

Summary
Hypoxia-inducing factor-1 alpha (HIF-1α), is a major survival factor for tumor cells growing in a low oxygen environment. The anti-cancer agent imexon binds thiols and causes accumulation of reactive oxygen species (ROS) in pancreatic cancer cells. Unlike many cytotoxic agents, imexon is equi-cytotoxic in human MiaPaCa-2 and Panc-1 cells grown in normoxic (21% O2) and hypoxic (1% O2) conditions. Western blot analyses of imexon treated cells demonstrated that imexon reduces HIF-1α protein levels in both normoxic and hypoxic conditions in a time- and concentration-dependant fashion. Gemcitabine did not similarly affect HIF-1α levels. Imexon did not reduce transcription of new HIF-1α mRNA, but did reduce the synthesis of new proteins, including HIF-1α, measured by 35S methionine/cysteine (Met/Cys) incorporation. Concurrently, the half-life of existing HIF-1α protein was increased by imexon, in association with a marked inhibition of chymotryptic activity in the 20S proteasome. The inhibition of HIF-1α translation was not specific, rather it was part of a general decrease in protein translation caused by imexon. This inhibitory effect on translation did not involve phosphorylation of eukaryotic initiation factor-2α (eIF-2α) and was not closely correlated to cell growth inhibition by imexon, suggesting that mechanisms other than protein synthesis inhibition contribute to the drug’s cytotoxic effects. In summary, imexon blocks the translation of new proteins, including HIF-1α, and this effect overcomes an increase in the stability of preformed HIF-1α due to proteasome inhibition by imexon. Because net HIF-1α levels are reduced by imexon, combination studies with other drugs affected by HIF-1α survival signaling are warranted.
Keywords: Hypoxia, Imexon, Normoxia, Pancreas, Protein synthesis
Introduction
Imexon is an aziridine-containing small molecule which has shown signs of clinical activity in phase I studies of several refractory tumor types [1], as well as in a phase I study in patients with metastatic pancreatic cancer [2]. The mechanism of action for this agent has been shown to involve binding to thiols [3] leading to a buildup of reactive oxygen species and apoptosis mediated largely through the intrinsic (mitochondrial) pathway [4–6]. These effects have also been observed in human pancreatic cancer cell lines in vitro [7]. Previous mechanistic studies in human lymphoma cell lines showed that imexon-induced protein synthesis inhibition occurred at concentrations within the range shown to inhibit lymphoma cell growth in vitro [8]. The reduction in protein synthesis in the lymphoma cell lines was much greater than inhibitory effects on the synthesis of DNA and RNA, suggesting that protein synthesis inhibition was one mechanism contributing to imexon’s cytotoxicity. Recognizing that the initiation of protein translation may be due to the phosphorylation (inactivation) of the redox-sensitive translation factor eIF-2α in oxidatively stressed cells [9–11], in this report we investigated whether this may be one mechanism for imexon’s inhibitory effects on protein synthesis.
The clinical activity noted for imexon in pancreas cancer in a phase I trial [1], and the observed cytotoxic effects of the drug in pancreas cancer cell lines in vitro has led to the initiation of a randomized phase II trial of gemcitabine and imexon in metastatic pancreatic cancer at several sites across the US. This trial, which started in March 2008, will attempt to overcome the lack of activity seen with other gemcitabine combinations in pancreatic cancer, including negative phase III trials of gemcitabine with fluorouracil, oxaliplatin, capecitabine, and most recently, with the VEGF-inhibitor bevacizumab and the EGF1 receptor inhibitor, cetuximab [12, 13]. Even the recently-approved combination of gemcitabine with the EGF-receptor kinase inhibitor erlotinib (Tarceva®), improved median overall survival by only 2 weeks [14, 15]. This suggests that new agents with novel mechanisms of action are needed for pancreatic cancer.
One explanation for the lack of highly effective agents to treat advanced pancreatic cancer is the observation from surgical specimens that human pancreatic adenocarcinomas typically contain large areas of hypoxia, and this involves not just the hypo-perfused core of large solid tumor masses [16], but also a ring of hypoxic tissue at the expanding cancer/normal tissue interface [17]. Hypoxic cancer cells activate the survival protein, hypoxia-inducing factor-1 alpha (HIF-1α), as a means of adapting to growth in conditions of low oxygen tension [18–21]. This cytoplasmic protein is rapidly degraded in normoxia, but under hypoxia, it is stabilized against proteasomal degradation by inhibition of oxygen-dependent proline hydroxylases [22]. This allows HIF-1α levels to accumulate in the cytoplasm of hypoxic cells and then translocate to the nucleus where they can dimerize with the constitutively expressed nuclear subunit partner, HIF-1β, (also known as the aryl hydrocarbon nuclear translocator, or ARNT). The resultant complex can then bind to hypoxia response elements (HREs) within the promoters of target genes, and initiate transcription of a variety of cell survival genes. Cell survival genes regulated by HIF-1α include angiogenic growth factors such as vascular endothelial growth factor (VEGF) and its receptor FLT1, a series of glycolysis proteins (GLUT1, GLUT3, aldolase A and C, lactate dehydrogenase, hexokinase and phosphofructokinase), hematopoietic factors such as erythropoietin alpha, and metabolic survival factors such as insulin-like growth factor (IGF) factor 1 and the IGF binding proteins 1, 2 and 3 [22, 23]. HIF-1α transactivation has been associated with resistance to apoptosis induced by both chemotherapy and radiotherapy [24]. These hypoxic cells are also capable of developing new vasculature which promotes both local tumor growth and distant metastatic spread [25].
