

Abstract
(−)-Sparteine and TMEDA dramatically lower both enthalpy and entropy of activation for the barrier to enantiomerization of N-Boc-2-lithiopyrrolidine in diethyl ether, whereas N,N'-diisopropylbispidine has little effect. The entropy of activation for enantiomerization is zero in the presence of TMEDA and slightly negative in the presence of sparteine. These data suggest a subtle change in mechanism of enantiomerization in the presence of TMEDA and sparteine.
The role of (−)-sparteine (1) in promoting asymmetric deprotonation with lithium alkyls is legendary,1 as is the role of TMEDA (2) in “activating” alkyllithiums in diethyl ether.2 Several bispidines were studied by Beak in the context of seeking alternatives to sparteine as a lithium ligand in asymmetric deprotonations,3 and N,N'-diisopropyl bispidine, 3, has recently been championed by O'Brien as an exchangeable ligand in asymmetric deprotonations, so that chiral ligands can be used in substoichiometric quantities.4
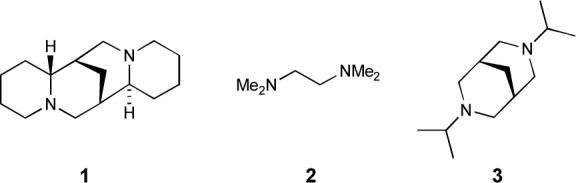
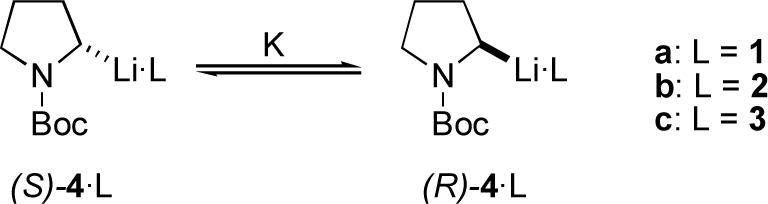
One of the more interesting developments in organolithium chemistry over the past decade has been the emergence of dynamic resolution of racemic organolithiums as a means of asymmetric synthesis.5, 6 Central to both asymmetric deprotonations and dynamic resolutions is the issue of enantiomerization: in an asymmetric deprotonation, it must be minimized, while in a dynamic resolution it must be controlled. Although the dynamics of enantiomerization of several rapidly inverting organolithiums have been determined (usually by dynamic NMR),7 there is relatively little data on enantiomerization dynamics in systems with high barriers. As part of an investigation into the structure and dynamic properties of chiral organolithiums,8 and on the dynamics of the resolution process in lithiated heterocycles, we have studied the effect of ligands 1−3 on the barrier to enantiomerization of N-Boc-2-lithiopyrrolidine, 4. This organolithium is particularly appealing for study due to the extraordinarily high entropy of activation for enantiomerization of 4 in the absence of any ligands (Table 1, entry 1).8
Table 1.
| Entry | Diamine | ΔH‡(kcal/mol) | ΔS‡(cal/mol·K) |
|---|---|---|---|
| 1 | Noneb | 29 ± 3 | 40 ± 8 |
| 2 | Bispidine 3 | 28 ± 1 | 32 ± 2 |
| 3 | TMEDA, 2 | 19 ± 1 | 0 ± 2 |
| 4 | (−)-Sparteine, 1 | 18 ± 1 | −6 ± 2 |
Errors expressed at two standard deviations.
Taken from reference 8.
Diamines 2 and 3 are achiral, so the enantiomer ratio (er) of Scheme 1 is 50:50 for these ligands (i.e., K = 1). Somewhat surprisingly, K = 1 for (−)-sparteine as well, over a wide temperature range.6
Scheme 1.
The effect of TMEDA, 2, on the rate of inversion of chiral organolithiums is not straightforward. TMEDA accelerates the epimerization of diastereomeric oxazolidinones and imidazolidinones,9 whereas in N-alkyl piperidines and pyrrolidines,10 and in N,N-dialkylaminobenzyllithium,11 2 retards racemization. Previous studies on the effect of diamines such as (−)-sparteine, 1, or bispidine 3 on the barrier to enantiomerization of chiral organolithiums are rare.12
Starting with (S)-N-Boc-2-lithiopyrrolidine, the progress of racemization was followed by quenching the organolithiums with trimethylsilyl chloride and determination of the er by chiral stationary phase gas chromatography.† From these data, the enthalpic and entropic barriers to enantiomerization were determined as described previously.8
Table 1 gives activation parameters for the enantiomerization illustrated in Scheme 1, determined over the temperature range −5 to −33 °C. Excellent fit of the data to first order kinetic plots was observed in all runs.
