBT (11). Interestingly, the activity of only one SOD protein could be detected on native gels in all intracellular protein samples. These results indicate that G. polyisoprenivorans VH2 contains only a single SOD. In contrast, no extracellular SOD activity was detectable in native gels from cultures grown with water-soluble substrates as carbon sources. After 1 week of incubation at 30°C, the extracellular protein fractions of latex cultures already showed SOD activity, and after 2 weeks the extracellular protein fractions of poly(cis-1,4-isoprene) cultures also exhibited SOD activity (Fig. 8). All results obtained from native PAGE were confirmed by Western blotting employing the antibodies raised against SodA.
FIG. 8.

Comparison of SOD activities in G. polyisoprenivorans VH2 growing on various carbon sources. Cells were incubated at 30°C in MSM containing 0.5% (vol/vol) latex, 0.2% (wt/vol) poly(cis-1,4-isoprene), 0.2% (wt/vol) glucose, 0.2% (wt/vol) gluconate, 0.2% (wt/vol) propionate, or 0.2% (wt/vol) acetate. Proteins of soluble cell fractions and supernatants were tested. Lanes: 1, 1-day incubation with latex (soluble cell fraction); 2, 1-day incubation with latex (supernatant); 3, 2-day incubation with latex (soluble cell fraction); 4, 2-day incubation with latex (supernatant); 5, 3-day incubation with latex (soluble cell fraction); 6, 3-day incubation with latex (supernatant); 7, 12-day incubation with poly(cis-1,4-isoprene) (soluble cell fraction); 8, 12-day incubation with poly(cis-1,4-isoprene) (supernatant); 9, 13-day incubation with poly(cis-1,4-isoprene) (soluble cell fraction); 10, 13-day incubation with poly(cis-1,4-isoprene) (supernatant); 11, 14-day incubation with poly(cis-1,4-isoprene) (soluble cell fraction); 12, 14-day incubation with poly(cis-1,4-isoprene) (supernatant); 13, 1-day incubation with glucose (soluble cell fraction); 14, 1-day incubation with gluconate (soluble cell fraction); 15, 1-day incubation with propionate (soluble cell fraction); 16, 1-day incubation with acetate (soluble cell fraction).
DISCUSSION
Biochemical studies at a molecular level of biodegradation of the polyisoprenoids natural rubber and gutta percha started only recently. Whereas almost nothing is still known about the microbial degradation of poly(trans-1,4-isoprene) and the first axenic cultures only recently became available (55), two extracellular enzymes capable of cleaving poly(cis-1,4-isoprene) were identified (31, 45). RoxA (rubber oxygenase A) is secreted by the gram-negative Xanthomonas sp. strain 35Y during growth of cells on natural rubber latex (30). Isolated dioxygenase RoxA cleaves poly(cis-1,4-isoprene) at the double bonds, yielding 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al as the major cleavage product (13, 14). Lcp (latex clearing protein) cleaves poly(cis-1,4-isoprene) in Streptomyces sp. strain K30 (45). A dicarbonyl isoprenoid with keto and aldehyde terminal groups resulted from oxidative cleavage by Nocardia farcinica S3 harboring an Lcp homologue (30). Oxidative degradation of cis- and trans-polyisoprenes and of vulcanized natural rubber was also demonstrated in vitro by two different artificial enzyme mediator systems. One applied lipoxygenase/linoleic acid and horseradish peroxidase/1-hydroxybenzotriazole, yielding polyisoprenoid molecules with low molecular weight (23). Degradation of vulcanized and nonvulcanized polyisoprenes by lipid peroxidation catalyzed by oxidative enzymes and transition metals was also demonstrated (47).
