

Abstract
Hypertrophic cardiomyopathy is caused by mutations in the genes that encode sarcomeric proteins and is primarily characterized by unexplained left ventricular hypertrophy, impaired cardiac function, reduced exercise tolerance, and a relatively high incidence of sudden cardiac death, especially in the young. The extent of left ventricular hypertrophy is one of the major determinants of disease prognosis. Angiotensin II has trophic effects on the heart and plays an important role in the development of myocardial hypertrophy. Here in a double-blind, placebo-controlled, randomized study, we show that the long-term administration of the angiotensin II type 1 receptor antagonist candesartan in patients with hypertrophic cardiomyopathy was associated with the significant regression of left ventricular hypertrophy, improvement of left ventricular function, and exercise tolerance. The magnitude of the treatment effect was dependent on specific sarcomeric protein gene mutations that had the greatest responses on the carriers of ß-myosin heavy chain and cardiac myosin binding protein C gene mutations. These data indicate that modulating the role of angiotensin II in the development of hypertrophy is specific with respect to both the affected sarcomeric protein gene and the affected codon within that gene. Thus, angiotensin II type 1 receptor blockade has the potential to attenuate myocardial hypertrophy and may, therefore, provide a new treatment option to prevent sudden cardiac death in patients with hypertrophic cardiomyopathy.
Hypertrophic cardiomyopathy (HCM) is a primary cardiac disease characterized by unexplained cardiac hypertrophy and a relatively high incidence of sudden cardiac death, especially in young people.1,2 The extent of left ventricular hypertrophy is one of the major determinants of symptoms and prognosis.3,4
Angiotensin II has trophic effects on the heart and plays an important role in the development of myocardial hypertrophy.5,6 Inhibition of angiotensin-converting enzyme (ACE) or the angiotensin II type 1 receptor (AT1-R) induced regression of myocardial hypertrophy in patients with hypertension or after myocardial infarction.7,8,9 In HCM, ACE and AT1-R gene polymorphisms have been shown to be associated with severity of hypertrophy, a high incidence of atrial fibrillation and the risk of sudden cardiac death.10,11,12,13,14,15,16,17 Therefore, we designed a double-blind, placebo-controlled, randomized, multicenter study to test the safety and effects of AT1-R antagonist candesartan in patients with nonobstructive HCM. We hypothesized that long-term use of candesartan would be associated with regression of left ventricular (LV) hypertrophy and improvement of LV function.
Materials and Methods
Patients
This was a double-blind, placebo-controlled, randomized multicenter study. The study population consisted of 24 consecutive, genetically independent, adult (≥18 years) patients (age 43 ± 13 yrs; 46% males) with nonobstructive HCM, and normal ejection fraction (≥60%) and sinus rhythm, who visited the participating institutions for routine follow-up. HCM had been diagnosed on the basis of echocardiography showing a nondilated, hypertrophied LV (any wall thickness >15 mm) in the absence of known causes of LV hypertrophy, hypertension, or valvular disease.18,19 Exclusion criteria were as follows1: hypertrophic obstructive cardiomyopathy defined as presence of a resting gradient in LV outflow tract ≥30 mmHg or in right ventricular outflow tract ≥15 mm Hg at Doppler echocardiography2; atrial fibrillation3; treatment with ACE inhibitors or AT1-R antagonists at any time in the past4; contraindications to AT1-R antagonists5; coronary artery disease, renal failure, hepatic disorders or serious intercurrent illness limiting survival; and6 poor echocardiographic image quality. The study protocol was approved by the Medical Ethics Committees of all participating institutions and informed consent was obtained from all patients. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the institutions human research committee.
Protocol
At baseline, patients provided a blood sample for molecular genetic testing. The presence of a putative HCM-causing variant was determined by comprehensive mutational analysis of the ß-myosin heavy chain (ß-MHC), cardiac myosin binding protein C (cMYBPC), cardiac troponin T and cardiac troponin I genes that account for between 75% and 99% of all identified HCM-associated genotypes.10,20,21,22,23 Patients were randomly assigned in a 1:1 ratio to candesartan (n = 12) or matching placebo (n = 12). The initial dose of the study drug was 8 mg once daily. Study drug dose was then doubled as tolerated every 2 weeks while aiming for a target dose of 32 mg once daily.24 Blood pressure, serum creatinine, serum potassium, and pressure gradient in the LV outflow tract were monitored during dose titration. Patients were observed clinically at 3, 6, and 12 months after the maintenance dose was reached. Exercise tolerance by bicycle ergometry, the presence of malignant arrhythmias by Holter monitoring, the extent of LV hypertrophy by 2-dimensional echocardiography, and LV outflow tract pressure gradient by Doppler echocardiography were all assessed at baseline and 12-month follow-up.