Hypoxic cancer cells express broad resistance to many different chemotherapy agents, including those used in the treatment of pancreatic cancer [26]. For example, hypoxia markedly increases the resistance to gemcitabine in L3.6pl human pancreatic cancer cells, possibly through activation of the MAPK (Erk) pathway [27]. However, the effects of gemcitabine and imexon on HIF-1α activity are unknown. The effect of imexon on HIF-1α activity in hypoxic pancreatic cancer cells had also not been explored nor the effect of imexon on protein synthesis in pancreatic cancer cells. While the effects of imexon on hypoxic cell signaling were unknown, reactive oxygen species (ROS) generated by imexon might alter hypoxia signaling since it has previously been reported that ROS tend to stabilize HIF-1α levels [28–30, 31]. If this occurred with imexon, it might theoretically enhance HIF-1α transcriptional activity [28, 29] and improve cancer cell survival in hypoxic conditions [30]. Overall, these observations suggested some testable hypotheses involving imexon’s activity in hypoxic pancreatic cancer cells: (1) that imexon might block protein synthesis and thereby reduce HIF-1α levels, or (2) that imexon might alternatively cause an ROS-mediated increase in HIF-1α activity commensurate with a loss of cytotoxic potency in hypoxic pancreatic cancer cells. Our findings show that imexon remains active in hypoxic cells, and does increase HIF-1α protein stability by inhibiting proteasomal degradation. However, there is an over-riding inhibition of new protein translation by imexon which causes a net decrease in global protein synthesis, including HIF-1α protein in both normoxic and hypoxic pancreatic cancer cells in vitro. These results contrast with the lack of an effect on HIF-1α expression or cytotoxic sensitivity of hypoxic pancreas cancer cells with gemcitabine.
Materials and methods
Cell culture
MiaPaCa-2 and Panc-1 and cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured in a humidified incubator at 37°C, 5% CO2, in CellGro RPMI 1640 (Mediatech, Herndon, VA) supplemented with 10% heat-inactivated bovine calf serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA).
The MiaPaCa-2 cells (CRL-1420) are an adherent epithelial human pancreatic cancer cell line with a doubling time of approximately 40 h [32]. The Panc-1 cells (CRL-1469) are an undifferentiated human pancreatic epithelial cell line with a doubling time of approximately 52 h [33].
For experiments requiring hypoxia, cells were trypsinized, replated, and allowed to adhere before placing under hypoxic conditions for 16 h, when drug treatment was initiated. Cells under hypoxia were cultured in a Ruskinn InVivo 4000 hypoxia chamber (BioTrace International, Bothell, WA) set at 1% O2.
Drugs and reagents
Imexon (4-imino-1,3-diazabicyclo-[3.1.0]-hexan-one) was generously provided by AmpliMed Corporation (Tucson, AZ). Gemcitabine was obtained from Eli Lilly (Indianapolis, IN). Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), menadione sodium bisulfate, anti-actin antibody, 3-(4,5-dimethylthiozol-3-yl)-2,5-diphenyltetrozolium bromide (MTT dye) and all other chemicals not specifically listed were obtained from Sigma Chemical Company (St. Louis, MO).
Monoclonal antibody to HIF-1α was purchased from BD Biosciences (San Jose, CA), and monoclonal antibodies to Lamin A/C, total eIF-2α and phospho-eIF-2α (Ser51) were purchased from Cell Signaling Technology (Danvers, MA). The synthesis of HIF-1α was evaluated using dynamic labeling of new proteins via the Express 35S Protein Labeling Mix® (Perkin Elmer, Wellesley, MA). The proteasome inhibitor MG132 [34] was obtained from BioMol International (Plymouth Meeting, PA).
Cytotoxicity assays
Cytotoxicity assays were performed according to the microculture tetrazolium dye method of Mosmann, which measures the activity of mitochondrial reductases [35]. Briefly, cells are seeded in 96 well plates and incubated for the indicated time in the presence or absence of imexon, under normoxic or hypoxic conditions. The MTT dye is added and the cells incubated an additional 4 h prior to solubilizing the formazan complex with DMSO and reading the absorbance at OD565. Cell growth inhibition data are expressed as percent survival, compared to untreated cells. The IC50 is defined as the drug concentration required to produce 50% growth inhibition.
Western blots
Panc-1 or MiaPaCa-2 cells were cultured with imexon or gemcitabine, or in subsequent experiments with MG132, for 24 h. For studies which involved proteasome inhibition, 50–500 nM of MG132 was added to cultures 15 min prior to adding imexon. Whole cell lysates or nuclear lysates were prepared using Active Motif’s nuclear extract kit (Carlsbad, CA) and protein quantitated using a bicinchoninic acid (BCA) colorimetric assay (Pierce Biotechnology Inc., Rockford, IL. For chemiluminescent western blots, proteins were separated using SDS-polyacrylamide gel electrophoresis, transferred to Immobilon-P PVDF® membranes (Millipore, Bedford, MA), and immunoblotted for HIF-1α or Lamin A/C. Proteins were detected by standard immunoblot techniques using SuperSignal West Dura or Pico chemiluminescent substrate following manufacturer’s instructions (Pierce Biotechnology, Inc.). Analysis of total and phosphorylated eIF-2α was carried out using the Odyssey Infared (IR) Imaging System (LI-COR, Lincoln, Nebraska), using IR secondary antibodies (IRDye 680 or IRDye 800). Due to the separate emission spectra of these dyes (710 λ and 795 λ, respectively), we are able to visualize total and phosphorylated eIF-2α concurrently.
