

Abstract
Here, we describe the JAK2 mutation profile in a series of approximately 20,000 blood samples from patients with clinically suspected myeloproliferative neoplasias. Using a sensitive reverse transcription-PCR and direct sequencing approach on RNA rather than DNA, we detected JAK2 mutations in exons 12–15 in approximately 20% of these patients. We identified new mutations in addition to the known V617F and exon 12 mutations, which were the most common. Most of the novel mutations are located in the pseudokinase domain and therefore are expected to relieve the autoinhibitory function of this domain on JAK2 kinase activity. Our data suggest that molecular testing of JAK2 mutations should not be restricted to the V617F and exon 12 mutations, but perhaps should extend to most of the pseudokinase domain coding region as well. Furthermore, mutation screening using RNA is highly sensitive and could replace DNA-based testing because of the relative abundance of target transcripts and the ease in detecting deletion of the entire exon.
Myeloproliferative neoplasias (MPNs) are clonal malignancies that are thought to arise from deregulated expansion of hematopoietic progenitors. MPNs are characterized by overproduction of mature, functional blood cells and a long clinical course. Disruption of protein tyrosine kinase signaling by mutations or other genetic alterations is postulated to be associated with these disorders.1 Mutant protein tyrosine kinases such as BCR-ABL and Janus kinase 2 (JAK2), derived from chromosomal translocation and gene mutations, respectively, can lead to constitutive activity in most patients with distinct MPNs.2 Among JAK2 mutations, the discovery of the JAK2 V617F allele represents a milestone in unraveling the molecular pathogenesis of MPNs. This single-point mutation is frequently present in three clinically distinct MPNs: polycythemia vera (∼95%), essential thrombocythemia (∼50%), and primary myelofibrosis (∼50%).2,3,4
The JAK2 V617F substitution, located in the pseudokinase domain of JAK2, relieves the autoinhibition of its kinase activity; the resulting constitutively active kinase augments downstream JAK2-STAT signaling pathways.2 Other JAK2 mutations in humans include translocations, point mutations, deletions, and insertions. However, the most frequent mutations are those seen in patients with JAK2 V617F-negative polycythemia vera or idiopathic erythrocytosis, the exon 12 mutations.5 Documented high-frequency JAK2 exon 12 mutations include in-frame deletions, missense, and tandem point mutations such as del/F537-K539ins/L, del/N542-E543, K539L, and H538QK539L.5,6 Whereas JAK2 V617F mutations are typically homozygous (by mitotic recombination), exon 12 mutations are often heterozygous in patients with polycythemia vera. In addition, exon 12 mutations can induce cytokine-independent hypersensitive proliferation in erythropoietin-expressing cell lines and are sufficient for the development of a polycythemia vera-like phenotype in a murine model.5
Although identification of the activating JAK2 mutations, such as V617F and exon 12 mutations, represents a major breakthrough for understanding the molecular pathology of MPN, it also raises the major question of how a single mutation can give rise to disease heterogeneity. To date, it is still unclear which mechanisms are responsible for the wide range of clinical features exemplified by the different MPN subtypes in the V617F-positive and V617F-negative populations.
To identify novel mutations with potential clinical relevance and establish a broad JAK2 gene mutation database as a roadmap, we performed mutation analysis on approximately 20,000 peripheral blood samples from patients with suspected MPN, using reverse transcription-PCR (RT-PCR) and direct DNA sequencing methods. Our findings from a large group of patients confirmed the prevalence of JAK2 V617F mutation. We also confirmed exon 12 as a target for mutations.5,6 In addition, we reported the discovery of several new mutations that have not been reported before. Point mutations were detected as the predominant mutation type in exons 13, 14, and 15, which encompass the N-terminal part of the JAK2 pseudokinase domain. Our results highlight the JAK2 pseudokinase domain as a potential hotspot for mutations and suggest that future JAK2 mutation testing should extend to all exons coding for this regulatory domain.
Materials and Methods
Patient Samples
Peripheral blood samples sent to Quest Diagnostics for JAK2 mutation testing were received daily. Five milliliters of blood in lavender (purple)-top tubes containing EDTA anticoagulant were required for analysis. Blood samples were refrigerated (not frozen) or kept at room temperature no more than 48 hours if immediate nucleic acid extraction is not possible. Over a period of 7 months, approximately 20,000 blood samples from patients with suspected MPN were collected and screened for JAK2 gene mutation, through the entire region of exons 12 though 15.