Bicycle Ergometry
Patients cycled in the upright position starting with a workload of 25 W. The workload was increased in 25-W steps every 2 minutes until the patient was unable to continue because of dyspnea or fatigue. A 12-lead ECG and blood pressure were recorded during the last 10 seconds of each step of exercise and 1, 3, and 5 minutes after exercise testing. Total exercise time was measured to assess exercise tolerance.
Echocardiography
All studies were performed with a commercially available system equipped with tissue Doppler imaging (Hewlett-Packard Sonos 5500, Philips Medical Systems, Andover, MA). Transthoracic images were obtained in standard cross-sectional planes. M-mode echocardiograms were derived from two-dimensional images under direct anatomical visualization. LV diameters (end-diastolic and end-systolic) were assessed in parasternal long-axis view.25 LV ejection fraction was calculated in apical 4-chamber and 2-chamber views using the biplane Simpson methods.25 Peak LV outflow tract gradient at rest was estimated using the modified Bernoulli equation, P = 4V2, where P is the pressure gradient and V is the velocity determined by Doppler echocardiography. LV filling pattern was assessed from the transmitral flow in apical views. Tissue Doppler was performed in pulsed wave mode. In apical 4-chamber view, peak mitral annular velocities during systole (Sa) and early diastole (Ea) were assessed as the average from two corners of the mitral annulus (septal and lateral). Gain and filters were adjusted to obtain an optimal tissue signal. Annular velocities were recorded during end-expiratory apnea at a sweep speed of 100 mm/s.
Analysis of LV Hypertrophy
End-diastolic LV wall thickness was assessed in all LV segments (16-segment model) in parasternal short-axis views.25 Mean wall thickness was calculated in each patient. In addition, LV mass was determined according to the method described by Devereux27: LV mass = 0.8(1.04[(IVS+LVEDd + LVPW)3 − LVEDd3])+0.6 g, where LVEDd is the LV end-diastolic diameter, and IVS and LVPW are the end-diastolic thickness of the interventricular septum and posterior wall, respectively. All studies were stored in digital format and on S-VHS videotape for off-line analysis. The mean from three consecutive beats was taken for each measurement. Digitized baseline and 12-month follow-up echocardiograms were analyzed side by side by two echocardiographers (M.P., J.K.) blinded to patient group assignment.
Statistical Analysis
All analyses were conducted according to the intention-to-treat principle. Continuous data are presented as mean ± SD Categorical data are presented as numbers and percentages. Two-sided paired and unpaired Student's t-test, Fisher's exact test and the Pearson test were used as appropriate. For all tests, P < 0.05 was considered significant.
The CHANCE investigators developed the study protocol with the sponsor. The sponsor had no access to the database. All data analyses were performed by investigators independently of the sponsor.
Results
Baseline Characteristics
Table 1 shows the baseline characteristics for patients assigned to candesartan and placebo. There were no differences between the two groups for any characteristic. None of the patients had isolated apical hypertrophy, an implantable cardioverter-defibrillator, pacemaker or a history of surgical septal myectomy or alcohol ablation.
Table 1.
Baseline Characteristics
| Candesartan group (n = 12) | Placebo group (n = 12) | P | |
|---|---|---|---|
| Age, y | 41 ± 15 | 45 ± 13 | 0.56 |
| Male sex, n (%) | 5 (42) | 6 (50) | NS |
| Cardiac symptoms, n (%) | 8 (67) | 7 (58) | NS |
| Family history | |||
| HCM, n (%) | 7 (58) | 8 (67) | NS |
| SCD, n (%) | 3 (25) | 3 (25) | NS |
| Myofilament genotype | |||
| ß-MHC, n (%) | 5 (42) | 4 (33) | |
| cMYBPC, n (%) | 3 (25) | 3 (25) | |
| cTnT, n (%) | 0 | 0 | NS |
| cTnI, n (%) | 2 (17) | 1 (8) | |
| Myofilament genotype-negative, n (%) | 2 (17) | 2 (17) | |
| Medication | |||
| ß-blockers, n (%) | 4 (33) | 4 (33) | |
| Verapamil, n (%) | 4 (33) | 4 (33) | NS |
| ß-blockers and verapamil, n (%) | 1 (8) | 1 (8) |
ß-MHC = ß-myosin heavy chain, cMYBPC = cardiac myosin binding protein C, cTnT = cardiac troponin T, cTnI = cardiac troponin I, HCM = hypertrophic cardiomyopathy, SCD = sudden cardiac death.
