

Abstract
The BCR-ABL tyrosine kinase produced by the t(9;22)(q34;q11) translocation, also known as the Philadelphia chromosome, is the initiating event in chronic myeloid leukemia (CML) and Ph+ acute lymphoblastic leukemia (ALL). Targeting of BCR-ABL with tyrosine kinase inhibitors (TKIs) has resulted in rapid clinical responses in the vast majority of patients with CML and Philadelphia chromosome+ ALL. However, long-term use of TKIs occasionally results in emergence of therapy resistance, in part through the selection of clones with mutations in the BCR-ABL kinase domain. We present here an overview of the current practice in monitoring for such mutations, including the methods used, the clinical and laboratory criteria for triggering mutational analysis, and the guidelines for reporting BCR-ABL mutations. We alsopresent a proposal for a public database for correlating mutational status with in vitro and in vivo responses to different TKIs to aid in the interpretation of mutation studies.
Treatment of chronic myeloid leukemia (CML) and Philadelphia chromosome (Ph)+ acute lymphoblastic leukemia (ALL) represents a model for targeted cancer therapy, with the demonstration that ATP-competitive kinase inhibitors that block BCR-ABL kinase activity, particularly imatinib mesylate (Gleevec), can induce durable responses in the vast majority of patients. However, the emergence of resistant leukemia clones bearing mutations in the BCR-ABL kinase domain (KD) represent a major mechanism of disease recurrence that can be treated by changing therapy, often to another tyrosine kinase inhibitor (TKI) that differs with respect to pharmacokinetics and kinase inhibitory properties.
Although differences still exist between laboratories in the methodology and timing of molecular monitoring in CML, they are becoming increasingly standardized. In most centers, reverse transcription quantitative polymerase chain reaction (RQ-PCR) assessment for the BCR-ABL transcript, a fusion of the BCR and ABL1 genes, has become the standard monitoring assay for residual disease with testing done every 3 to 6 months over the course of disease. The treatments and the algorithms for monitoring responses in Ph+ ALL are more variable, with more intensive monitoring by both multiparameter flow cytometry and RQ-PCR commonly used in the first year after treatment has begun.
To further standardization efforts, we present here guidelines for BCR-ABL mutational analysis including considerations of triggers for analysis, assay performance, and reporting, and include a summary of current practice in clinical laboratories in the United States and Canada. Although we do not intend to fully define standards of practice in this article, the suggested guidelines contribute to this effort and point out areas that need further development.
What Is the Clinical Rationale for Detection of BCR-ABL Point Mutations in CML and Ph+ ALL?
In CML, most data on the frequency of BCR-ABL KD mutations and their clinical significance has been generated from patients with cytogenetic or hematological resistance or relapse. Among patients with chronic phase CML who develop (secondary) resistance to imatinib, 30% to 50% will have one or more BCR-ABL KD mutations detectable by direct DNA sequencing,1,2 whereas mutation frequencies are higher in those with accelerated or blast phases of disease, especially in lymphoid blast phases.3 The absence of a BCR-ABL KD mutation does not exclude acquired drug resistance, since other less common mechanisms of resistance include BCR-ABL gene amplification, BCR-ABL overexpression, alterations in drug efflux kinetics, upregulation of other kinase pathways, and rare BCR-ABL mutations outside of the KD. Causes of therapy resistance unrelated to kinase activity are typically due to additional oncogenic activation or loss of tumor suppressor function, often manifested by additional karyotypic changes.
The prognostic significance of finding any BCR-ABL KD mutation, or any specific mutation such as T315I, is complex and is described in more detail below. Some studies, for example, have shown no differences in progression-free survival in TKI-resistant CML with or without BCR-ABL KD mutation.1,3,4,5 However, in those patients with imatinib resistance due to KD mutations, use of more potent kinase inhibitors, including dasatinib, nilotinib, and bosutinib can often overcome resistance in the subset of patients in which the specific acquired BCR-ABL KD mutation observed does not cause resistance to the alternate drug.6,7
As compared with CML, BCR-ABL KD mutations occur much more frequently (80% to 90% of cases) at the time of relapse in Ph+ ALL8,9 in those patients who have been treated with TKIs as initial or maintenance therapy. Lymphoid blast transformation of CML is also associated with a similar high rate of new BCR-ABL KD mutations.10 Using more sensitive detection methods, low-levels of a point mutation clone occasionally have even been detected in Ph+ ALL before exposure to TKIs, suggesting that resistant clones may precede TKI selection in some cases of ALL.8 The detection of a BCR-ABL KD mutation at relapse in Ph+ ALL usually is followed by a switch to a new TKI along with salvage polychemotherapy.
When Should BCR-ABL Mutational Analysis Be Performed?
Since BCR-ABL KD mutations in CML and Ph+ ALL can occasionally be found in patients without clinical evidence of resistant disease,11,12 the question remains when to test for mutations and by what method. An international consensus group was convened to develop guidelines for use of BCR-ABL transcript monitoring and mutation testing in CML, formalizing its recommendations at a meeting at the National Institutes Health in 2005 and subsequently in a publication in 2006.13 Following these recommendations, BCR-ABL KD mutation screening in chronic phase CML is only recommended for those patients with inadequate initial response to TKIs or those with evidence of loss of response. Mutation screening is also recommended at the time of progression to accelerated or blast phase CML. The National Comprehensive Cancer Network adopted these guidelines in 2007 (Practice Guidelines in Oncology: Chronic myelogenous leukemia, http://www.nccn.org/professionals/physician_gls/PDF/cml.pdf, accessed 9/1/08).