Although rubber-degrading Gordonia species possess genes coding for functional active Lcp (18), the functions of these homologues and the enzymes in these bacteria initiating rubber degradation are not known. As in other bacteria, cleavage of poly(cis-1,4-isoprene) occurred at the double bonds by oxygen addition (21, 53), yielding cleavage products with aldehyde and keto groups at the surface of degrading rubber materials (35). During growth on poly(cis-1,4-isoprene), the rubber-degrading bacteria G. polyisoprenivorans and G. westfalica expressed 25-kDa extracellular proteins (Fig. 1) which were unequivocally identified as Mn SODs. A Mn SOD was also observed in two-dimensional gels in supernatants obtained from rubber-degrading Nocardia farcinica IFM10152 cells (Q. Arenskötter, unpublished data).
Although little is known about the function of extracellular SODs, they are obviously important for certain lifestyles. For pathogenic microorganisms, an involvement in resistance against immune defense has been discussed (2). Some organisms express a SOD during degradation of certain compounds; one of these is the lignin degrader Phanerochaete chrysosporium, which expressed a Mn SOD during exposure to dibenzo-p-dioxin (33). The diatom Skeletonema costatum increased SOD activity during exposure to 2,4-dichlorophenol (57). In rat liver, copper/zinc SOD activity was shown to stimulate anthranilamide hydroxylation by a microsomal monooxygenase system (41), and in Phanerochaete chrysosporium BKM-F-1767 SOD activity enhanced the rate of veratryl alcohol oxidation by lignin peroxidase (9). Since SOD activity produces hydrogen peroxide from superoxide anions, it may in this case provide the substrate for the lignin peroxidase reaction and thereby increase enzyme activity. Additionally, during oxidation of certain extracellular compounds, large amounts of cell-damaging ROS arise.
Multiple pieces of evidence for the involvement of SodA in rubber degradation were obtained in this study. (i) Its formation is induced in the presence of poly(cis-1,4-isoprene). (ii) Regular sodAΔKm mutants, which were obtained by a homogenotization approach using the knockout cassette sodAΔKm (Fig. 3), mineralized rubber to CO2 much slower than the wild type. (iii) The typical Mn SOD inhibitor NaN3 affected growth of G. westfalica significantly at low concentrations during cultivation on poly(cis-1,4-isoprene) but not on nonisoprenoid carbon sources. (iv) SodA restored enhanced paraquat resistance in the E. coli sodA sodB double mutant QC779. (v) SOD enzyme activity could be measured in recombinant E. coli cells expressing SodA of G. polyisoprenivorans. (vi) SOD enzyme activity was also demonstrated by activity staining in polyacrylamide gels. Detailed analyses of the function of Sod in these Gordonia species were mostly done with G. polyisoprenivorans because it is much more genetically accessible than G. westfalica (4). All mutants with confirmed sodA disruption exhibited almost no extracellular SOD activity, and also no SodA was detectable by SDS-PAGE, Western blotting, and native PAGE analysis (Fig. 1). The phenotype of the “irregular” mutants and how they still synthesize a functional SodA (Fig. 1) are not understood. Surprisingly, disruption sodA mutants were distinguishable from the wild type due to an altered, rougher colony morphology. This indicated not only that SodA affects growth on poly(cis-1,4-isoprene) but also that it is important for other functions. However, whereas growth of G. polyisoprenivorans VH2 sodAΔKm on synthetic poly(cis-1,4-isoprene) was clearly reduced (Fig. 5, right panel) in comparison to that of the wild type, no negative effect was observed during growth on acetate or succinate (Fig. 5, left panel).