In the placebo group, two individuals refused genetic screening. A putative HCM-causing myofilament variant was identified in 18 (82%) out of the 22 remaining patients who underwent genetic screening. The prevalence of specific HCM genotypes was similar between groups.
Effects of Candesartan on LV Hypertrophy and Function at 12-Month Follow-Up
Follow-up was completed by 23 patients (candesartan n = 12, placebo n = 11). One patient in the placebo group withdrew informed consent and was excluded from the analysis. Resting systolic blood pressure, heart rate, and LV outflow tract gradient did not differ between baseline and follow-up examination. At baseline, symptoms, exercise tolerance, LV systolic, and diastolic function and the magnitude of LV hypertrophy were similar between groups (Table 2). At 12-month follow up, patients in the candesartan group showed significant reductions of mean LV wall thickness and LV mass compared with patients receiving placebo, while LV end-diastolic diameter did not change (Figure 1). In the candesartan group, three patients showed a decrease in LV mass >100g and eight patients had a reduction >50g between baseline and follow-up. Of note, all patients with a marked decrease (>100g) had a mutation in ß-MHC. In contrast, no regression of hypertrophy was observed in patients with a cardiac troponin I gene mutation. Carriers of the cMYBPC genotype showed moderate responses (Table 3). In 10 study subjects, the inter- and intraobserver variability for assessment of LV mass was 6.4% (27 g) and 5.1% (22 g), respectively. Patients on candesartan showed significant increases in LV contractile (Sa) and diastolic function (Ea) and decreases in LV filling pressures (E/Ea) during follow-up (all P < 0.01). In contrast, no improvements in these parameters were observed in the placebo group. LV ejection fraction remained similar regardless of treatment assignment. Furthermore, six (50%) patients receiving candesartan showed ≥1 point decrease in New York Heart Association class compared with only one (9%) patient receiving placebo (P = 0.07). Finally, total exercise time increased only in patients on candesartan, and was associated with reduction of LV mass and improvements in LV diastolic and systolic function (Figure 2). The systolic blood pressure at peak exercise tended to be lower in the candesartan versus placebo group (166 ± 21 vs. 177 ± 18, P = 0.08).
Table 2.
Symptoms, Exercise Tolerance, and Echocardiographic Indices of LV Hypertrophy and Function at Baseline and 12-Month Follow-Up
| Candesartan group (n = 12)* |
Placebo group (n = 11) |
||||
|---|---|---|---|---|---|
| Baseline | Follow-up | Baseline | Follow-up | P (follow-up) Candesartan versus Placebo | |
| NYHA, n | |||||
| I | 4 | 8 | 4 | 4 | |
| II | 4 | 4 | 4 | 5 | 0.07 |
| III | 4 | 0 | 3 | 2 | |
| Systolic blood pressure, mmHg | 113 | 114 | 119 | 119 | NS |
| Heart rate, bpm | 65 | 66 | 66 | 67 | NS |
| Total exercise time at ergometry, s | 574 ± 151 | 751 ± 161‡ | 629 ± 149 | 603 ± 162 | 0.049 |
| Peak LV outflow tract gradient, mmHg | 7.5 ± 3.1 | 8.2 ± 5.1 | 9.2 ± 6.3 | 8.6 ± 5.8 | NS |
| LV end-diastolic diameter, mm | 46 ± 4 | 46 ± 5 | 46 ± 7 | 46 ± 8 | NS |
| LV ejection fraction, % | 69 ± 5 | 68 ± 6 | 70 ± 6 | 69 ± 4 | NS |
| Sa, cm/s | 6.1 ± 1.5 | 8.2 ± 2.3† | 6.6 ± 2.0 | 6.9 ± 2.2 | 0.04 |
| Ea, cm/s | 5.5 ± 1.8 | 8.0 ± 3.1† | 5.3 ± 1.9 | 5.9 ± 3.2 | 0.01 |
| E/Ea | 13.5 ± 3.5 | 9.3 ± 1.3† | 12.9 ± 3.9 | 12.3 ± 3.5 | <0.01 |
| Mean LV wall thickness, mm | 20.0 ± 3.6 | 16.2 ± 3.0‡ | 20.1 ± 2.5 | 20.2 ± 2.8 | 0.006 |
| LV mass, g | 407 ± 139 | 344 ± 102‡ | 451 ± 228 | 449 ± 232 | 0.04 |
E = early transmitral filling peak velocity, Ea = mitral annular velocity during early diastole, LV = left ventricular, Sa = mitral annular velocity during systole.