Criteria for inadequate initial response (ie, primary resistance) include lack of complete hematological response, minimal cytogenetic response or lack of major cytogenetic response at 3, 6, and 12 months respectively and are similar to the criteria adopted by the European LeukemiaNet.14 Criteria for loss of response to TKI (ie, secondary resistance) are also based on cytogenetic and/or hematological relapse, with variable use of molecular relapse criteria. One proposed molecular trigger for mutation testing is a tenfold or greater increase in BCR-ABL transcript levels, although smaller rises in BCR-ABL transcript levels may also be predictive of mutation development.15 However, use of increasing BCR-ABL transcripts levels as the sole criterion for triggering a mutation screen are not yet universally adopted, in part because a universal standard for normalizing BCR-ABL RQ-PCR is not yet available making values obtained at different centers difficult to compare.
There are no widely adopted guidelines as yet for the use of mutation screening in Ph+ ALL, although more intensive screening based solely on RQ-PCR levels may be warranted. Screening samples for BCR-ABL KD mutations from patients with Ph+ ALL who have never received TKI therapy is not warranted, except perhaps as a baseline for subsequent TKI treatment.9
Which Techniques Are Used to Detect BCR-ABL KD Mutations?
The particular methods used to detect BCR-ABL KD mutations will obviously have a great influence on the detection frequency, analytical sensitivity, and in turn the clinical impact of such testing (Table 1). The various mutation detection methods available have widely differing analytical sensitivities, from the least sensitive direct Sanger sequencing method, detecting a mutation present in approximately 1 in 5 BCR-ABL transcripts, to the highly sensitive mutation-specific quantitative PCR methods, which can reliably detect a mutant transcript down to 1 in 10,000 BCR-ABL transcripts. Because the detection of low levels of mutant clones may not be clinically significant, direct sequencing of the BCR-ABL transcript by the Sanger method is currently the most appropriate screening test, and was recommended by an international consensus panel.16
Table 1.
Comparison of Methods for BCR-ABL Mutation Detection
| Method | Sensitivity | Advantages | Disadvantages |
|---|---|---|---|
| Direct sequencing (Sanger) | 20% to 25% | Bidirectional confirmation | May require nested PCR to obtain enough product |
| Routine method available in most labs | Expensive and time-consuming | ||
| Not quantitative | |||
| Pyrosequencing | 1% to 5% | Lower cost | Shorter read lengths require more PCR amplicons for full BCR-ABL KD coverage |
| Higher sensitivity | |||
| Quantitative | |||
| Mutation-specific RQ-PCR | 0.01% to .1% | Highest sensitivity | Need different primers and/or probes for each mutation |
| Quantitative | |||
| Could be multiplexed | |||
| Liquid bead-array (Luminex) | 5% to 10% | Can be multiplexed to detect multiple mutations | Largely qualitative |
| Lower cost |
Other screening methods for BCR-ABL KD mutations that have been reported include denaturing high performance liquid chromatography (with follow-up definitive sequencing of abnormal cases), targeted microarrays, and liquid bead arrays. Several quantitative mutation detection methods that have been developed to track the level or proportion of a mutated clone after therapy switch,17 including PCR-based pyrosequencing18 and mutation-specific quantitative PCR, have been the most widely adopted but digital PCR applications using microfluidic separation have also been tried.19 These quantitative assays are most clearly relevant for therapy with novel agents against the pan-resistant T315I mutation, and several laboratories now offer this testing as a stand-alone assay. This type of directed approach is not likely to replace the less sensitive full BCR-ABL KD mutation screens in the near future.
How Should the Finding of a Particular BCR-ABL Mutation Be Interpreted?
At least 70 different mutations involving 57 different amino acids have been reported in the BCR-ABL kinase domain. However, many of these mutations are quite rare in imatinib-treated clinical samples, given that 15 amino acid substitutions account for 80% to 90% of all reported imatinib-resistant mutations, and 7 mutated codons (G250, Y253, E255, T315, M351, F359, and H396) account for a cumulative 60% to 70% (Table 2).16,20,21,22,23,24,25,26,27,28,29 The more common mutations cluster to one of four “hot spots” within the BCR-ABL KD (Figure 1), namely: 1) the ATP-binding P-loop (amino acids 248–256); 2) the imatinib binding region (amino acids 315–317); 3) the catalytic domain (amino acids 350–363); and 4) the activation (A)-loop (amino acids 381–402). The A-loop is a major regulator of BCR-ABL kinase activity by adopting either a closed (inactive) or open (active) conformation, and A-loop mutations often destabilize the inactive conformation that is required for imatinib binding.
Table 2.