35S-methionine/cysteine incorporation studies
The 35S-labeling method of Shain et al. was used to evaluate the dynamics of HIF-1α protein synthesis and degradation in imexon-treated cells [36]. Cells were pre-treated with 1,000 μM of imexon for 2 h prior to depletion in methionine- and cysteine-free media, (which included 10% dialyzed FBS) for 1 h. The 35-sulfur labeled methionine/cysteine (35S-Met/Cys) Protein Labeling Mix (100 μCi/ml) was then added for 1 h, after which time the “pulse” media was removed. Cells were washed, and incubated with complete media for up to 3 h. All labeling conditions were done in the presence of 1,000 μM imexon. Cells were collected at 0, 1, 2, and 3 h, washed with ice cold PBS, and lysates immunoprecipitated for HIF-1α protein. Immunoprecipitated proteins were separated by SDS-polyacrylamide gel electrophoresis. The gels were dried and densitometric analysis of bands performed using Molecular Dynamics ImageQuant software version 5.1 (Amersham Biosciences Corp, Piscataway, NJ).
Proteasome activity assay
20S Proteasome activity was measured using BioMol’s QuantiZyme Assay System (BioMol International, Plymouth Meeting, PA). The assay uses a fluorogenic, non-radioactive substrate which measures chymotrypsin-like protease activity of purified 20S proteasome. Epoxomicin was used to provide rapid, potent and irreversible inhibition of 20S proteasome chymotrypsin-like activity [37]. Purified human erythrocyte 20S proteasome enzyme (0.2 μg) was incubated with various concentrations of imexon or epoxomicin, and 75 μM Suc-LLVY-AMC substrate, in the provided assay buffer. Fluorescence readings (excitation 360 λ, emission 460 λ) were taken at 1 min intervals for 1 h, after allowing an initial 10 min equilibration at room temperature to allow inhibitor/enzyme interaction. Seven-amino-4-methylcoumarin (AMC) was used as a calibration standard. The specific activity (SA) of the samples were determined from the linear portion of the curve, and calculated as follows: SA (pmol/min)=slope (AFU/min)×conversion factor (μM/AFU)×assay volume (μl), where the conversion factor=1/slope of the prepared standard curve. (AFU=arbitrary flourescence units)
To measure 20S proteasome activity in imexon treated cells, cells were treated with 0–750 μM imexon or 0–500 nM MG132 for 24 h, lysed, and equivalent amounts of total cellular protein assayed for 20S proteasome activity. As with all studies comparing normoxia and hypoxiatreated cells, cells were plated and allowed to adhere before placing into the hypoxia chamber overnight (16 h), prior to drug treatment.
Determination of HIF-1α half-life
Cells were treated with or without imexon concurrently with 20 μg/ml cycloheximide, a standard inhibitor of protein synthesis. After 30, 60, 90 or 120 min of cycloheximide exposure, cells were harvested and nuclear lysates prepared. Equivalent amounts of nuclear lysate were separated using SDS-polyacrylamide gel electrophoresis and probed for HIF-1α protein using standard immunoblot detection techniques.
14-C valine incorporation studies
Total protein synthesis was analyzed by 14C valine incorporation studies. Cells were plated in 96 well plates and exposed to imexon for 24 h, followed by the addition of 0.5 μCi/ml of 14C valine (Amersham Biosciences, Piscataway, NJ). Cells which incorporated radioactivity were harvested using a Packard FilterMate Harvester (Perkin Elmer, Waltham, MA). Data are expressed as percent control of untreated cells. For each experiment, duplicate plates were prepared and cell death measured by MTT concurrently.
Results
Hypoxia does not alter the cytotoxic potency of imexon in vitro
To investigate the effects of hypoxia on imexon cytotoxicity, pancreatic cancer cell lines were treated with imexon under normoxic or hypoxic conditions. No significant difference was identified in the growth-inhibitory potency of imexon in hypoxic MiaPaCa-2 cells, IC50 373± 102 μM (mean±SEM) compared to normoxic MiaPaCa-2 cells, IC50 426±72 μM (p<0.711), or hypoxic Panc-1 cells, IC50 263±22 μM compared to normoxic Panc-1 cells, IC50 303±14 μM (p<0.205). These results demonstrate a marked contrast between the effects of imexon and most other anti-cancer agents which are reported to lose activity under hypoxic conditions [26].
Effect of imexon and gemcitabine on HIF-1α levels
To investigate the effect of imexon on HIF-1α expression, pancreatic cell lines were exposed to imexon or gemcitabine for 24 h and HIF-1α expression was measured by Western blotting. Imexon-treated pancreatic cancer cells showed a concentration-dependant decrease in nuclear HIF-1α protein levels in both Panc-1 cells (Fig. 1a and c) and MiaPaCa-2 cells (Fig. 1b and d). Because HIF-1α activity requires nuclear localization and dimerization with HIF-1β, we present results obtained from nuclear lysates, although results obtained using total cell lysates revealed a similar pattern (data not shown). The 24 h exposure time was selected for these studies based on prior data demonstrating that imexon-induced apoptosis in Panc-1 cells became prominent after 24 h and increased to a maximum after 48–72 h of drug exposure [7]. Thus, at the 24 h time point, the cells are beginning to undergo apoptosis but it is not yet detectable in the majority of cells evaluated by flow cytometry for Annexin V binding [7]. Figure 1 also illustrates that imexon-induced loss of HIF-1α protein levels occurs under both normoxic and hypoxic conditions, whereas gemcitabine does not affect HIF-1α levels, under either normoxia or hypoxia in both Panc-1 and MiaPaCa-2 cells (Fig. 1).
Fig. 1.

Concentration dependent decrease in active HIF-1α protein after 24 h imexon under normoxic (a and b) and hypoxic (c and d) conditions. Cells were cultured under normoxic or hypoxic conditions (1% O2) for 16 h prior to exposing to imexon or gemcitabine for 24 h, harvested and nuclear lysates prepared. Nuclear lysates were separated by SDS-PAGE and immunoblotted for HIF-1α protein. Lamin A/C serves as a loading control. a Panc-1, normoxia; b MiaPaCa-2, normoxia; c Panc-1, hypoxia; d MiaPaCa-2, hypoxia
Effect of imexon on proteasomal degradation of HIF-1α
HIF-1α stability is regulated by the ubiquitin proteasome degradation pathway, which is inhibited under hypoxic conditions [22]. Our preliminary studies confirmed that MiaPaCa-2 cells do possess a functioning proteasomal degradation pathway and that HIF-1α is rapidly degraded under normoxic conditions (data not shown). When we added MG132 to MiaPaCa-2 cells prior to co-culture with imexon, we saw a reversal of the loss of HIF-1α protein normally seen upon imexon addition, suggesting that the imexon-induced loss of HIF-1α protein might involve proteasomal degradation (Fig. 2).