The 12-month follow-up data are reported in 23 patients who completed follow up.
P < 0.01,
P < 0.001 baseline versus follow-up.
Figure 1.
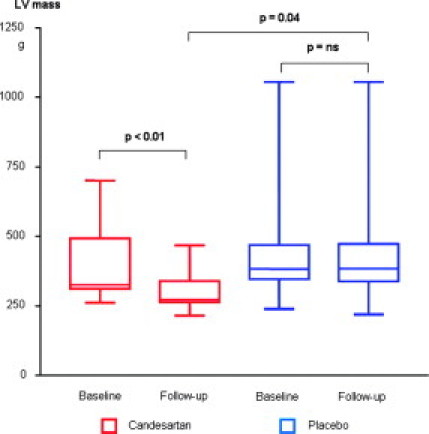
Left ventricular mass at baseline and 12-month follow-up in the candesartan (red) and placebo (blue) groups. The plots display the medians (horizontal bars), 25th and 75th percentiles (lower and upper limits of the boxes), and lowest and highest values (error bars).
Table 3.
Change in LV Mass in the Candesartan Group between Baseline and 12-Month Follow-Up According to Specific HCM-Causing Gene Mutation
| LV mass change, g (%) | |
|---|---|
| ß-MHC (n = 5) | |
| Arg403Trp | [−]94 ([−]29) |
| Arg719Trp | [−]99 ([−]25) |
| Arg403Gln | [−]145 ([−]21) |
| Ile263Thr | [−]105 ([−]20) |
| Arg249Gln | [−]121 ([−]20) |
| Average | [−]113 ([−]23) |
| cMYBPC (n = 3) | |
| Arg502Gln | [−]51 ([−]16) |
| SDSint23 | [−]50 ([−]13) |
| SASint20 | [−]41 ([−]13) |
| Average | [−]47 ([−]14) |
| Other genotyped patients (n = 4) | |
| TnI: Arg141Gln | [−]10 ([−]4) |
| TnI: Ala157Val | +10 (+4) |
| Myofilament genotype-negative | [−]51 ([−]16) |
| Myofilament genotype-negative | [−]59 ([−]13) |
| Average | [−]28 ([−]7) |
| P (ANOVA) | < 0.001 |
Abbreviations: ß-MHC = ß-myosin heavy chain, cMYBPC = cardiac myosin binding protein C, cTnI = cardiac troponin I.
Figure 2.
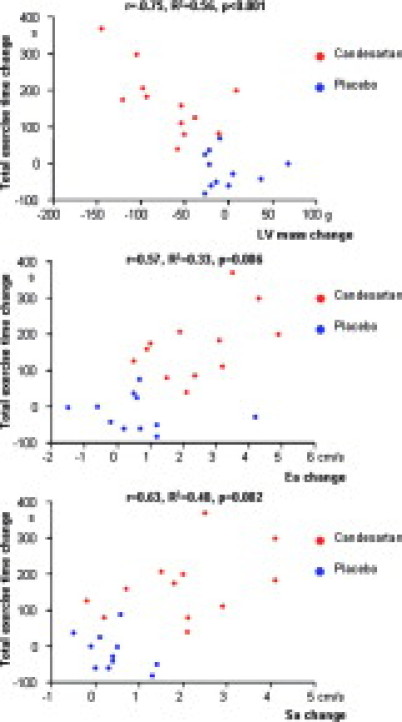
Relationship between change in LV mass (upper panel), change in peak mitral annular velocity at early diastole (Ea; middle panel) and systole (Sa; lower panel), and change in total exercise time by bicycle ergometry between baseline and 12-month follow-up. Red circles indicate the candesartan group, and blue circles indicate the placebo group.