Prevalence and Drug Sensitivity of Common Imatinib-Resistant BCR-ABL Mutations
| Amino acid change* | Prevalence in imatinib-resistant CML† | Imatinib IC50 (nM) | Nilotinib IC50 (nM) | Dasatinib IC50 (nM) |
|---|---|---|---|---|
| Wild-type | 260 | 13 | 0.8 | |
| M237I | 3% | 1550 | 35 | NA |
| M244V | 4% | 2000 | 38 | 1.3 |
| L248V | 2% | 2100 | 100 | NA |
| G250E | 5% to 9% | 1350 | 48 | 1.8 |
| Q252H | 2% to 3% | 1325 | 70 | 3.4 |
| Y253F | 6% | 3475 | 125 | 1.4 |
| Y253H | 5% | >6400 | 450 | 1.3 |
| E255K | 9% to 14% | 5200 | 200 | 5.6 |
| E255V | 2% to 3% | >6400 | 430 | 11 |
| D276G | 2% | 2500 | 80 | NA |
| F311L | 1% to 2% | 480 | 23 | 1.3 |
| F311I | 1% | NA | NA | NA |
| F311V | 1% | 3500 | 160 | NA |
| T315I | 13% to 16% | >6400 | >2000 | >200 |
| F317L | 3% to 4% | 1050 | 50 | 7.4 |
| M351T | 10% to 13% | 880 | 15 | 1.1 |
| E355G | 2% to 3% | 2400 | 47 | 1.8 |
| F359V | 4% to 5% | 1825 | 175 | 2.2 |
| F359C | 1% | 2400 | 290 | NA |
| V379I | 1% | 1600 | 51 | 0.8 |
| L387M | 1% | NA | NA | NA |
| M388L | 1% | 530 | 20 | NA |
| H396R | 4% | 1750 | 41 | 1.3 |
| H396P | 1% | 850 | 41 | 0.6 |
| S417Y/T | 1% | NA | NA | NA |
| E450G | 1% to 2% | NA | NA | NA |
| E453K/V/D | 1% | NA | NA | NA |
| E459L/K/G | 1% | NA | NA | NA |
| F486S | 2% | NA | NA | NA |
In vitro TKI sensitivity cutoffs.
In vitro TKI sensitivity was determined in proliferation assays of cells transfected with each BCR-ABL variant as per reference 21, with “high” sensitivity defined as an IC50 of <1000 nM imatinib, <50 nM nilotinib, and <3 nM dasatinib. “Low” sensitivity was defined as an IC50 of >3000 nM imatinib, >500 nM nilotinib, and >60 nM dasatinib. “Intermediate” sensitivity would be defined as an IC50 value in between the “high” and “low” cutoffs.
Achievable plasma trough levels are approximately (depending on dose) 1500 nM for imatinib,22 2000 nM for nilotinib,23 and 100 nM for dasatinib.24
The IC50 values for each drug for in vitro inhibition of proliferation of particular BCR-ABL mutants are somewhat variable depending on the study and the assay method.21,24,25,26,27,28 The IC50values in the table are from reference,21 except for mutants M237I, L248V, D276G, F311V, F359C, and M388L, which are from reference 28.28 The IC50values for mutant E355G are from reference 29 for imatinib and dasatinib, and from reference 28 for nilotinib.28,29
NA, IC50values not available.
Only the most common BCR-ABL KD mutations in imatinib-resistant patients are listed. The positions are those of the GenBank sequence (accession number AAB60394).
Figure 1.
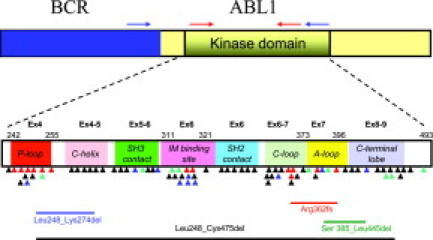
Schematic of the BCR-ABL fusion transcript and the location of reported KD mutations and alternate transcripts. Common areas for location of BCR (blue box) and ABL1 (yellow box including KD) primers for nested PCR (blue arrows) and BCR-ABL KD PCR (red arrows) used for direct sequencing assays are indicated. Colored triangles indicate the location of KD mutations reported in TKI-resistant samples (black for imatinib, green for nilotinib/imatinib, blue for dasatinib/imatinib, and red for all three TKIs). The KD subdomains, exons, and amino acid numbers are shown. P-loop: phosphate binding loop; IM binding site: imatinib binding region; C-loop: kinase catalytic domain; and A-loop: activation loop. The locations of four deletions in the BCR-ABL transcript that were most commonly reported in our laboratory survey are included in the lower part of the figure.
Specific mutation types are also becoming closely associated with newer generation TKIs, with dasatinib use often selecting for mutations at amino acids 299 (V299L), 315 (T315I), and 317 (F317L/I),6,7,30 and nilotinib preferentially selecting for certain mutations in the P-loop (G250E, Y253H, E255K), T315I, or F311I.6 The spectrum of mutations in patients being treated with dasatinib or nilotinib is closely mimicked by the pattern of clones that evolve from in vitro exposure of BCR-ABL-expressing cell lines to these same drugs.
The clinical interpretation and significance of finding a particular BCR-ABL KD mutation can be complex. The relative degree of imatinib resistance, defined by in vitro drug inhibition of kinase activity or growth of mutant-expressing cell lines, is quite variable for different BCR-ABL KD mutations, with some mutations conferring only low-level resistance that may respond to imatinib dose escalation (eg, M351T), and others conferring high-level resistance to imatinib and other TKIs (eg, T315I, Y253H, E255K), thus implying imatinib “failure” and the need for a change in therapy (Table 2).