The genomes of most bacteria closely related to G. polyisoprenivorans encode at least two SODs; typically, they synthesize both a copper/zinc SOD and an iron/manganese SOD. Various analyses clearly indicated that the investigated G. polyisoprenivorans strain possesses only one SOD, which belongs to the latter group. SODs isolated from extracellular fractions of actinomycetes also usually belong to the iron/manganese SOD family. Although Western blot analysis and native PAGE also demonstrated expression of SodA during growth on nonpolyisoprenoid substrates like acetate, SodA is not essential for aerobic growth of G. polyisoprenivorans VH2, as revealed by the phenotype of the sodA mutant. SodA was present in the wild type only intracellularly after growth on these substrates. Since the extracellular SOD expressed during growth on poly(cis-1,4-isoprene) and the intracellular SOD expressed during growth on propionate exhibited identical N-terminal amino acid sequences, only one SOD, rather than two SODs which are differently expressed during growth on substrates that have, e.g., rubber on one side and a nonisoprenoid on the other side, is present in G. polyisoprenivorans. Since sodA disruption mutants of G. polyisoprenivorans degraded poly(cis-1,4-isoprene) at a reduced rate, a lack of SodA cannot be compensated for by another SOD enzyme, and other oxidant defense enzymes like glutathione reductase, catalase, and peroxidase (49) must compensate for the lack of SOD in G. polyisoprenivorans. Since also almost no extracellular SOD activity was measured in supernatants of the sodA mutants, SodA is unlikely to produce hydrogen peroxide as a substrate for a rubber-cleaving enzyme. Otherwise, inactivation of sodA would have led to a total loss of rubber-degrading ability. This clearly indicates that SodA functions as an extracellular radical scavenger enzyme during cleavage of poly(cis-1,4-isoprene) when toxic ROS superoxide anions are probably released at elevated concentrations.
Tullius et al. (54) proposed that extracellular SODs from mycobacteria are not actively secreted but appear extracellularly due to bacterial leakage or autolysis; the extracellular abundance of these enzymes resulted from their high level of expression and extracellular stability. N-terminal analysis of SOD from G. westfalica and G. polyisoprenivorans VH2 gave no hints that SodA is secreted actively, as described for M. tuberculosis (16), but rather indicated that it was detectable due to long incubation periods and the stability of the enzyme. Otherwise, SodA should be also be detected extracellularly when cultivated on water-soluble substrates. Possibly, SodA released from perforated and/or dying cells during the long incubation times required for rubber degradation may protect the surviving cells from oxidative damage. Secretion of SodA by M. tuberculosis depends on an alternative SecA pathway. This pathway is not based on a signal peptide (17). Whether Gordonia also possesses such an alternative Sec pathway is not known. In this case, intracellular SodA could protect cells during growth on water-soluble substrates and is secreted only after induction of the alternative pathway, e.g., in the presence of poly(cis-1,4-isoprene). This has to be investigated in the future.
Acknowledgments
We are indebted to Bernhard Schmidt (Institut für Biochemie II der Georg-August-Universität Göttingen, Germany) and Barbara Schedding (Institut für Physiologische Chemie und Pathobiochemie, Universitätsklinikum Münster, Germany) for N-terminal sequence analyses. MALDI-TOF analyses by Simone König (Integrated Functional Genomics, Interdisciplinary Center for Clinical Research, Westfälische Wilhelms-Universität Münster) are gratefully acknowledged.
Footnotes
Published ahead of print on 24 October 2008.
REFERENCES
- 1.Alamuri, P., and R. J. Maier. 2006. Methionine sulfoxide reductase in Heliobacter pylori: interaction with methionine-rich proteins and stress-induced expression. J. Bacteriol. 188:5839-5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alcendor, D. J., G. D. Chapman, and B. L. Beaman. 1995. Isolation, sequencing and expression of the superoxide dismutase-encoding gene (sod) of Nocardia asteroides strain GUH-2. Gene 164:143-147. [DOI] [PubMed] [Google Scholar]
- 3.Almiron, M., A. Link, D. Furlong, and R. Kolter. 1992. A novel DNA binding protein with regulatory and protective roles in starved E. coli. Genes Dev. 6:2646-2654. [DOI] [PubMed] [Google Scholar]
- 4.Arenskötter, M., D. Baumeister, R. Kalscheuer, and A. Steinbüchel. 2003. Identification and application of plasmids suitable for transfer of foreign DNA to members of the genus Gordonia. Appl. Environ. Microbiol. 69:4971-4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arenskötter, M., D. Bröker, and A. Steinbüchel. 2004. Biology of the metabolically diverse genus Gordonia. Appl. Environ. Microbiol. 70:3195-3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1987. Current protocols in molecular biology, 1st ed., vol. 1. John Wiley & Sons, New York, NY.