Safety
Neither adverse events nor side effects were observed during the up-titration phase or the entire follow-up period. No patient developed LV outflow tract obstruction, malignant arrhythmias or showed an increase in serum creatinine and potassium levels. In the placebo group, one patient had an episode of atrial fibrillation that needed electrical cardioversion during follow up. The target dose of study drug (32 mg daily) was reached by eight (67%) patients in the candesartan group and nine (75%) patients in the placebo group (NS). The remaining patients received 16 mg of study drug daily. The principal reason for stopping uptitration was a subjective feeling of lightheadedness. Once titration was completed, the maintenance dose remained unchanged throughout the follow-up period.
Discussion
The present study demonstrated that use of the AT1-R antagonist candesartan in patients with non obstructive HCM, is safe. Furthermore, candesartan use was associated with significant regression of LV hypertrophy, and improvement of LV function, symptoms and exercise tolerance. Finally, the magnitude of treatment effect was dependent on specific sarcomeric protein gene mutation with the greatest response in carriers of ß-MHC gene mutations.
Role of Angiotensin II in HCM
HCM is genetic disease, which is characterized by LV hypertrophy, impaired myocardial contractile and LV diastolic function, reduced exercise tolerance and risk of sudden death.1,2 HCM is caused by mutations in genes encoding sarcomeric proteins. The phenotypic expression of HCM depends both on specific mutations and modifying factors. Angiotensin II is believed to be an important modulator of cardiac hypertrophy and even prognosis in HCM.10,11,12,13,14,15,16,17 Several studies have demonstrated an association between the ACE insertion/deletion polymorphism and the AT1-R adenine/cytosine1166 polymorphism and expression of LV hypertrophy.10,12,13,14,15,16 Carriers of ACE D (increased myocardial angiotensin II level) and AT1-R C (increased angiotensin II effect) alleles showed higher penetrance and degree of LV hypertrophy compared with patients with the ACE I and AT1-R A alleles. Only three studies reported a HCM-causing genotype with controversial results.13,14,16 Perkins et al16 showed a significant pro-LV hypertrophic effect of ACE polymorphism only in patients with the cMYBPC gene mutation, but not in carriers of the ß-MHC mutation. In contrast, Tesson et al14 observed a significant association between the ACE D allele and hypertrophy only in subjects carrying a mutation in the Arg403 codon of ß-MHC, but not for other ß-MHC or cMYBPC gene mutations. In the present study, patients in the candesartan group with either ß-MHC-HCM and cMYBPC-HCM mutations showed regression of hypertrophy, but the greatest effect was seen in the former group. The inconsistency in results may be accounted for by the heterogeneity of the underlying mutation. Several hundreds of HCM-causing mutations have been described,28,29 and one may speculate that the modulating role of angiotensin II in the development of hypertrophy is specific both with respect to the affected sarcomeric protein gene and the affected codon within that gene. Yet, we observed a treatment effect of candesartan in the two most common genetic subtypes of HCM (ß-MHC and cMYBPC).
Angiotensin II Blockade in HCM
In a mouse model of human HCM, treatment with the AT1-R antagonist losartan decreased the extent of interstitial fibrosis and collagen synthesis compared with placebo.30 In a hemodynamic study, intracoronary administration of the ACE inhibitor enalaprilat was associated with improvement of LV diastolic function and coronary blood flow as well as a decrease in LV filling pressures.31 Three studies reported effects of AT1-R antagonists in patients with non obstructive HCM.32,33,34 Kawano et al32 randomized 23 patients to valsartan (maximal dose 80 mg/d) or conventional therapy. At 12-month follow-up, valsartan decreased collagen synthesis, while no favorable effects on LV diastolic function, filling pressures and LV hypertrophy were observed. In the study by Araujo,33 losartan use was significantly associated with improvement of LV diastolic function, decrease in LV filling pressure and New York Heart Association functional class but not in LV hypertrophy at 6-month follow-up. Corroborating these results, we demonstrated significant increases in LV systolic and diastolic function and decreases in LV filling pressures, with concomitant improvements in symptoms and exercise tolerance in patients receiving candesartan. Since the Doppler-echocardiography was not performed immediately after exercise, the mechanisms of improvement in exercise tolerance by candesartan can be only speculative. Corroborating the previous results,4,35 in the present study, increase in exercise tolerance correlated with improvement in myocardial systolic