The increasing utilization of the second-generation kinase inhibitors, particularly dasatinib and nilotinib, has further complicated the interpretation of BCR-ABL KD mutation analyses. It appears that the spectrum of resistance mutations seen following use of these more powerful TKIs are more restricted than those seen following imatinib treatment, but often have complex dynamics dependent on the specific treatment regimen and the prior therapy. Common scenarios include 1) (complete or incomplete) clonal replacement of an imatinib-selected mutation with a completely different dasatinib or nilotinib-selected clone; 2) new emergence of a BCR-ABL KD mutation only after exposure to a second-generation agent (but not with prior imatinib); and 3) persistence of an imatinib-selected mutation plus the acquisition of an additional mutation after dasatinib/nilotinib exposure; sometimes even on the same transcript.6
For most individual BCR-ABL KD mutations, there is good correlation between demonstration of resistance to TKIs in vitro and development of resistance in vivo. Many of the mutations elicited by in vitro treatment with one of the TKIs have subsequently been identified in patients with clinical resistance to that TKI.6,7 In addition, there is good correlation between in vitro sensitivity and clinical response. For example, the V299L mutation, which is associated with resistance to dasatinib, remains sensitive to imatinib in vitro25 and has demonstrated response clinically to imatinib and to the imatinib analog nilotinib.7
However, because there are multiple mechanisms of resistance to TKIs in CML and ALL, and many different mutations can emerge during therapy, the identification of a mutation while on TKI therapy does not necessarily correlate with clinical resistance.5,31 Although the presence of a BCR-ABL KD mutation is often only evaluated in a patient who is failing (or at risk of failing) TKI therapy, the development of these mutations may not be dependent on the presence of these drugs, but may, instead, be part of the natural history of the disease.26 In support of this hypothesis, mutations are more prevalent in patients with a more advanced disease phase,2,3 and are still often detectable both in pre-therapeutic samples (by sensitive allele-specific PCR methods),32,33 and in patients with a complete cytogenetic response to TKI therapy.11,12 Furthermore, the presence of a mutation per se, even the highly pan-resistant T315I mutation (at least at a low clonal burden), may not impart a growth advantage,34 leading to inconsistent association with resistance to therapy.33
How Should BCR-ABL Mutation Studies Be Reported?
Following the molecular checklist guidelines of the College of American Pathologists, we propose that all reports on BCR-ABL mutational analysis should include pre- and post-analytical elements, in addition to the assay result, as summarized in Table 3. Methodology should be briefly summarized including the region of the BCR-ABL KD that was evaluated and the nominal analytic sensitivity of the assay, defined as the lowest fraction of BCR-ABL transcripts with a mutation that can be reliably detected. The report should also include a statement that BCR-ABL KD mutation analysis does not always provide a full explanation for apparent resistance to TKI therapy.
Table 3.
Reporting Recommendations for BCR-ABL KD Mutational Analysis
| Pre-analytic |
| Clinical indication (including current and/or past TKI therapy, if available) |
| Tissue source (e.g. peripheral blood, bone marrow aspirate) |
| Most recent BCR-ABL RQ-PCR results or trend data (if available) |
| Analytic |
| Methods description including region that is sequenced, with assay sensitivity and controls |
| BCR-ABL amplification and control results (acceptable/fail) |
| Mutation not detected |
| Mutation detected: amino acid change (e.g., T315I), and relative abundance of mutation (e.g., mixed mutated/unmutated) [inclusion of nucleotide change is optional] |
| Other sequence alterations (e.g., ins/del, known SNP) |
| Post-analytic |
| Comment if mutation is known to confer clinical and/or in vitro resistance to a particular TKI, with appropriate reference |
| Comment if novel mutation(s) identified and denote lack of published clinical or laboratory data concerning its TKI resistance profile |
If a mutation is detected, this should be indicated according to standard amino acid substitution nomenclature.35 If a non-quantitative mutation detection method such as Sanger sequencing is used, an estimate of the relative quantity of the mutation can also be provided (eg, only mutated sequence, mixed mutated/unmutated sequences or low level mutation detected). If more than one mutation is identified, this type of semiquantitative estimate is important to compare the relative shifts seen in follow-up samples. If novel mutations or other genetic findings (eg, insertion/deletion events, known or previously unreported single nucleotide polymorphisms) are identified, a statement should be added indicating that the impact of the alteration on TKI resistance is not currently known.
A brief interpretation could also be included to state if the detected mutation(s) has been associated with in vitro or in vivo TKI, with an appropriate citation such as references 16 and 36. Other information that could be included in a report concern the clinical indications for testing (eg, loss of BCR-ABL molecular response, cytogenetic relapse, etc), the current TKI therapy, and the most recent BCR-ABL transcript level. Although the ultimate goal of mutation testing is to guide therapeutic decisions, the report should not contain specific recommendations concerning which therapies are optimal for any given patient.
What Are the Needs for Standardization and Proficiency Testing?
As described above, direct sequencing of the BCR-ABL transcript is the predominant method for BCR-ABL KD mutation detection in the clinical setting, however a range of other qualitative and quantitative methods are now available. This proliferation of methodologies raises the urgent but as yet unmet need for standards, calibrators, and proficiency testing programs, as required for all clinical laboratories that are accredited through Clinical Laboratory Improvement Amendments and/or the College of American Pathologists mechanisms.