RT-PCR and Direct Sequence Analysis
Total nucleic acids were isolated from peripheral blood cells by the NucliSens extraction kit (bioMerieux Inc., Durham, NC) according to the manufacturer's instructions. First strand cDNAs were then prepared by reverse transcription of total RNAs with random primers at 55°C for 30 minutes, followed by PCR reactions using SuperSript III one-step RT-PCR system with Platinum TaqDNA polymerase (Invitrogen, Carlsbad, CA). For amplification of JAK2 exons 12 through 15, the following primer set and conditions were used: 5′-TGTAAAACGACGGCCAGTCTAAATGCTGTCCCCCAAAG-3′ (forward) and 5′-CAGGAAACAGCTATGACCCCATGCCAACTGTTTAGCAA-3′ (reverse); initial step of 2 minutes at 94°C, followed by 40 cycles of 94°C for 15 seconds, 60°C for 30 seconds, and 68°C for 1 minute, and ending with one step of 68°C for 7 minutes. The 491-bp amplified products were filter-purified by Multiscreen PCR plates (Millipore, Billerica, MA) and sequenced in both directions using the ABI Prism BigDye Terminator v3.1 Cycle Sequencing Kit with detection by an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence data were then base-called, assembled, and analyzed by ABI Prism SeqScape software (Applied Biosystems) using the JAK2 sequence (accession number NM004972) as a reference.
Results and Discussion
JAK2 V617F was the most prevalent mutation detected among the ∼20,000 patient samples tested (Table 1), in agreement with previous reports.3,4 Among cases with exon 12 mutations, the mutation hotspots F537, H538, K539 and N542, E543, D544 were also identified, with mutation types similar to those of recent reports5,6 (Table 2). The concordance with previous results indicates that our testing approach using RNA is sensitive, reliable, and accurate. In addition to these known acquired gain-of-function mutations, we also discovered novel mutations in exon 12, 13, 14, and 15. These novel mutations included T514M, N533Y, L545V, F547L in exon 12; R564L, R564Q, V567A, G571S, G571R, L579F, H587N, S591L, and F557L (with frameshift and early termination) in exon 13; H606Q, H608Y, V617I, C618R in exon 14; and L624P, I645V in exon 15 (Table 1; Figure 1A, B). The mutations detected in this study are most likely not polymorphisms since they are absent from the JAK2 mutation-negative samples, are extremely rare (frequencies <0.01%), and are not recorded in the databases of single-nucleotide polymorphisms. Some may represent an actual polymorphism, however, and may not contribute to the disease. It is noteworthy that most of the novel mutations are point (missense) mutations, except for a deletion that eliminated the entire exon 14 and the one nucleotide deletion at F557. The deletion of exon 14 is interesting and most likely due to alternative splicing (exon skipping). This mutation was not detected when genomic DNA was tested using primers to detect mutations in the V617 locus, which is the approach used by most investigators for testing mutations in the JAK2 gene. The deletion of one nucleotide (T) in exon 13 leads to a F557L (TTT to TTA) mutation along with a frameshift and early termination at residue 567 (FTKIFKGVRE to LQRFLKAYEEK-ter). This mutation is particularly interesting because it causes significant changes to the JAK2 protein and confirms the importance of testing for exon 13 mutations.
Table 1.