and diastolic function. Regression of LV mass appeared to be one of the major mechanisms underlying this improvement. Moreover, decrease in myocardial collagen synthesis associated with AT1-R antagonists30,32 could also contribute to increased myocardial function. In the candesartan group, patients showed slightly lower systolic blood pressure at peak exercise compared with the placebo group. Blunted blood pressure response to exercise might have partly explained the positive effect of candesartan on exercise tolerance. In the study by Yamazaki et al,34 nineteen patients with non obstructive HCM were randomized to 50 mg of losartan or conventional therapy. At 1-year follow up, patients on losartan showed a trend (p 0.07) toward regression of LV mass at magnetic resonance imaging. The mean reduction of LV mass by losartan was 6.4%. In the present study, we observed a larger regression of LV hypertrophy by 15.5% in the candesartan group. The explanations for conflicting effects of AT1-R blockade on LV hypertrophy between studies may be several. It appears that the magnitude of modulating effects of angiotensin II differ in various HCM-causing mutations.14,16 It is likely that frequencies of putative mutations differ between continents. Corroborating this explanation, in the study by Yamazaki, the mean baseline LV mass was twofold lower (190 ± 55 cm3 = 199.5 ± 58 g) compared with the present study (407 ± 139 g). This strongly suggests a heterogeneous genetic substrate in patients included in these studies, which may account for differences in the extent of treatment effect on LV hypertrophy. The primary HCM-causing genetic substrate was not reported in either the Japanese32,34 or the Brazilian33 studies. Furthermore, in previous reports, the dose of study drug was lower32,34 or the follow-up was shorter33 than in the present study. This suggests that higher doses of AT1-R antagonist for a longer time may be required to decrease the magnitude of LV hypertrophy. Finally, in contrast to the present study, the open, non-placebo-controlled design of previous studies may hamper interpretation of results.32,33 Nevertheless, these findings suggest that AT1-R blockade has the potential to attenuate myocardial fibrosis and hypertrophy, two major predictors of sudden cardiac death in patients with HCM.3,36
Safety of AT1-R Antagonists in HCM
ACE inhibitors and AT1-R blockers have been discouraged in HCM, because of their vasodilating properties.1 In the present study, we did not observe any adverse events related to candesartan use and the predefined target dose was reached in the majority of patients, regardless of treatment arm assignment. These findings are consistent with the previous reports.32,33
Limitations
In the present study, the sample size both in general and for individual mutated genes was too small to draw a confident conclusion on the mutation-specific effects. The present study did not investigate specific molecular mechanisms underlying the effect of candesartan in HCM. The assessment of circulating angiotensin II, catecholamines or markers of oxidative stress could have provided more mechanistic insight since all these parameters affect both LV structure and function. Neither ACE nor AT1-R polymorphisms were assessed in the present study. Since the carriers of the ACE D or AT1-R C alleles may experience the greatest benefit from angiotensin II blockade, the information on these polymorphisms could help in selecting patients for AT1-R antagonist therapy. Thus, findings of the present study should be considered as preliminary and interpreted with caution.
The presented cohort showed higher prevalence of ß-MHC gene mutations than reported by other authors.23 These discrepant findings might be explained by the selection bias. In the present study, only patients with LV wall thickness >15 mm were included. Of note, severe LV hypertrophy is the hallmark of ß -MHC hypertrophic cardiomyopathy, while carriers of other gene mutations (cMYBPC, cardiac troponin T, or cardiac troponin I) have milder hypertrophy.
Conclusions
In a double-blind, placebo-controlled, randomized study, candesartan induced regression of LV hypertrophy, and improved LV function and exercise tolerance with no side effects in patients with non obstructive HCM. The magnitude of treatment effects of AT1-R antagonists is likely to be dependent both on specific HCM-causing mutations and specific angiotensin II system polymorphisms. These findings call for a prospective trial to assess the effects of AT1-R antagonists on clinical outcome in a large cohort of patients with non obstructive HCM.
Acknowledgements
We are indebted to Dr. Anders Ljunggren and to Martina Havlikova for their contribution to the trial.
Footnotes
Supported by research grant IGA NR 9164–3 awarded by the Czech Ministry of Health. ClinicalTrials.gov number, NCT00430833.
Placebo was provided by AstraZeneca.