Several studies have raised a number of problematic quality control issues for BCR-ABL transcript and mutation tests that should be considered in future efforts.16,37 The most important pre-analytical consideration is the quality of the extracted RNA. Many laboratories use blood collected in EDTA tubes where RNA quality may be compromised, compared with RNA-stabilizing tubes. This is an important consideration if there is a long delay between acquiring the specimen and RNA preparation. Depending on the extraction method used, RNA quality can vary significantly. Therefore, as a pre-analytical control, many laboratories establish a cutoff for the minimum level of control gene amplification required before reporting RQ-PCR assays for BCR-ABL level or mutation status.
In the analytic phase of BCR-ABL testing, it has been shown that cDNA synthesis (reverse transcription) is the most important cause of assay variation.16 Additionally in those laboratories that use a nested PCR strategy to amplify the BCR-ABL transcript before sequencing to avoid amplifying the non-translocated ABL1 transcript, variations in the efficiency of the two PCR steps can dramatically influence BCR-ABL KD mutation detection.
A common issue for proficiency testing in the molecular pathology arena is the lack of standardization of reagents and technology platforms. A typical scenario in molecular pathology testing begins with individual laboratories independently developing testing strategies, followed by industry development of analyte-specific reagents and, eventually and only in a minority of cases, kits approved by the Food and Drug Administration for clinical use. In the initial phase, each laboratory's assay is different, often with unknown strengths and weaknesses. Currently, as with BCR-ABL RQ-PCR assays, there is a need for reference material that can be used to assess the sensitivity, dynamic range and normalized values for each assay.37
As standards for quantitative BCR-ABL RQ-PCR testing are made available, the goal should be to include levels of BCR-ABL transcript normalized to the international major molecular response scale as a criteria for triggering BCR-ABL KD mutation testing.13,16
What Alterations in BCR-ABL Have Been Reported in Resistant Samples besides Point Mutations?
A number of laboratories that routinely sequence the BCR-ABL transcript have found that point mutations are not the only frequently seen genetic alteration. In our survey of clinical laboratories performing BCR-ABL mutation screening, 7 of 12 (58%) observed alternate splicing, insertions, deletions and/or duplications. A 35-bp intronic insertion, which occurred at the exon 8/9 junction after amino acid 474, was the most commonly reported, seen by five laboratories at a frequency of 2% to 10%, but was also seen by two laboratories in the ABL1 transcript in BCR-ABL negative samples. Translation of this mutant would produce a BCR-ABL protein with an insertion of 10 amino acids followed by a stop codon.38,39 Alternatively spliced products with loss of entire exons 4, 7, and 8 were reported by five laboratories. Deletions described in a clinical laboratory survey (see below) included Leu248_Cys475del (found by three laboratories with frequency of ∼2%), Arg326fs reported by two laboratories, and Leu248_Lys274del, Met318_Thr319delinsLeu, and Ser385_Leu445del reported by one laboratory each.
The significance of such grossly altered transcripts is unclear, but many would be predicted to lack active BCR-ABL kinase activity. A recent publication suggests that such deletions and proteins arising from alternatively spliced transcripts may act as dominant-negative inhibitors of the full-length BCR-ABL.40
What Is the State of Current Practice in Clinical Laboratories for BCR-ABL KD Mutation Testing?
To assess how the current state of clinical testing conforms to recommended practice, we conducted a survey of American and Canadian accredited clinical laboratories performing routine BCR-ABL KD mutational analysis. Fourteen laboratories responded and all performed testing on RNA extracted from blood or bone marrow aspirate material followed by cDNA conversion before mutation detection. Direct Sanger sequencing using Applied Biosystems BigDye Terminator chemistry on the ABI 3100, 3130, or 3730 genetic analyzers was used in 11/14 (79%) labs with most using a nested approach with BCR-ABL PCR amplification followed by ABL KD PCR amplification in a second round; pyrosequencing (directed against codon 315) was used in two laboratories (including one also using Sanger sequencing), and microarray or liquid-bead array approaches for specific mutation panels were used in one laboratory each. Quantification of the T315I mutation was available in three laboratories. The reported turn-around times for reporting the test results were less than 7 days (23%), 8 to 13 days (46%), or 14 to 28 days (23%). Nine of 14 (64%) laboratories had no preference with regards to sample type; RNA was extracted from bone marrow or peripheral blood.
The majority of laboratories (11/13, 85%) reported screening the entire KD (ABL1 exons 4 to 9, see schematic in Figure 1) for mutations, while two laboratories only tested for a specific panel of known mutations. Most labs performed bidirectional sequencing and reported positive results only when detecting a mutation in both forward- and reverse-strand chromatograms, with a commonly reported sensitivity of 10% to 20%. All clinical laboratories surveyed currently report only BCR-ABL KD point mutations producing amino acid shifts. Only a minority of laboratories reported whether the mutation was previously reported to confer resistance to kinase inhibitors, either based on clinical experience or based on data from in vitro screens. Most laboratories, while observing alternate splice products and insertion/deletions (see previous section), synonymous mutations or single nucleotide polymorphisms, do not include this finding on their reports because of limited information regarding their clinical significance.
What are the Future Directions in BCR-ABL Mutation Reporting?
There is a clear need for progress in implementing standards for reporting the results of BCR-ABL mutation studies, and (as demonstrated by our survey) also a need for tools to aid in the clinical interpretation of these results. As the number of known BCR-ABL KD mutations increase, and the number of TKIs increase, there is a greater need for a publicly-available comprehensive database to serve as a reference for interpreting the clinical significance of the results of mutation screens, as has been done in infectious diseases (HIV Drug Resistance Database, http://hivdb.stanford.edu, accessed 9/1/08) and genetic syndromes (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac, accessed 9/1/08). Such a database would be invaluable in differentiating benign polymorphisms/passenger mutations from resistance mutations and assisting in predicting response to a different TKI to help in selecting an alternate therapy.