Mutation Analysis of JAK2 Gene Exons 12 through 15 in ∼20,000 Samples from Patients with Suspected Myeloproliferative Neoplasias
| Exon | Mutation | DNA alteration | Codon change | Number of cases | Pattern on sequencing |
|---|---|---|---|---|---|
| 12 | Silent | T1593C | H531 | 1 | Heterozygous |
| 12 | Duplication | 1606-1641dupl36 | dupl/V536-F547 | 2 | Heterozygous |
| 12 | Deletion | 1612-1614del3 | del H538 | 1 | Heterozygous |
| 12 | Deletion | 1612-1617del6 | del/H538-K539 | 1 | Heterozygous |
| 12 | Deletion | 1624-1629del6 | del/N542-E543 | 5 | Heterozygous |
| 12 | Deletion | 1627-1632del6 | del/E543-D544 | 3 | Heterozygous |
| Indels | 1609-1617del9 | del/F537-K539 | 1 | ||
| 12 | ins/K | Heterozygous | |||
| Indels | 1609-1617del9 | del/F537-K539 | 2 | ||
| 12 | ins/L | Heterozygous | |||
| Indels | 1612-1617del6 | del/H538-K539 | 4 | ||
| 12 | ins/L | Heterozygous | |||
| Indels | 1621-1629del9 | del/R541-E543 | 1 | ||
| 12 | ins/K | Heterozygous | |||
| Indels | 1618-1632del15 | del/I540-D544 | 1 | ||
| 12 | ins/MK | Heterozygous | |||
| Indels | 1624-1632del9 | del/N542-D544 | 1 | ||
| 12 | ins/N | Heterozygous | |||
| 12 | Missense | C1541T | T514 M | 2 | Heterozygous |
| 12 | Missense | T1598C | N533Y | 1 | Heterozygous |
| 12 | Missense | A1615C, A1616T | K539L | 2 | Heterozygous |
| Missense | C1614A, A1616T | H538Q | 1 | ||
| 12 | K539L | Heterozygous | |||
| Missense | A1615T, T1633G | K539L | 1 | ||
| 12 | L545V | Heterozygous | |||
| 12 | Missense | T1639C | F547L | 1 | Heterozygous |
| 13 | Silent | C1686T | G562 | 2 | Heterozygous |
| 13 | Silent | C1710T | Y570 | 4 | Heterozygous |
| 13 | Silent | T1668C | F556 | 1 | Heterozygous |
| 13 | Frameshift | C1671del | F557L# | 1 | Heterozygous |
| 13 | Missense | G1691T | R564L | 3 | Heterozygous |
| 13 | Missense | G1691A | R564Q | 2 | Heterozygous |
| 13 | Missense | T1700C | V567A | 1 | Heterozygous |
| 13 | Missense | G1711A | G571S | 3 | Heterozygous |
| 13 | Missense | G1711C | G571R | 1 | Heterozygous |
| 13 | Missense | C1735T | L579F | 1 | Heterozygous |
| 13 | Missense | C1759A | H587N | 1 | Heterozygous |
| 13 | Missense | C1772T | S591L | 1 | Heterozygous |
| 14 | Deletion | 1777-1864del88 | S593-N622 | 2 | Heterozygous |
| 14 | Missense | C1818A | H606Q | 1 | Heterozygous |
| 14 | Missense | C1824T | H608Y | 1 | Heterozygous |
| 14 | Missense | G1849T | V617F | ∼4280 | Hetero/Homo |
| 14 | Missense | G1849A | V617I | 1 | Heterozygous |
| 14 | Missense | G1849T, T1852C | V617F | 2 | Hetero/Homo |
| C618R | |||||
| 14 | Missense | T1852C | C618R | 1 | Heterozygous |
| 15 | Missense | T1871C | L624P | 1 | Heterozygous |
| 15 | Missense | A1933G | I645V | 1 | Heterozygous |
Deletion of one nucleotide (T) resulting in F557L mutation and frameshift leading to insertion of 10 residues and premature termination at residue 567.
Table 2.
Comparison of exon 12 mutations in the JAK2 gene discovered in this study (RNA-based approaches) with previously reported somatic mutations (genomic DNA-based methods)
| Exon | Mutation | Identified in this study (RNA-based) | Reported mutations (DNA-based) | Reference |
|---|---|---|---|---|
| 12 | Duplication | dupl./V536-F547 | dupl/V536-I546 | 6 |
| − | dupl/F537-I546 | |||
| F547L | 6 | |||
| 12 | Deletion | delH538 | − | |
| 12 | Deletion | del/H538-K539 | − | |
| 12 | Deletion | del/N542-E543 | del/N542-E543 | 5,6 |
| 12 | Deletion | del/E543-D544 | del/E543-D544 | 6 |
| 12 | Indels | del/F537-K539 | del/F537-K539 | 5,6 |
| ins/L | Ins/L | |||
| 12 | Indels | del/F537-K539 | − | |
| ins/K | ||||
| 12 | Indels | del/H538-K539 | del/H538-K539 | 6 |
| ins/L | Ins/L | |||
| 12 | Indels | del/R541-E543 | del/R541-E543 | 6 |
| ins/K | ins/K | |||
| 12 | Indels | del/N542-D544 | − | 6 |
| ins/N | ||||
| 12 | Indels | del/I540-D544 | del/I540-E543 | 6 |
| ins/MK | ins/MK | |||
| 12 | Missense | T514 M | − | |
| 12 | Missense | N533Y | − | |
| 12 | Missense | K539L | K539L | 5,6 |
| 12 | Missense | H538Q K539L | H538Q | 5,6 |
| K539L | ||||
| 12 | Missense | K539L L545V | − | |
| 12 | Missense | F547L | − |
Figure 1.