- 7.Banh, Q., M. Arenskötter, and A. Steinbüchel. 2005. Establishment of Tn5096-based transposon mutagenesis in Gordonia polyisoprenivorans. Appl. Environ. Microbiol. 71:5077-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnard, D., and P. M. Lewis. 1988. Oxidative ageing, p. 621-673. In A. D. Roberts (ed.), Natural rubber: science and technology, part II. Oxford University Press, New York, NY.
- 9.Barr, D. P., and S. D. Aust. 1994. Effect of superoxide and superoxide-dismutase on lignin peroxidase-catalyzed veratryl alcohol oxidation. Arch. Biochem. Biophys. 311:378-382. [DOI] [PubMed] [Google Scholar]
- 10.Beaman, B. L., S. M. Scates, S. E. Moring, R. Deem, and H. P. Misra. 1983. Purification and properties of a unique superoxide dismutase from Nocardia asteroides. J. Biol. Chem. 258:91-96. [PubMed] [Google Scholar]
- 11.Beauchamp, C., and I. Fridovich. 1971. Superoxide dismutase: improved assay and an assay applicable to acrylamide gels. Anal. Biochem. 44:276-287. [DOI] [PubMed] [Google Scholar]
- 12.Birnboim, H. C., and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7:1513-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braaz, R., P. Fischer, and D. Jendrossek. 2004. Novel type of heme-dependent oxygenase catalyzes oxidative cleavage of rubber (poly-cis-1,4-isoprene). Appl. Environ. Microbiol. 70:7388-7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braaz, R., W. Armbruster, and D. Jendrossek. 2005. Heme-dependent rubber oxygenase RoxA of Xanthomonas sp. cleaves the carbon backbone of poly(cis-1,4-isoprene) by a dioxygenase mechanism. Appl. Environ. Microbiol. 71:2473-2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradford, M. M. 1976. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 16.Braunstein, M., A. M. Brown, S. Kurtz, and W. R. Jacobs, Jr. 2001. Two nonredundant SecA homologues function in Mycobacteria. J. Bacteriol. 183:6979-6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braunstein, M., B. J. Espinosa, J. Chan, J. T. Belisie, and W. R. Jacobs. 2003. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol. Microbiol. 48:453-464. [DOI] [PubMed] [Google Scholar]
- 18.Bröker, D., D. Dietz, M. Arenskötter, and A. Steinbüchel. 2008. The genomes of the non-clearing-zone-forming and natural rubber-degrading species Gordonia polyisoprenivorans and Gordonia westfalica harbor genes expressing Lcp activity in Streptomyces strains. Appl. Environ. Microbiol. 74:2288-2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bullock, W. O., J. M. Fernandez, and J. M. Short. 1987. XL1-Blue: high efficiency plasmid transforming recA Escherichia coli strain with β-galactosidase selection. BioTechniques 5:376-378. [Google Scholar]
- 20.Carlioz, A., and D. Touati. 1986. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 5:623-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cundell, A. M., and A. P. Mulcock. 1974. The biodegradation of vulcanized rubber. Dev. Ind. Microbiol. 16:88-96. [Google Scholar]
- 22.Dong, Y., S. Demaria, X. Sun, F. R. Santori, B. M. Jesdale, A. S. De Groot. W. N. Rom, and Y. Bushkin. 2004. HLA-A2-restricted CD8+-cytotoxic-T-cell responses to novel epitopes in Mycobacterium tuberculosis superoxide dismutase, alanine dehydrogenase, and glutamine synthetase. Infect. Immun. 72:2412-2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enoki, M., Y. Doi, and T. Iwata. 2003. Oxidative degradation of cis- and trans-polyisoprenes and vulcanized natural rubber with enzyme-mediator systems. Biomacromolecules 4:314-320. [DOI] [PubMed] [Google Scholar]