Such a database should present information on the in vivo context in which specific mutations have previously developed (eg, which resistance mutation are found in patients relapsing after treatment with dasatinib or nilotinib)6,7 but also summarize the in vitro sensitivity of particular mutations to each TKI (Table 2).21 There is an increasingly large amount of published data on the effects of particular TKIs on inhibiting KD-mutated BCR-ABL in kinase assays, on inhibiting growth of cell lines expressing particular KD-mutated BCR-ABL proteins, or in promoting outgrowth of certain mutations in long-term in vitro culture (mutagenesis screens).24 All of these data elements provide corroborating evidence of the pattern of drug resistance for each particular mutation under controlled conditions. The type of database we outline would provide easy access to a set of laboratory information needed for clinical decision making. To be truly useful the database would need to be updated frequently with new information and be carefully curated for accuracy. It would also need to be free of commercial influences.
In silico modeling of the effect of a mutation on kinase function based on structural protein data (including co-crystallization of the ABL kinase domain with specific TKIs) can also predict which inhibitors will be effective against which BCR-ABL KD mutations in vivo.18,41 This approach has elucidated the mechanism of resistance for the BCR-ABL pan-resistant mutation T315I, which is a key contact residue for TKIs,41 and of imatinib-resistance mutations that destabilize the inactive conformation of BCR-ABL.
Summary
Given our evolving understanding of the molecular events mediating resistance in CML and Ph+ ALL, standards for reporting of BCR-ABL mutational studies would benefit from a greater degree of uniformity. Commercially-available reference samples and calibrators as well as a publicly available BCR-ABL mutation database are the currently needed resources to allow laboratories and clinicians to interpret the significance of BCR-ABL KD mutation studies. While these standardization efforts are proceeding, mutation studies should be based on the already developed criteria for clinical resistance (summarized above) to better ensure appropriate utilization. As shared databases become more widely available, the most appropriate statements regarding the clinical significance of specific mutations will be better defined and allow more precise guidance to be given.
Acknowledgements
We thank Michael Langley and Tong Zhang for help in figure and table preparation and Terence Dunn for manuscript review.
Footnotes
The ABL Mutation Working Group is a subcommittee of the AMP Clinical Practice Committee.
The 2007–2008 AMP Clinical Practice Committee consisted of Aaron Bossler, William Funkhouser, Julie Gastier-Foster, Dan Jones, Victoria M. Pratt (Chair), Daniel Sabath, and Antonia Sepulveda.
Standard of practice is not being defined by this article, and there may be alternatives.
Address reprint requests to the Association for Molecular Pathology, 9650 Rockville Pike, Bethesda, MD 20814. E-mail: amp@amp.org
References
- 1.Jabbour E, Kantarjian H, Jones D, Talpaz M, Bekele N, O'Brien S, Zhou X, Luthra R, Garcia-Manero G, Giles F, Rios MB, Verstovsek S, Cortes J. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20:1767–1773. doi: 10.1038/sj.leu.2404318. [DOI] [PubMed] [Google Scholar]
- 2.Branford S, Rudzki Z, Walsh S, Parkinson I, Grigg A, Szer J, Taylor K, Herrmann R, Seymour JF, Arthur C, Joske D, Lynch K, Hughes T. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–283. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 3.Soverini S, Colarossi S, Gnani A, Rosti G, Castagnetti F, Poerio A, Iacobucci I, Amabile M, Abruzzese E, Orlandi E, Radaelli F, Ciccone F, Tiribelli M, di Lorenzo R, Caracciolo C, Izzo B, Pane F, Saglio G, Baccarani M, Martinelli G. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12:7374–7379. doi: 10.1158/1078-0432.CCR-06-1516. [DOI] [PubMed] [Google Scholar]
- 4.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP, Hughes TP. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475. doi: 10.1182/blood.v99.9.3472. [DOI] [PubMed] [Google Scholar]
- 5.Jabbour E, Kantarjian H, Jones D, Breeden M, Garcia-Manero G, O'Brien S, Ravandi F, Borthakur G, Cortes J. Characteristics and outcomes of patients with chronic myeloid leukemia and T315I mutation following failure of imatinib mesylate therapy. Blood. 2008;112:53–55. doi: 10.1182/blood-2007-11-123950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortes J, Jabbour E, Kantarjian H, Yin CC, Shan J, O'Brien S, Garcia-Manero G, Giles F, Breeden M, Reeves N, Wierda WG, Jones D. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110:4005–4011. doi: 10.1182/blood-2007-03-080838. [DOI] [PubMed] [Google Scholar]