Fig. 2.
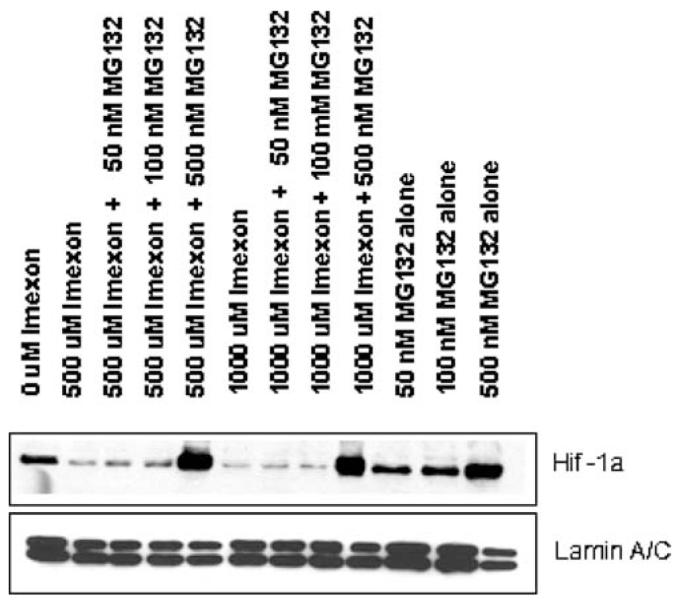
Proteasome inhibitor MG132 prevents imexon induced decrease in HIF-1α protein in MiaPaCa-2 cells. Cells were pre-treated with 50–500 nM of MG132 for 15 min prior to incubation with imexon for 24 h. Nuclear lysates were separated by SDS-PAGE and immunoblotted for HIF-1α protein. Lamin A/C serves as a loading control
Next, we evaluated the specific inhibition of 20S proteasome chymotrypsin-like activity using Suc-LLVY-AMC fluorogenic substrate in both a cell free system and in MiaPaCa-2 cells treated with imexon. To evaluate direct inhibition of 20S proteasome activity by imexon, purified human erythrocyte 20S proteasome enzyme was incubated with Suc-LLVY-AMC substrate and imexon in a cell-free system. The results, shown in Fig. 3a, demonstrate a concentration dependent inhibition of 20S proteasome activity by imexon, and this inhibition was additive when imexon was combined with the proteasome inhibitor epoxomicin. To determine if imexon inhibition of the 20S proteasome also occurred in cells, MiaPaCa-2 cells were incubated with imexon (24 h) or MG132 (positive control), and cell lysates were assayed for 20S proteasomal activity. Lysates were prepared from cells treated under both normoxic and hypoxic conditions (Fig. 3b). As with direct (cell-free) inhibition of 20S proteasome activity (Fig. 3a), imexon also reduced 20S proteasome activity in treated MiaPaCa-2 cells (Fig. 3b).
Fig. 3.

a Direct inhibition of 20S proteasome activity by imexon in a cell-free system. Purified human erythrocyte 20S proteasome enzyme (0.2 μg) was incubated with increasing concentrations of imexon or epoxomicin, and 75 μM Suc-LLVY-AMC substrate. Fluorescence readings were taken at 1 min intervals and the specific activity (SA) was calculated as follows: SA (pmol/min)=slope (AFU/min)×conversion factor (μM/AFU)×assay volume (μl), where the conversion factor=1/slope of the standard curve, which was prepared using 7-amino-4-methylcoumarin (AMC). b Inhibition of 20S proteasome activity in imexon treated MiaPaCa-2 cells. Cells were treated with increasing concentrations of imexon or MG132 for 24 h, lysed, and equivalent amounts of total cellular protein assayed for 20S proteasome activity. Cells cultured under normoxic conditions (left) are compared to cells cultured under hypoxic conditions (right)
Effects of imexon on HIF-1α transcription, translation, and degradation
Using real time PCR, we evaluated whether imexon altered the transcription of HIF-1α message and found no change in the levels of HIF-1α-specific mRNA in imexon treated cells (data not shown). We then asked whether the reduction in HIF-1α levels was due to changes in protein translation or degradation. The effect of imexon on new protein synthesis and degradation was examined using 35S-Met/Cys pulse chase labeling. The results show that imexon not only reduces the synthesis of newly formed HIF-1α compared to untreated cells, but the degradation of this newly formed HIF-1α is also delayed (Fig. 4). The slope (decay) of newly synthesized HIF-1α protein in imexon treated cells (y = –12.25x + 104.47 compared to control cells (y = –20.224x + 115.44 confirms that HIF-1α is degraded more slowly in the presence of imexon. The total amount of 35S-Met/Cys labeled HIF-1α was significantly lower (p<0.0474) for 2 h after imexon treatment, but gradually recovered to control levels by 3 h of incubation in the non-drug containing “chase” media. These data show that imexon markedly blocks the translation of new HIF-1α protein, but conversely delays the degradation of preformed HIF-1α in human pancreatic cancer cells.
Fig. 4.