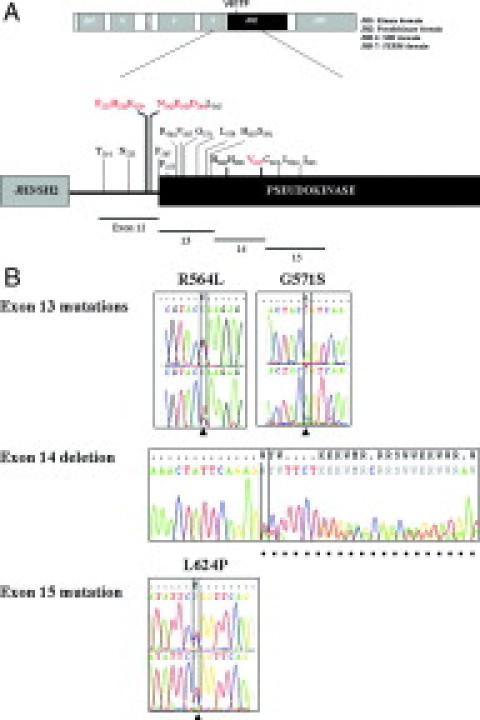
Schematic diagram of the JAK2 gene structure. A: JAK2 protein contains JAK homology domains 1 through 7 (JH1-JH7) with the JH2 pseudokinase domain highlighted in black. The corresponding exon regions are also shown. All mutated residues in exons 12–15 discovered in this report are indicated with mutation hotspots labeled in red. B: Representative sequencing chromatograms of some novel mutations. R564L and G571S in exon 13, the deletion of entire exon 14, and L624P in exon 15 are shown. Positions of mutant nucleotides are indicated by arrowheads and deleted nucleotides by dots.
All of the novel mutations we found are located either near or at the N-terminal part of the JAK2 pseudokinase domain (JH2) (Figure 1A). This domain negatively regulates the activity of JAK2, raising the possibility that at least some of these mutations could be activating mutations. A considerable portion of activating mutations in tyrosine kinases associated with human cancers are loss-of-function alleles affecting the autoinhibitory domains. For example, point mutations and deletions that result in constitutive tyrosine kinase activation have been identified in FLT3,7 c-KIT,8 and epidermal growth factor receptor.9
Clearly, functional studies are needed to demonstrate the role of these mutations in deregulating JAK2 activity and their contribution to leukemogenesis. It has been suggested from homology modeling approaches that residues 537 through 543 (mutation hotspots in exon 12) lie within a loop region bridging the SH2 and JH2 domains of JAK2. In the predicted model, positive polar or hydrophobic interactions between D407-K655, S411-E653, K415-E685, and F408-H608 from the SH2 and JH2 domains, respectively, are closely packed in the predicted interface that further supported the JH2-JH1 interaction.10 Therefore, mutations in this loop area may disrupt the JH2-JH1 interaction, leading to constitutive kinase activation. For example, del/F537-K539ins/L, del/N542-E543, H538QK539L, and K539L all display increased JAK2 activation, cytokine-independent hypersensitive proliferation, and in the case of K539L, a myeloproliferative phenotype.5 Each of these four known somatic mutations of exon 12 were identified in our mutation profiling, with a relatively high frequency for del/N542-E543, del/H538-K539ins/L, and K539L (Table 1). We also discovered T514M, a novel point mutation in exon 12, which presumably resides in the activation loop region.