Both the enthalpy and entropy of activation for the enantiomerization of N-Boc-2-lithiopyrrolidine are high in the absence of amine ligands (Entry 1).8 In contrast, thermodynamic enantiomerization parameters for twenty different chiral organolithiums, summarized in a recent review,7 are revealing: eighteen had negative ΔS‡. values, and only two had small positive values (+2 and +3 cal·mol−1K−1). This is expected, since charge separation should require additional solvation in the transition state. The enantiomerization parameters of 4 were rationalized8 by invoking a conducted tour mechanism, in which the lithium atom is escorted between enantiomeric faces of the carbanion by the carbonyl oxygen. This movement is necessarily accompanied by the movement of the bulky tert-butoxy group, which would disrupt the solvent cage. Another contributing factor could be that binding of the carbonyl oxygen to the lithium restricts conformational motion in the ground state which is then restored in going to the TS.
In the presence of 3, which O'Brien employs as a ligand that can readily exchange with 1, ΔH‡ or ΔS‡ for enantiomerization (Entry 2) are changed only slightly from Entry 1. The enthalpy of activation is lowered dramatically in the presence of TMEDA (Entry 3) or sparteine (Entry 4), and the entropic benefit is completely erased. One possible the entropic benefit is completely erased. One possible explanation is that these diamines weaken the C—Li bond, perhaps by coordinating strongly to the lithium, and that the conducted tour mechanism may no longer be operative in the presence of 1 and 2.
Bispidine 3 has virtually no effect on the enthalpy of activation, perhaps because it is only weakly coordinated to the lithium, consistent with O'Brien's observation that excess bispidine readily exchanges Li+ with sparteine.4 Further, 3 induces only a slight lowering of the entropy term, consistent with weak binding of 3 to the lithium and more important involvement of the solvent.
Free energies of activation for inversion of 4 at −78 °C are calculated from the parameters of Table 1, and listed in Table 2. These data suggest that 3 would have no effect on the enantiomerization barrier of 4, but predict that both 1 and 2 would lower the barrier to inversion, consistent with the early low temperature observations of Beak.13
Table 2.
Calculated barriers for the racemization of Scheme 1
| Diamine | ΔG‡/kcal·mol−1 calculated at 195K (−78 °C) |
|---|---|
| None (ether) | 21.3 ± 5.6 |
| None (ether/hexane) | 21.4 ± 3.0 |
| Bispidine 3 (ether) | 21.3 ± 1.5 |
| TMEDA, 2 (ether) | 19.1 ± 1.0 |
| (−)-Sparteine, 1 (ether) | 19.4 ± 1.5 |
Supplementary Material
Acknowledgments
We thank the US National Science Foundation (CHE 0616352) for support of this work, and the Royal Society of Chemistry for a Travel Grant in support of our collaboration. RLW was supported as a summer REU student under NSF CHE-0552947.
Footnotes
Electronic Supplementary Information (ESI) available: [First order rate plots for the racemisation, Eyring plots, and a representative chromatogram of the chiral silanes]. See DOI: 10.1039/b??????x/
Typical Experiment: A stock solution of N-Boc-2-(tributylstannyl)-pyrrolidine, typically 0.04M, was prepared in diethyl ether and 2 mL transferred to each of six 10 mL tubes via septum seal (N2 atmosphere). The tubes were cooled to −78 °C, and 0.1 mL of a 2.5M solution of ligand in ether were added to each tube followed by 0.1 mL of a 2.5M solution of n-BuLi in hexane. The dull yellow color of the organolithium was seen within seconds. The tubes were thermostatted at the reaction temperature, and a stopwatch was started. Internal temperature was monitored in a separate tube in the same bath. Tubes were removed at various times, cooled to −78 °C, and quenched with 0.2 mL of a 2.5M solution of TMSCl in hexane for ca. 16 h. Water (2mL) was added to each tube, and the organics extracted into ether which was then dried (MgSO4) and concentrated to ca. 0.3 mL, and purified by prep TLC (2% EtOAc/hexane). The silanes (Rf = 0.65 ) were scraped off, extracted from silica into ether and then concentrated to one drop, of which 0.1 μL was subjected to CSP-GC analysis (β-cyclodextrin stationary phase). The column temperature was programmed as follows: T = 70 °C for 5 min, then 5 °C/min to T = 200 °C, then maintained for 10 min. Rt = 14.3 min and 16.1 min for 4-(S)- and 4-(R)- N-Boc-2-(trimethylsilyl)-pyrrolidine, respectively.