- 7.Shah NP, Skaggs BJ, Branford S, Hughes TP, Nicoll JM, Paquette RL, Sawyers CL. Sequential ABL kinase inhibitor therapy selects for compound drug-resistant BCR-ABL mutations with altered oncogenic potency. J Clin Invest. 2007;117:2562–2569. doi: 10.1172/JCI30890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeifer H, Wassmann B, Pavlova A, Wunderle L, Oldenburg J, Binckebanck A, Lange T, Hochhaus A, Wystub S, Bruck P, Hoelzer D, Ottmann OG. Kinase domain mutations of BCR-ABL frequently precede imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) Blood. 2007;110:727–734. doi: 10.1182/blood-2006-11-052373. [DOI] [PubMed] [Google Scholar]
- 9.Jones D, Thomas D, Yin CC, O'Brien S, Cortes JE, Jabbour E, Breeden M, Giles FJ, Zhao W, Kantarjian HM. Kinase domain point mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia emerge after therapy with BCR-ABL kinase inhibitors. Cancer. 2008;113:985–994. doi: 10.1002/cncr.23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones D, Luthra R, Cortes J, Thomas D, O'Brien S, Bueso-Ramos C, Hai S, Ravandi F, de Lima M, Kantarjian H, Jorgensen J. BCR-ABL fusion transcript types and levels and their interaction with secondary genetic changes in determining the phenotype of Philadelphia chromosome-positive leukemias. Blood. 2008 doi: 10.1182/blood-2008-04-148791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherbenou DW, Wong MJ, Humayun A, McGreevey LS, Harrell P, Yang R, Mauro M, Heinrich MC, Press RD, Druker BJ, Deininger MW. Mutations of the BCR-ABL-kinase domain occur in a minority of patients with stable complete cytogenetic response to imatinib. Leukemia. 2007;21:489–493. doi: 10.1038/sj.leu.2404554. [DOI] [PubMed] [Google Scholar]
- 12.Chu S, Xu H, Shah NP, Snyder DS, Forman SJ, Sawyers CL, Bhatia R. Detection of BCR-ABL kinase mutations in CD34+ cells from chronic myelogenous leukemia patients in complete cytogenetic remission on imatinib mesylate treatment. Blood. 2005;105:2093–2098. doi: 10.1182/blood-2004-03-1114. [DOI] [PubMed] [Google Scholar]
- 13.Hughes T. ABL kinase inhibitor therapy for CML: baseline assessments and response monitoring. Hematology Am Soc Hematol Educ Prog. 2006:211–218. doi: 10.1182/asheducation-2006.1.211. [DOI] [PubMed] [Google Scholar]
- 14.Baccarani M, Saglio G, Goldman J, Hochhaus A, Simonsson B, Appelbaum F, Apperley J, Cervantes F, Cortes J, Deininger M, Gratwohl A, Guilhot F, Horowitz M, Hughes T, Kantarjian H, Larson R, Niederwieser D, Silver R, Hehlmann R. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108:1809–1820. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 15.Branford S, Rudzki Z, Parkinson I, Grigg A, Taylor K, Seymour JF, Durrant S, Browett P, Schwarer AP, Arthur C, Catalano J, Leahy MF, Filshie R, Bradstock K, Herrmann R, Joske D, Lynch K, Hughes T. Real-time quantitative PCR analysis can be used as a primary screen to identify patients with CML treated with imatinib who have BCR-ABL kinase domain mutations. Blood. 2004;104:2926–2932. doi: 10.1182/blood-2004-03-1134. [DOI] [PubMed] [Google Scholar]
- 16.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, Baccarani M, Cortes J, Cross NC, Druker BJ, Gabert J, Grimwade D, Hehlmann R, Kamel-Reid S, Lipton JH, Longtine J, Martinelli G, Saglio G, Soverini S, Stock W, Goldman JM. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vivante A, Amariglio N, Koren-Michowitz M, Ashur-Fabian O, Nagler A, Rechavi G, Cohen Y. High-throughput, sensitive and quantitative assay for the detection of BCR-ABL kinase domain mutations. Leukemia. 2007;21:1318–1321. doi: 10.1038/sj.leu.2404635. [DOI] [PubMed] [Google Scholar]
- 18.Giles FJ, Cortes J, Jones D, Bergstrom D, Kantarjian H, Freedman SJ. MK-0457, a novel kinase inhibitor, is active in patients with chronic myeloid leukemia or acute lymphocytic leukemia with the T315I BCR-ABL mutation. Blood. 2007;109:500–502. doi: 10.1182/blood-2006-05-025049. [DOI] [PubMed] [Google Scholar]
- 19.Oehler VG, Qin J, Ramakrishnan R, Facer G, Ananthnarayan S, Cummings C, Deininger M, Shah N, McCormick F, Willis S, Daridon A, Unger M, Radich JP. Absolute quantitative detection of ABL tyrosine kinase domain point mutations in chronic myeloid leukemia using a novel nanofluidic platform and mutation-specific PCR. Leukemia. 2008 doi: 10.1038/leu.2008.183. (Letter to the Editor) Epub ahead of print, doi: 10.1038leu.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Apperley JF. Part I: mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007;8:1018–1029. doi: 10.1016/S1470-2045(07)70342-X. [DOI] [PubMed] [Google Scholar]