In exon 13, the relatively high frequency of mutations at arginine 564 (R564L, R564Q) and glycine 571 (G571S, G571R) is of special interest. The point mutation at R564 has not been reported in the literature, and its impact on the JAK2 kinase activity is currently unknown. The residue adjacent to G571, tyrosine 570 (Y570), is one of the most important tyrosine phosphorylation sites (of all 49 tyrosine residues) in JAK2; autophosphorylation of Y570 plays an important role in the mechanism that down-regulates JAK2 kinase activity.11 Thus, the G571 mutation may cause a conformational change such that the adjacent Y570 is no longer accessible and phosphorylated, resulting in a constitutively active kinase.
We were surprised to discover an unusual deletion involving the entire 30-residue region comprising exon 14 (containing V617), with direct linking of exon 13 to 15. This was most likely generated by alternative splicing via exon skipping. Ours is the first report to document such a deletion, which would have been missed if DNA rather than RNA was used for testing. Another exon 14 mutation worthy of mention is V617I, which, like V617F, was shown to induce cytokine independence and constitutive downstream signaling.12 Mutation at cysteine 618 (C618R) also occurs. We also detected two novel point mutations in exon 15, L624P and I645V.
Because of the wide spectrum of mutations across the pseudokinase domain, we recommend that JAK2 mutation testing in patients with MPNs cover all exons in this region. Further, by using an RNA approach, the entire region involving exons 12 to15 can be easily and accurately sequenced and tested. While sequencing is not as sensitive as other methods (allele-specific/PCR-based), testing plasma as compared with cells increases sensitivity significantly.13 We believe that using plasma makes up for the lower sensitivity of sequencing. More importantly, when the level of the JAK2 mutant allele is low, the risk of false positive is very high when allele-specific/PCR-based approach is used. In contrast, this risk is minimal or non-existent when bidirectional sequencing is used. In addition, the current clinical practice recommends investigating JAK2 for additional mutations if V617 is not mutated. Therefore, in our opinion, testing by sequencing the plasma RNA, which allows us to encompass the entire pseudokinase domain is very valid approach. However, for this approach to be practical, the laboratory must be experienced and diverse enough in sequencing. Finally, establishment of a JAK2 mutation database will likely identify other potential disease alleles, and permit a new classification of these diseases, and hopefully, help the development of novel therapeutic approaches.
References
- 1.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 2.Morgan KJ, Gilliland DG. A role for JAK2 mutations in myeloproliferative diseases. Ann Rev Med. 2008;59:213–222. doi: 10.1146/annurev.med.59.061506.154159. [DOI] [PubMed] [Google Scholar]
- 3.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 4.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 5.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, Skoda R, Cazzola M. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111:1686–1689. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 7.Reindl C, Bagrintseva K, Vempati S, Schnittger S, Ellwart JW, Wenig K, Hopfner KP, Hiddemann W, Spiekermann K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood. 2006;107:3700–3707. doi: 10.1182/blood-2005-06-2596. [DOI] [PubMed] [Google Scholar]
- 8.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet. 1996;12:312–314. doi: 10.1038/ng0396-312. [DOI] [PubMed] [Google Scholar]
- 9.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 10.Giordanetto F, Kroemer RT. Prediction of the structure of human Janus kinase 2 (JAK2) comprising JAK homology domains 1 through 7. Protein Eng Des Sel. 2002;15:727–737. doi: 10.1093/protein/15.9.727. [DOI] [PubMed] [Google Scholar]
- 11.Feener EP, Rosario F, Dunn SL, Stancheva Z, Myers MG., Jr Tyrosine phosphorylation of Jak2 in the JH2 domain inhibits cytokine signaling. Mol Cell Biol. 2004;24:4968–4978. doi: 10.1128/MCB.24.11.4968-4978.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dusa A, Staerk J, Elliot J, Pecquet C, Poirel HA, Johnston JA, Constantinescu SN. Substitution of pseudokinase domain residue Val-617 by large non-polar amino acids causes activation of JAK2. J Biol Chem. 2008;283:12941–12948. doi: 10.1074/jbc.M709302200. [DOI] [PubMed] [Google Scholar]
- 13.Ma W, Kantarjian H, Zhang X, Sun W, Buller AM, Jilani I, Schwartz JG, Giles F, Albitar M. Higher detection rate of JAK2 mutation using plasma. Blood. 2008;111:3906–3907. doi: 10.1182/blood-2008-02-139188. [DOI] [PubMed] [Google Scholar]