References
- 1.Roberts R, Sigwart U. Current concepts of the pathogenesis and treatment of hypertrophic cardiomyopathy. Circulation. 2005;112:293–296. doi: 10.1161/01.CIR.0000146788.30724.0A. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 3.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 4.Wu WC, Bhavsar JH, Aziz GF, Sadaniantz A. An overview of stress echocardiography in the study of patients with dilated or hypertrophic cardiomyopathy. Echocardiography. 2004;21:467–475. doi: 10.1111/j.0742-2822.2004.03083.x. [DOI] [PubMed] [Google Scholar]
- 5.Lee YA, Lindpaintner K. Role of the cardiac renin-angiotensin system in hypertensive cardiac hypertrophy. Eur Heart J. 1993;(14 Suppl J):42–48. [PubMed] [Google Scholar]
- 6.Kawano H, Do YS, Kawano Y, Starnes V, Barr M, Law RE, Hsueh WA. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130–1137. doi: 10.1161/01.cir.101.10.1130. [DOI] [PubMed] [Google Scholar]
- 7.Thurmann PA, Kenedi P, Schmidt A, Harder S, Rietbrock N. Influence of the angiotensin II antagonist valsartan on left ventricular hypertrophy in patients with essential hypertension. Circulation. 1998;98:2037–2042. doi: 10.1161/01.cir.98.19.2037. [DOI] [PubMed] [Google Scholar]
- 8.Johnson DB, Foster RE, Barilla F, Blackwell GG, Roney M, Stanley AW, Jr, Kirk K, Orr RA, van der Geest RJ, Reiber JH, Dell'Italia LJ. Angiotensin-converting enzyme inhibitor therapy affects left ventricular mass in patients with ejection fraction >40% after acute myocardial infarction. J Am Coll Cardiol. 1997;29:49–54. doi: 10.1016/s0735-1097(96)00451-2. [DOI] [PubMed] [Google Scholar]
- 9.Dahlof B. Regression of left ventricular hypertrophy–are there differences between antihypertensive agents? Cardiology. 1992;81:307–315. doi: 10.1159/000175821. [DOI] [PubMed] [Google Scholar]
- 10.Lechin M, Quinones MA, Omran A, Hill R, Yu QT, Rakowski H, Wigle D, Liew CC, Sole M, Roberts R. Angiotensin-I converting enzyme genotypes and left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Circulation. 1995;92:1808–1812. doi: 10.1161/01.cir.92.7.1808. [DOI] [PubMed] [Google Scholar]
- 11.Marian AJ, Yu QT, Workman R, Greve G, Roberts R. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet. 1993;342:1085–1086. doi: 10.1016/0140-6736(93)92064-z. [DOI] [PubMed] [Google Scholar]
- 12.Osterop AP, Kofflard MJ, Sandkuijl LA, ten Cate FJ, Krams R, Schalekamp MA, Danser AH. AT1 receptor A/C1166 polymorphism contributes to cardiac hypertrophy in subjects with hypertrophic cardiomyopathy. Hypertension. 1998;32:825–830. doi: 10.1161/01.hyp.32.5.825. [DOI] [PubMed] [Google Scholar]
- 13.Ortlepp JR, Vosberg HP, Reith S, Ohme F, Mahon NG, Schroder D, Klues HG, Hanrath P, McKenna WJ. Genetic polymorphisms in the renin-angiotensin-aldosterone system associated with expression of left ventricular hypertrophy in hypertrophic cardiomyopathy: a study of five polymorphic genes in a family with a disease causing mutation in the myosin binding protein C gene. Heart. 2002;87:270–275. doi: 10.1136/heart.87.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tesson F, Dufour C, Moolman JC, Carrier L, al-Mahdawi S, Chojnowska L, Dubourg O, Soubrier E, Brink P, Komajda M, Guicheney P, Schwartz K, Feingold J. The influence of the angiotensin I converting enzyme genotype in familial hypertrophic cardiomyopathy varies with the disease gene mutation. J Mol Cell Cardiol. 1997;29:831–838. doi: 10.1006/jmcc.1996.0332. [DOI] [PubMed] [Google Scholar]
- 15.Doolan G, Nguyen L, Chung J, Ingles J, Semsarian C. Progression of left ventricular hypertrophy and the angiotensin-converting enzyme gene polymorphism in hypertrophic cardiomyopathy. Int J Cardiol. 2004;96:157–163. doi: 10.1016/j.ijcard.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Perkins MJ, Van Driest SL, Ellsworth EG, Will ML, Gersh BJ, Ommen SR, Ackerman MJ. Gene-specific modifying effects of pro-LVH polymorphisms involving the renin-angiotensin-aldosterone system among 389 unrelated patients with hypertrophic cardiomyopathy. Eur Heart J. 2005;26:2457–2462. doi: 10.1093/eurheartj/ehi438. [DOI] [PubMed] [Google Scholar]