- 21.O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, Druker BJ. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–4505. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 22.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 23.Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 24.Bradeen HA, Eide CA, O'Hare T, Johnson KJ, Willis SG, Lee FY, Druker BJ, Deininger MW. Comparison of imatinib mesylate, dasatinib (BMS-354825), and nilotinib (AMN107) in an N-ethyl-N-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood. 2006;108:2332–2338. doi: 10.1182/blood-2006-02-004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgess MR, Skaggs BJ, Shah NP, Lee FY, Sawyers CL. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc Natl Acad Sci USA. 2005;102:3395–3400. doi: 10.1073/pnas.0409770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110:2242–2249. doi: 10.1182/blood-2007-03-066936. [DOI] [PubMed] [Google Scholar]
- 27.Ray A, Cowan-Jacob SW, Manley PW, Mestan J, Griffin JD. Identification of BCR-ABL point mutations conferring resistance to the Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study. Blood. 2007;109:5011–5015. doi: 10.1182/blood-2006-01-015347. [DOI] [PubMed] [Google Scholar]
- 28.von Bubnoff N, Manley PW, Mestan J, Sanger J, Peschel C, Duyster J. Bcr-Abl resistance screening predicts a limited spectrum of point mutations to be associated with clinical resistance to the Abl kinase inhibitor nilotinib (AMN107) Blood. 2006;108:1328–1333. doi: 10.1182/blood-2005-12-010132. [DOI] [PubMed] [Google Scholar]
- 29.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 30.Soverini S, Martinelli G, Colarossi S, Gnani A, Rondoni M, Castagnetti F, Paolini S, Rosti G, Baccarani M. Second-line treatment with dasatinib in patients resistant to imatinib can select novel inhibitor-specific BCR-ABL mutants in Ph+ ALL. Lancet Oncol. 2007;8:273–274. doi: 10.1016/S1470-2045(07)70078-5. [DOI] [PubMed] [Google Scholar]
- 31.Khorashad JS, Anand M, Marin D, Saunders S, Al-Jabary T, Iqbal A, Margerison S, Melo JV, Goldman JM, Apperley JF, Kaeda J. The presence of a BCR-ABL mutant allele in CML does not always explain clinical resistance to imatinib. Leukemia. 2006;20:658–663. doi: 10.1038/sj.leu.2404137. [DOI] [PubMed] [Google Scholar]
- 32.Roche-Lestienne C, Deluche L, Corm S, Tigaud I, Joha S, Philippe N, Geffroy S, Lai JL, Nicolini FE, Preudhomme C. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL+ leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood. 2008;111:3735–3741. doi: 10.1182/blood-2007-07-102533. [DOI] [PubMed] [Google Scholar]
- 33.Willis SG, Lange T, Demehri S, Otto S, Crossman L, Niederwieser D, Stoffregen EP, McWeeney S, Kovacs I, Park B, Druker BJ, Deininger MW. High-sensitivity detection of BCR-ABL kinase domain mutations in imatinib-naive patients: correlation with clonal cytogenetic evolution but not response to therapy. Blood. 2005;106:2128–2137. doi: 10.1182/blood-2005-03-1036. [DOI] [PubMed] [Google Scholar]
- 34.Miething C, Feihl S, Mugler C, Grundler R, von Bubnoff N, Lordick F, Peschel C, Duyster J. The Bcr-Abl mutations T315I and Y253H do not confer a growth advantage in the absence of imatinib. Leukemia. 2006;20:650–657. doi: 10.1038/sj.leu.2404151. [DOI] [PubMed] [Google Scholar]
- 35.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 36.Baccarani M, Pane F, Saglio G. Monitoring treatment of chronic myeloid leukemia. Haematologica. 2008;93:161–169. doi: 10.3324/haematol.12588. [DOI] [PubMed] [Google Scholar]
- 37.Saldanha J, Silvy M, Beaufils N, Arlinghaus R, Barbany G, Branford S, Cayuela JM, Cazzaniga G, Gonzalez M, Grimwade D, Kairisto V, Miyamura K, Lawler M, Lion T, Macintyre E, Mahon FX, Muller MC, Ostergaard M, Pfeifer H, Saglio G, Sawyers C, Spinelli O, van der Velden VH, Wang JQ, Zoi K, Patel V, Phillips P, Matejtschuk P, Gabert J. Characterization of a reference material for BCR-ABL (M-BCR) mRNA quantitation by real-time amplification assays: towards new standards for gene expression measurements. Leukemia. 2007;21:1481–1487. doi: 10.1038/sj.leu.2404716. [DOI] [PubMed] [Google Scholar]
- 38.Quigley NB, Henley DC, Hubbard RA, Laudadio J, Press RD. ABL kinase domain pseudoexon insertion is not uncommon in BCR-ABL transcripts. J Mol Diagn. 2008;10:475–476. doi: 10.2353/jmoldx.2008.080055. author reply 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laudadio J, Deininger MW, Mauro MJ, Druker BJ, Press RD. An intron-derived insertion/truncation mutation in the BCR-ABL kinase domain in chronic myeloid leukemia patients undergoing kinase inhibitor therapy. J Mol Diagn. 2008;10:177–180. doi: 10.2353/jmoldx.2008.070128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherbenou DW, Hantschel O, Turaga L, Kaupe I, Willis S, Bumm T, Press RD, Superti-Furga G, Druker BJ, Deininger MW. Characterization of BCR-ABL deletion mutants from patients with chronic myeloid leukemia. Leukemia. 2008;22:1184–1190. doi: 10.1038/leu.2008.65. [DOI] [PubMed] [Google Scholar]
- 41.Young MA, Shah NP, Chao LH, Seeliger M, Milanov ZV, Biggs WH, 3rd, Treiber DK, Patel HK, Zarrinkar PP, Lockhart DJ, Sawyers CL, Kuriyan J. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006;66:1007–1014. doi: 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]