- 24.Farr, S. B., and T. Kogoma. 1991. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol. Rev. 55:561-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridovich, I. 1986. Superoxide dismutases. Adv. Enzymol. 58:61-97. [DOI] [PubMed] [Google Scholar]
- 26.Fridovich, I. 1995. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 64:97-112. [DOI] [PubMed] [Google Scholar]
- 27.Hassan, H. M., and I. Fridovich. 1978. Regulation and role of superoxide dismutase. Biochem. Soc. Trans. 6:356-361. [DOI] [PubMed] [Google Scholar]
- 28.Hein, S., H. Tran, and A. Steinbüchel. 1998. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch. Microbiol. 170:162-170. [DOI] [PubMed] [Google Scholar]
- 29.Hjelm, H., K. Hjelm, and J. Sjöquist. 1972. Protein A from Staphylococcus aureus. Its isolation by affinity chromatography and its use as an immunosorbent for isolation of immunoglobulins. FEBS Lett. 28:73-76. [DOI] [PubMed] [Google Scholar]
- 30.Ibrahim, E. M. A., M. Arenskötter, H. Luftmann, and A. Steinbüchel. 2006. Identification of poly(cis-1,4-isoprene) degradation intermediates during growth of moderately thermophilic actinomycetes on rubber and cloning of a functional lcp homologue from Nocardia farcinica strain E1. Appl. Environ. Microbiol. 72:2275-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jendrossek, D., and S. Reinhardt. 2003. Sequence analysis of a gene product synthesized by Xanthomonas sp. during growth on natural rubber latex. FEMS Microbiol. Lett. 224:61-65. [DOI] [PubMed] [Google Scholar]
- 32.Juhnke, S., N. Peitzsch, N. Hübener, C. Große, and D. H. Nies. 2002. New genes involved in chromate resistance in Ralstonia metallidurans strain CH34. Arch. Microbiol. 179:15-25. [DOI] [PubMed] [Google Scholar]
- 33.Kurihara, H., H. Wariishi, and H. Tanaka. 2002. Chemical stress-responsive genes from the lignin-degrading fungus Phanerochaete chrysosporium exposed to dibenzo-p-dioxin. FEMS Microbiol. Lett. 212:217-220. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 227:680-685. [DOI] [PubMed] [Google Scholar]
- 35.Linos, A., M. M. Berekaa, R. Reichelt, U. Keller, J. Schmitt, H. C. Flemming, R. M. Kroppenstedt, and A. Steinbüchel. 2000. Biodegradation of cis-1,4-polyisoprene rubbers by distinct actinomycetes: microbial strategies and detailed surface analysis. Appl. Environ. Microbiol. 66:1639-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linos, A., and A. Steinbüchel. 2001. Biodegradation of natural and synthetic rubbers, p. 321-359. In T. Koyama and A. Steinbüchel (ed.), Biopolymers, vol. 2. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 37.Loprasert, S., P. Vattanaviboon, W. Praituan, S. Chamnongpol, and S. Mongkolsuk. 1996. Regulation of the oxidative stress protective enzymes, catalase and superoxide dismutase in Xanthomonas—a review. Gene 179:33-37. [DOI] [PubMed] [Google Scholar]
- 38.Martinez, A., and R. Kolter. 1997. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 179:5188-5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mishra, R. N., S. L. Singla-Pareek, S. Nair, S. K. Sopory, and M. K. Reddy. 2002. Directional genome walking using PCR. BioTechniques 33:830-832. [DOI] [PubMed] [Google Scholar]
- 40.Misra, H. P., and I. Fridovich. 1978. Inhibition of superoxide dismutases by azide. Arch. Biochem. Biophys. 189:317-322. [DOI] [PubMed] [Google Scholar]