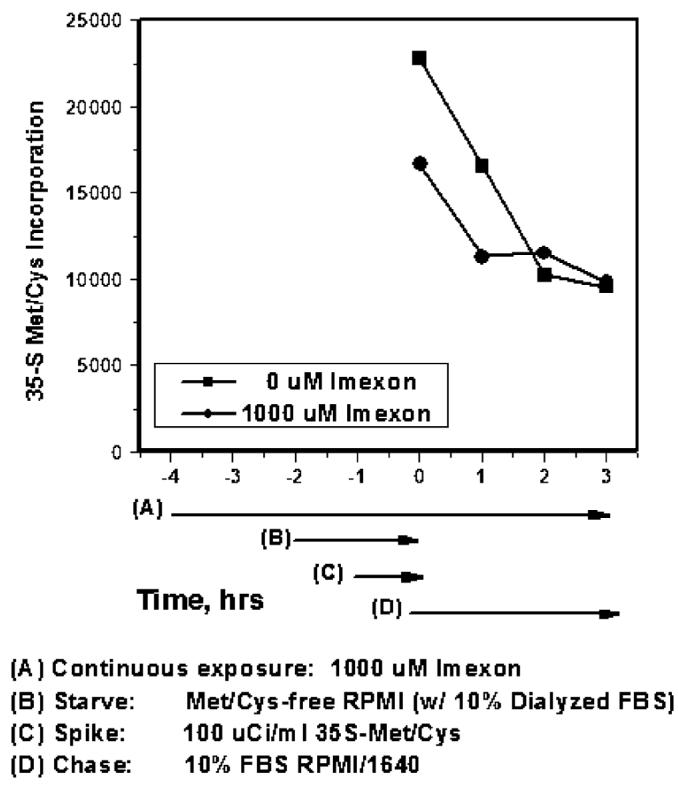
Imexon inhibits the synthesis and delays the degradation of HIF-1α protein. Cells were pre-treated with 1,000 μM of imexon for 2 h prior to depletion in methionine- and cysteine-free media (“starve”) for 1 h. 100 μCi/ml of 35S-Met/Cys protein labeling mix was added (“pulse”) for 1 h, after which time the “pulse” media was removed, cells washed, and replaced by “chase” media containing 10% FBS/RPMI for up to 3 h. Exposure to imexon was maintained throughout the experiment. Cells were collected at 0, 1, 2, and 3 h, and lysates immunoprecipitated for HIF-1α protein. Immunoprecipitated proteins were separated by SDS-PAGE, gels dried, and band density quantitated using ImageQuant. Data shown is the densitometric quantitation of HIF-1α protein normalized to Lamin A/C
Effects of imexon on HIF-1α half-life
Because of previous reports that ROS stabilize HIF-1α levels against degradation, the effects of imexon on pre-existing HIF-1α levels were determined [28, 29]. Cycloheximide, a standard inhibitor of protein synthesis, was used to calculate the half-life of preformed HIF-1α protein in MiaPaCa-2 cells. MiaPaCa-2 cells were cultured under normoxic or hypoxic conditions (1% O2) for up to 24 h with 500 μM imexon, and HIF-1α protein was measured by Western blotting after 30, 60, 90, or 120 min of cycloheximide treatment. Under normoxic conditions (without imexon), the HIF-1α half-life was approximately 20 min, compared to approximately 60 min under hypoxic conditions. Following the addition of 500 μM of imexon, HIF-1α half-life was extended to greater than 120 min (Fig. 5).
Fig. 5.

Imexon increases HIF-1α protein half-life. MiaPaCa-2 cells were treated under normoxic or hypoxic conditions, with or without imexon (0–24 h) concurrently with cycloheximide (20 μg/ml), a standard inhibitor of protein synthesis. After 30, 60, 90 or 120 min of cycloheximide exposure, cells were harvested and nuclear lysates prepared, proteins separated using SDS-PAGE, and immunoblotted for HIF-1α protein. Data shown is the densitometric quantitation of HIF-1α protein normalized to Lamin A/C, and presented as percent control (0 h). Filled squares normoxia control (0 μM imexon), t1/2=20 min; filled circles hypoxia control (0 μM imexon), t1/2=60 min; filled triangles hypoxia (500 μM imexon×4 h), t1/2>120 min; filled diamonds hypoxia (500 μM imexon×24 h), t1/2>120 min
Inhibition of protein synthesis and effects on eIF-2α
To determine if the reduction of the HIF-1α translation is a specific effect of imexon exposure or due to a general inhibition of protein synthesis, 14C valine incorporation studies were carried out in both pancreatic cancer cell lines. As shown in Fig. 6, overall protein synthesis is significantly inhibited at 300 μM imexon within 24 h. This effect is not due to cell death, as concurrently performed MTT assays show that cells remain viable in the presence of substantial (≥50%) inhibition of protein synthesis by imexon. Finally, there were no changes in the degree of phosphorylation (inactivation) of the redox-sensitive protein translation factor eIF-2α at 1, 2, 4, 8 and 24 h (data not shown).
Fig. 6.