Notes and references
- 1.a Hoppe D, Hense T. Angew. Chem. Int. Ed. Engl. 1997;36:2283–2316. [Google Scholar]; b Hoppe D, Hintze F, Tebben P, Paetow M, Ahrens H, Schwerdtfeger J, Sommerfeld P, Haller J, Guarnieri W, Kolczewski S, Hense T, Hoppe I. Pure Appl. Chem. 1994;66:1479–1486. [Google Scholar]
- 2.Collum DB. Acc. Chem. Res. 1992;25:448–454. [Google Scholar]
- 3.Beak P, Gallagher DJ, Wu. S, Nikolic NA. J. Org. Chem. 1995;60:8148–8154. [Google Scholar]
- 4.a O'Brien P, McGrath MJ, Bilke JL. Chem. Commun. 2006:2607–2609. doi: 10.1039/b603804m. [DOI] [PubMed] [Google Scholar]; b O'Brien P, McGrath MJ. J. Am. Chem. Soc. 2005;127:16378–16379. doi: 10.1021/ja056026d. [DOI] [PubMed] [Google Scholar]; c McGrath MJ, O'Brien P. Synthesis. 2006:2233–2241. [Google Scholar]
- 5.a Beak P, Anderson DR, Curtis MD, Laumer JM, Pippel DJ, Weisenburger GA. Acc. Chem. Res. 2000;33:715–727. doi: 10.1021/ar000077s. [DOI] [PubMed] [Google Scholar]; b Coldham I, Dufour S, Haxell TFN, Howard S, Vennall GP. Angew. Chem. Int. Ed. 2002;41:3887–3889. doi: 10.1002/1521-3773(20021018)41:20<3887::AID-ANIE3887>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; c Coldham I, Dufour S, Haxell TFN, Patel JJ, Sanchez-Jimenez G. J. Am. Chem. Soc. 2006;128:10943–10951. doi: 10.1021/ja061963m. [DOI] [PubMed] [Google Scholar]; d Coldham I, Dufour S, Haxell TFN, Vennall GP. Tetrahedron. 2005;61:3205–3220. [Google Scholar]; e Park YS, Yum EK, Basu A, Beak P. Org. Lett. 2006;8:2667–2670. doi: 10.1021/ol0604015. [DOI] [PubMed] [Google Scholar]; f Nakamura S, Nakagawa R, Watanabe Y, Toru T. Angew. Chem. Int. Ed. 2000;39:353–355. doi: 10.1002/(sici)1521-3773(20000117)39:2<353::aid-anie353>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]; g Nakamura S, Nakagawa R, Watanabe Y, Toru T. J. Am. Chem. Soc. 2000;122:11340–11347. [Google Scholar]
- 6.Coldham I, Patel JJ, Sanchez-Jiminez G. Chem. Commun. 2005:3083–3085. doi: 10.1039/b504015a. [DOI] [PubMed] [Google Scholar]
- 7.Basu A, Thayumanavan S. Angew. Chem. Int. Ed. 2002;41:716–738. doi: 10.1002/1521-3773(20020301)41:5<716::aid-anie716>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 8.Ashweek NJ, Brandt P, Coldham I, Dufour S, Gawley RE, Hæffner F, Klein R, Sanchez-Jimenez G. J. Am. Chem. Soc. 2005;127:449–457. doi: 10.1021/ja048090l. [DOI] [PubMed] [Google Scholar]
- 9.Pearson WH, Lindbeck AC, Kampf JW. J. Am. Chem. Soc. 1993;1993:2622–2636. [Google Scholar]
- 10.a Gawley RE, Zhang Q. J. Am. Chem. Soc. 1993;115:7515–7516. [Google Scholar]; b Gawley RE, Zhang Q. Tetrahedron. 1994;50:6077–6088. [Google Scholar]
- 11.Ahlbrecht H, Harbach J, Hoffmann RW, Ruhland T. Liebigs Ann. 1995:211–216. [Google Scholar]
- 12.Heinl T, Retzow S, Hoppe D, Fraenkel G, Chow A. Chem. Eur. J. 1999;5:3464–3470. [Google Scholar]
- 13.a Beak P, Kerrick ST, Wu S, Chu J. J. Am. Chem. Soc. 1994;116:3231–3239. [Google Scholar]; b Bertini Gross KM, Beak P. J. Am. Chem. Soc. 2001;123:315–321. doi: 10.1021/ja002662u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.