- 41.Ohta, Y., I. Ishiguro, J. Naito, and R. Shinohara. 1984. Role of cytosolic superoxide dismutase as a stimulator in anthranilamide hydroxylation by a microsomal monooxygenase system in rat liver. J. Biochem. 96:1323-1336. [DOI] [PubMed] [Google Scholar]
- 42.Polle, A. 2001. Dissecting the superoxide dismutase-ascorbate-glutathione-pathway in chloroplasts by metabolic modeling. Computer simulations as a step towards flux analysis. Plant Physiol. 126:445-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porqué, P. G., A. Baldestein, and P. Reichards. 1970. The involvement of the thioredoxin system in the reaction of methionine sulfoxide and sulfate. J. Biol. Chem. 245:2371-2374. [PubMed] [Google Scholar]
- 44.Rose, K., and A. Steinbüchel. 2005. Biodegradation of natural rubber and related compounds: recent insights into a rare and hardly understood catabolic capability of microorganisms. Appl. Environ. Microbiol. 71:2803-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rose, K., K. B. Tenberge, and A. Steinbüchel. 2005. Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 6:180-188. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook, J. E., F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 47.Sato, S., Y. Honda, M. Kuwahara, and T. Watanabe. 2003. Degradation of vulcanized and nonvulcanized polyisoprene rubbers by lipid peroxidation catalyzed by oxidative enzymes and transition metals. Biomacromolecules 4:321-329. [DOI] [PubMed] [Google Scholar]
- 48.Schlegel, H. G., H. Kaltwasser, and G. Gottschalk. 1961. Ein Submersverfahren zur Kultur wasserstoffoxidierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch. Mikrobiol. 38:209-222.13747777 [Google Scholar]
- 49.Scott, M. D., S. R. Meshnick, and J. W. Eaton. 1987. Superoxide dismutase-rich bacteria. J. Biol. Chem. 262:3640-3645. [PubMed] [Google Scholar]
- 50.Snapper, S. B., R. E. Melton, S. Mustafa, T. Kieser, and W. R. Jacobs, Jr. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911-1919. [DOI] [PubMed] [Google Scholar]
- 51.Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X Windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsuchii, A., T. Suzuki, and K. Takeda. 1985. Microbial degradation of natural rubber vulcanizates. Appl. Environ. Microbiol. 50:965-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tullius, M. V., G. Harth, and M. A. Horwitz. 2001. High extracellular levels of Mycobacterium tuberculosis glutamine synthetase and superoxide dismutase in actively growing cultures are due to high expression and extracellular stability rather than to a protein-specific export mechanism. Infect. Immun. 69:6348-6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warneke, S., M. Arenskötter, K. B. Tenberge, and A. Steinbüchel. 2007. Bacterial degradation of poly(trans-1,4-isoprene) (gutta percha). Microbiology 153:347-356. [DOI] [PubMed] [Google Scholar]
- 56.Weber, K., and M. Osborn. 1969. The reliability of molecular weight determinations by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244:4406-4412. [PubMed] [Google Scholar]
- 57.Yang, S., R. S. Wu, and R. Y. Kong. 2002. Biodegradation and enzymatic responses in the marine diatom Skeletonema costatum upon exposure to 2,4-dichlorophenol. Aquat. Toxicol. 59:191-200. [DOI] [PubMed] [Google Scholar]
- 58.Yikmis, M., M. Arenskötter, K. Rose, N. Lange, H. Wernsmann, L. Wiefel, and A. Steinbüchel. 2008. Secretion and transcriptional regulation of the latex clearing protein Lcp by the rubber-degrading bacterium Streptomyces sp. strain K30. Appl. Environ. Microbiol. 74:5373-5382. [DOI] [PMC free article] [PubMed] [Google Scholar]