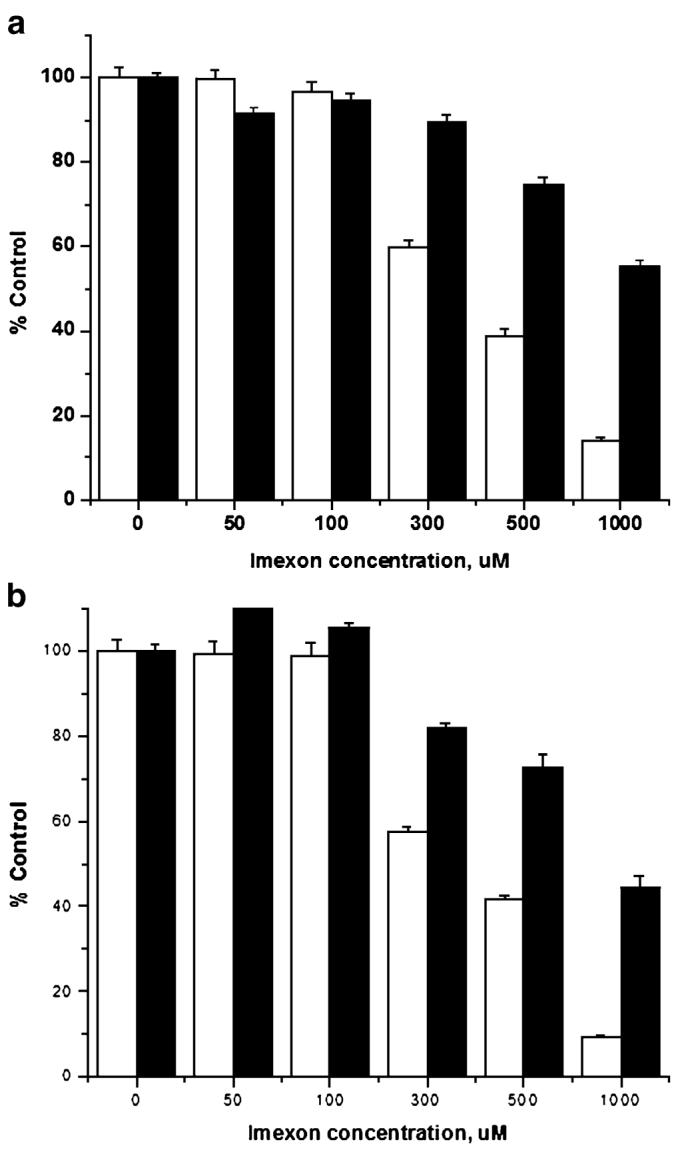
Imexon blocks cellular protein synthesis. Cells were seeded in duplicate 96 well plates and exposed to imexon for 24 h, followed by the addition of 0.5 μCi/ml of 14C valine (to measure protein synthesis) or MTT dye (to measure cytotoxicity) in tandem. Data are expressed as mean±SEM. a MiaPaCa-2, b Panc-1. Open bars represent 14C valine incorporation, closed bars represent cytotoxicity by MTT
Discussion
In the current report, the pro-oxidant small molecule imexon is shown to produce two opposing effects on HIF-1α in human pancreatic cancer cells in vitro: (1) a general decrease in protein translation characterized by a marked decrease in HIF-1α translation, and (2) an increase in the stability of previously synthesized HIF-1α protein due to proteasomal inhibition. However, the net effect of imexon in the pancreatic cancer cells was a marked reduction in total HIF-1α protein levels. Therefore, one can conclude that the inhibition of translation by imexon is the predominant effect of the drug on HIF-1α in human pancreatic cancer cells and this may explain the maintained cytotoxic potency of imexon in hypoxic pancreas cancer cells. In contrast, gemcitabine had no effect on HIF-1α levels, under either hypoxic or normoxic conditions.
The inhibition of HIF-1α translation by imexon is not specific to HIF-1α, as total protein synthesis was markedly reduced by imexon in these pancreatic cancer cells. This same inhibitory effect on protein synthesis was previously seen in human lymphoma and multiple myeloma cell lines [8]. Eukaryotic initiation factor 2α, a known redox-regulated factor for the initiation of protein translation, was unchanged by imexon. Thus, the molecular mechanism for the inhibition of protein translation by imexon is still unknown. In contrast, the mechanism mediating prolonged stability of preformed HIF-1α by imexon appears to involve direct inhibition of the 20S proteasome. These results are consistent with well-described effects of ROS on HIF-1α stability, wherein mitochondrial ROS are known to mediate a significant increase in HIF-1α stability [31]. And, the accumulation of mitochondrially-derived ROS following imexon has been well-documented in both lymphoid [4, 5] and pancreatic cancer cell lines [7]. However, this does not prove that imexon-induced ROS are responsible for increasing the stability of preformed HIF-1α proteins following drug exposure. Indeed, because imexon inhibited proteasome activity in a cell-free assay wherein significant ROS accumulation is unlikely, the inhibitory effects of imexon may be due to direct binding of imexon to critical sulfhydryl elements in the 20S proteasome. Similarly, the likelihood of an effect of imexon on prolyl hydroxylase labeling of HIF-1α by imexon is also unlikely since the cell-free proteasome assay system does not include a prolyl hydroxylase component for degradation of the peptide substrate. This again supports the possibility that imexon may be directly binding to sulfhydryl components within the proteasome’s enzymatic machinery, an effect consistent with imexon’s covalent binding to other thiols [3].
A wide variety of agents have been shown to experimentally inhibit HIF-1α [38, 39]. These include the heat shock protein-90 (HSP-90) inhibitor geldanamycin, which leads to HIF-1α breakdown [40], the transactivation (MEK1) inhibitor PD98059, transcription inhibitors such as the topoisomerase II poison GL331 [41], the histone deacetylase inhibitor FK228 [42] and the Ca++ blocker carboxyamidotriazole [43]. However, by far the largest group of experimental HIF-1α inhibitors act like imexon, via inhibition of translation. This includes agents such as topotecan (a TOPO-I inhibitor [44]), the anti-microtubule agents paclitaxel and 2-methoxyestradiol, the PI3-kinase pathway inhibitors wortmannin and LY294002 and the mTOR inhibitor rapamycin [45]. One caveat with all of these reports of HIF-1α inhibition is that drug concentrations used to inhibit HIF-1α were higher than necessary for cell growth inhibition. This raises the possibility that HIF-1α inhibition is not directly contributory to the cytotoxic effect of these agents [39]. Importantly, the concentrations of imexon used to inhibit HIF-1α translation in the current experiments are within the range of concentrations that can be achieved in cancer patients [1]. For example, at the maximally tolerated phase I dose of 875 mg/m2/day, imexon peak plasma levels of 715 μM were routinely obtained [1]. This is within the range of imexon concentrations (500–1,000 μM) which blocked HIF-1α translation in the current studies.