- 17.Ogimoto A, Hamada M, Nakura J, Miki T, Hiwada K. Relation between angiotensin-converting enzyme II genotype and atrial fibrillation in Japanese patients with hypertrophic cardiomyopathy. J Hum Genet. 2002;47:184–189. doi: 10.1007/s100380200021. [DOI] [PubMed] [Google Scholar]
- 18.Maron BJ, Epstein SE. Hypertrophic cardiomyopathy: a discussion of nomenclature. Am J Cardiol. 1979;43:1242–1244. doi: 10.1016/0002-9149(79)90160-7. [DOI] [PubMed] [Google Scholar]
- 19.Gregor P. Diagnosis of hypertrophic cardiomyopathy. In: Gregor P, editor. Hypertrophic Cardiomyopathy. 1st ed. Scientia Medica; Prague: 1992. pp. 75–77. [Google Scholar]
- 20.Ho CY, Seidman CE. A contemporary approach to hypertrophic cardiomyopathy. Circulation. 2006;113:e858–e862. doi: 10.1161/CIRCULATIONAHA.105.591982. [DOI] [PubMed] [Google Scholar]
- 21.Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation. 2003;108:445–451. doi: 10.1161/01.CIR.0000080896.52003.DF. [DOI] [PubMed] [Google Scholar]
- 22.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 23.Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 24.Young JB, Dunlap ME, Pfeffer MA, Probstfield JL, Cohen-Solal A, Dietz R, Granger CB, Hradec J, Kuch J, McKelvie RS, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Held P, Solomon SD, Yusuf S, Swedberg K, Candesartan in heart failure assessment of reduction in mortality and morbidity (CHARM) investigators and committees Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection fraction trials. Circulation. 2004;110:2618–2626. doi: 10.1161/01.CIR.0000146819.43235.A9. [DOI] [PubMed] [Google Scholar]
- 25.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 27.Devereux RB, Alonso DR, Lutas EM, Gottlieb GJ, Campo E, Sachs I, Reichek N. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450–458. doi: 10.1016/0002-9149(86)90771-x. [DOI] [PubMed] [Google Scholar]
- 28.Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- 29.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104:557–567. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 30.Lim DS, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, Roberts R, Marian AJ. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyriakidis M, Triposkiadis F, Dernellis J, Androulakis AE, Mellas P, Kelepeshis GA, Gialafos JE. Effects of cardiac versus circulatory angiotensin-converting enzyme inhibition on left ventricular diastolic function and coronary blood flow in hypertrophic obstructive cardiomyopathy. Circulation. 1998;97:1342–1347. doi: 10.1161/01.cir.97.14.1342. [DOI] [PubMed] [Google Scholar]
- 32.Kawano H, Toda G, Nakamizo R, Koide Y, Seto S, Yano K. Valsartan decreases type I collagen synthesis in patients with hypertrophic cardiomyopathy. Circ J. 2005;69:1244–1248. doi: 10.1253/circj.69.1244. [DOI] [PubMed] [Google Scholar]
- 33.Araujo AQ, Arteaga E, Ianni BM, Buck PC, Rabello R, Mady C. Effect of Losartan on left ventricular diastolic function in patients with nonobstructive hypertrophic cardiomyopathy. Am J Cardiol. 2005;96:1563–1567. doi: 10.1016/j.amjcard.2005.07.065. [DOI] [PubMed] [Google Scholar]
- 34.Yamazaki T, Suzuki J, Shimamoto R, Tsuji T, Ohmoto-Sekine Y, Ohtomo K, Nagai R. A new therapeutic strategy for hypertrophic nonobstructive cardiomyopathy in humans. A randomized and prospective study with an Angiotensin II receptor blocker. Int Heart J. 2007;48:715–724. doi: 10.1536/ihj.48.715. [DOI] [PubMed] [Google Scholar]
- 35.Lele SS, Thomson HL, Seo H, Belenkie I, McKenna WJ, Frenneaux MP. Exercise capacity in hypertrophic cardiomyopathy. Role of stroke volume limitation, heart rate, and diastolic filling characteristics. Circulation. 1995;92:2886–2894. doi: 10.1161/01.cir.92.10.2886. [DOI] [PubMed] [Google Scholar]
- 36.Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
Uncited references
- 26.Penicka M, Gregor P, Krupicka J, Jira M. Tumour necrosis factor-alpha soluble receptors type I are related to symptoms and left ventricular function in hypertrophic cardiomyopathy. Can J Cardiol. 2001;17:777–784. [PubMed] [Google Scholar]