Another observation from the current studies was that hypoxia does not affect the cytotoxic potency of imexon in pancreatic cancer cell lines in vitro. This stands in marked contrast to most other cytotoxic agents wherein cytotoxic potency is significantly reduced when cells are exposed under hypoxic conditions [26]. This observation suggests that imexon comprises a good candidate for combination therapy of human pancreatic cancers, wherein tumor hypoxia is clinically well documented [16, 46, 47, 48]. Furthermore, the combination of imexon and anti-metabolites including gemcitabine have been shown to be synergistic in other cells types in vitro [49]. Collectively, these studies support continued clinical evaluation of gemcitabine and imexon in gemcitabine-sensitive tumor types such as advanced cancer of the lung, breast and pancreas.
Acknowledgments
These studies were supported by grants CA17094 (RTD) and CA115626 (RTD) from the National Institutes of Health, Bethesda, MD.
References
- 1.Dragovich T, Gordon M, Mendeslon D, et al. Phase I trial of imexon in patients with advanced malignancy. J Clin Oncol. 2007;25:1779–1784. doi: 10.1200/JCO.2006.08.9672. [DOI] [PubMed] [Google Scholar]
- 2.Cohen SJ, Zalupski M, Modiano M, Conkling P, Patt Y, Davis P, Dorr R, Boytim M, Hersh E. A phase I study of Amplimexon (AMP, imexon inj.) plus gemcitabine (Gem) as first line therapy in advanced pancreatic cancer (PC) with preclinical mechanistic study of dose limiting toxicity (DLT). Proceedings of the ASCO Annual Meeting.2008. [Google Scholar]
- 3.Iyengar BS, Dorr RT, Remers WA. Chemical basis for the biological activity of imexon and related cyanoaziridines. J Med Chem. 2004;47:218–223. doi: 10.1021/jm030225v. [DOI] [PubMed] [Google Scholar]
- 4.Dvorakova K, Payne CM, Tome ME, Briehl MM, McClure T, Dorr RT. Induction of oxidative stress and apoptosis in myeloma cells by the aziridine-containing agent imexon. Biochem Pharmacol. 2000;60:749–758. doi: 10.1016/s0006-2952(00)00380-4. [DOI] [PubMed] [Google Scholar]
- 5.Dvorakova K, Waltmire CN, Payne CM, Tome ME, Briehl MM, Dorr RT. Induction of mitochondrial changes in myeloma cells by imexon. Blood. 2001;97:3544–3551. doi: 10.1182/blood.v97.11.3544. [DOI] [PubMed] [Google Scholar]
- 6.Dvorakova K, Payne CM, Landowski TH, Tome ME, Halperin DS, Dorr RT. Imexon activates an intrinsic apoptosis pathway in RPMI8226 myeloma cells. Anticancer Drugs. 2002;13:1–13. doi: 10.1097/00001813-200211000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Dorr RT, Raymond MA, Landowski TH, Roman NO, Fukushima S. Induction of apoptosis and cell cycle arrest by imexon in human pancreatic cancer cells lines. Int J Gastrointest Cancer. 2005;36(1):15–28. doi: 10.1385/IJGC:36:1:015. [DOI] [PubMed] [Google Scholar]
- 8.Hersh EM, Gschwind CR, Taylor CW, Dorr RT, Taetle R, Salmon SE. Antiproliferative and antitumor activity of the 2-cyanoaziridine compound imexon on tumor cell lines and fresh tumor cells in vitro. J Natl Cancer Inst. 1992;84:1238–1244. doi: 10.1093/jnci/84.16.1238. [DOI] [PubMed] [Google Scholar]
- 9.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34(Pt 1):7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 10.Tan S, Somia N, Maher P, Schubert D. Regulation of antioxidant metabolism by translation initiation factor 2 alpha. J Cell Biol. 2001;152:997–1006. doi: 10.1083/jcb.152.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang HY, Wek RC. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem. 2005;280(14):14189–14202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 12.Oettle H, Arnold D, Hempel C, Riess H. The role of gemcitabine alone and in combination in the treatment of pancreatic cancer. Anticancer Drugs. 2000;11:771–786. doi: 10.1097/00001813-200011000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Poplin E, Levy DE, Berlin M. Phase III trial of gemcitabine (30-minute infusion) versus gemcitabine (fixed-dose-rate infusion (FDR) versus gemcitabine + oxaliplatin (GEMOX) in patients with advanced pancreatic cancer (E6201) ASCO Proceedings. 2006;24(18s):180s. [Google Scholar]
- 14.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared to gemcitabine alone in patients with advanced pancreatic cancer. A phase III trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG) J Clin Oncol. 2007;25(15):1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 15.Senderowicz AM, Johnson JR, Sridhara R, Zimmerman P, Justice R, Pazdur R. Erlotinib/gemcitabine for first-line treatment of locally advanced or metastatic adenocarcinoma of the pancreas. Oncology. 2007;21:1696–1706. [PubMed] [Google Scholar]
- 16.Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48:919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 17.Megibow AJ. Pancreatic adenocarcinoma: designing the examination to evaluate the clinical questions. Radiology. 1992;183:297–303. doi: 10.1148/radiology.183.2.1561324. [DOI] [PubMed] [Google Scholar]
- 18.Duffy JP, Eibl G, Reber HA, Hines OJ. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol Cancer. 2003;2:12. doi: 10.1186/1476-4598-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitada T, Seki S, Sakaguchi H, Sawada T, Hirakawa K, Wakasa K. Clinicopathological significance of hypoxia-inducible factor-1a expression in human pancreatic carcinoma. Histopathology. 2003;43:550–555. doi: 10.1111/j.1365-2559.2003.01733.x. [DOI] [PubMed] [Google Scholar]
- 20.Buchler P, Reber HA, Buchler M, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26:56–64. doi: 10.1097/00006676-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Akakura N, Kobayashi M, Horiuchi I, et al. Constitutive expression of hypoxia-inducible factor-1a renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001;61:6548–6554. [PubMed] [Google Scholar]
- 22.Welsh SJ, Koh MY, Powis G. The hypoxic inducible stress response as a target for cancer drug discovery. Semin Oncol. 2006;33:486–497. doi: 10.1053/j.seminoncol.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 23.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 24.Brown JM. Tumor microenvironment and the response to anticancer therapy. Cancer Biol Ther. 2002;1:453–458. doi: 10.4161/cbt.1.5.157. [DOI] [PubMed] [Google Scholar]
- 25.Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1 alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–572. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- 26.Teicher BA. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994;13:139–168. doi: 10.1007/BF00689633. [DOI] [PubMed] [Google Scholar]
- 27.Yokoi K, Fidler IJ. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res. 2004;10:2299–2306. doi: 10.1158/1078-0432.ccr-03-0488. [DOI] [PubMed] [Google Scholar]
- 28.Sanjuan-Pla A, Cervera AM, Apostolova N, et al. A targeted antioxidant reveals the importance of mitochondrial reactive oxygen species in the hypoxic signaling of HIF-1alpha. FEBS Lett. 2005;579:2669–2674. doi: 10.1016/j.febslet.2005.03.088. [DOI] [PubMed] [Google Scholar]
- 29.Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1a during hypoxia: a mechanism of 02 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 31.Simon MC. Mitochondrial reactive oxygen species are required for hypoxic HIF alpha stabilization. Adv Exp Med Biol. 2006;588:165–170. doi: 10.1007/978-0-387-34817-9_15. [DOI] [PubMed] [Google Scholar]
- 32.Yunis A, Arimura GK, Russin DJ. Human pancreatic carcinoma (MiaPaCa-2) in continuous culture: sensitivity to asparaginase. Int J Cancer. 1977;19:128–135. doi: 10.1002/ijc.2910190118. [DOI] [PubMed] [Google Scholar]
- 33.Lieber M, Mazzetta J, Nelson-Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor-cell line (Panc-1) from a human carcinoma of the exocrine pancreas. Int J Cancer. 1975;15:741–747. doi: 10.1002/ijc.2910150505. [DOI] [PubMed] [Google Scholar]
- 34.Lightcap ES, McCormack TA, Pien CS, Chau V, Adams J, Elliott PJ. Proteasome inhibition measurements: clinical application. Clin Chem. 2000;46:673–683. [PubMed] [Google Scholar]
- 35.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 36.Shain KH, Landowski TH, Dalton WS. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J Immunol. 2002;168:2544–2553. doi: 10.4049/jimmunol.168.5.2544. [DOI] [PubMed] [Google Scholar]
- 37.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 38.Semenza GL. Targeting HIF-1 for cancer therapy. Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 39.Powis G, Kirkpatrick L. Hypoxia inducible factor-1a as a cancer drug target. Mol Cancer Ther. 2004;3:647–654. [PubMed] [Google Scholar]
- 40.Mabjeesh NJ, Post DE, Willard MT, et al. Geldanamycin induces degradation of hypoxia-inducible factor 1a protein via the proteosome pathway in prostate cancer cells. Cancer Res. 2002;62:2478–2482. [PubMed] [Google Scholar]
- 41.Chang H, Shyu KG, Lee CC, et al. GL331 inhibits HIF-1a expression in a lung cancer model. Biochem Biophys Res Commun. 2003;302:95–100. doi: 10.1016/s0006-291x(03)00111-6. [DOI] [PubMed] [Google Scholar]
- 42.Lee YM, Kim SH, Kim HS, et al. Inhibition of hypoxia-induced angiogenesis by FK228, a specific histone deacetylase inhibitor, via suppression of HIF-1a activity. Biochem Biophys Res Commun. 2003;300:241–246. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 43.Oliver VK, Patton AM, Desai S, Lorang D, Libutti SK, Kohn EC. Regulation of the pro-angiogenic microenvironment by carboxyamido-triazole. J Cell Physiol. 2003;197:139–148. doi: 10.1002/jcp.10350. [DOI] [PubMed] [Google Scholar]
- 44.Rapisarda A, Uranchimerg B, Sordet O, Pommier Y, Shoemaker PH, Belillo G. Topoisomerase 1-mediated inhibition of hypoxia-inducible factor 1:mechanism and therapeutic implications. Cancer Res. 2004;64:1475–1482. doi: 10.1158/0008-5472.can-03-3139. [DOI] [PubMed] [Google Scholar]
- 45.Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1a expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 46.Raijiman I, Levin B. Exocrine tumors of pancreas. In: Haubrich WS, Schaffner F, editors. Bockus gastroentrology. 5th Saunders; Philadelphia: 1995. pp. 2984–3001. [Google Scholar]
- 47.Ranniger K, Saldino RM. Arteriographic diagnosis of pancreas lesion. Radiology. 1966;86:470–474. doi: 10.1148/86.3.470. [DOI] [PubMed] [Google Scholar]
- 48.Yassa NA, Yang J, Stein S, Johnson M, Ralls P. Gray-scale and color flow sonography of pancreatic ductal adenocarcinoma. J Clin Ultrasound. 1997;25:473–470. doi: 10.1002/(sici)1097-0096(199711/12)25:9<473::aid-jcu2>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 49.Scott J, Dorr RT, Samulitis B, Landowski TH. Imexon-based combination chemotherapy in A375 human melanoma and RPMI 8226 human myeloma cell lines. Cancer Chemother Pharmacol. 2007;59:749–757. doi: 10.1007/s00280-006-0329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]