

Abstract
Several different primer-probe chemistries have been produced commercially for real-time PCR detection and quantification of cytomegalovirus, but there are few studies evaluating their relative performance. We assessed three such commercial reagents with respect to analytical and clinical operating characteristics. The samples included 149 clinical whole blood specimens that were de-identified and assayed in parallel with all primer–probe systems. Individual methods used TaqMan, dual fluorescence resonance energy transfer hybridization probes, and labeled primer chemistries. Method comparability was determined both qualitatively, based on pair-wise assessment of concordance, and quantitatively, based on pair-wise linear regression analysis. Analytical sensitivity and the lower end of the linear dynamic range reached 10 target copies per reaction for the TaqMan and labeled primer systems and 100 target copies per reaction for the dual fluorescence resonance energy transfer probe system. Quantitative linearity reached an upper limit of 105 copies per reaction for all methods. No assay cross-reactivity was seen with other common viral pathogens (100% analytical specificity). Pair-wise analysis of qualitative results from clinical samples showed no significant differences in sensitivity between the three sets of reagents, and linear regression analysis indicated that the quantitative values achieved were comparable in all positive specimens. The findings demonstrate that similar analytical and clinical performance characteristics can be demonstrated for quantitative detection of cytomegalovirus in clinical whole blood extracts using a wide variety of real-time PCR chemistries.
The detection of cytomegalovirus (CMV) has become increasingly important in the care of immunocompromised patients.1,2 Over time, molecular methods have been implemented as a primary means of quantitatively detecting this and other systemic viruses, particularly in peripheral blood specimens.3 Roughly in parallel with the increased use of such methods, molecular techniques have also evolved, with end-point PCR methods now being supplanted by real-time technologies. Real-time PCR can offer decreased turnaround time, improved ease of use and result interpretation, and an improved ability to quantify viral targets.4,5 Several commercial reagents (primarily analyte specific reagents [ASRs]) have recently been produced. While many of these rely on fluorescence resonance energy transfer (FRET) to produce real-time detection,6 several primer-probe chemistries have been used.5
Various real-time chemistries have been used to target CMV,4,7,8,9 including TaqMan, dual hybridization probes, and labeled primer systems. All of these FRET systems rely on either the production or quenching of fluorescence signal; all but the labeled primer chemistries require an initial PCR step with unlabeled primers followed by a separate probe hybridization step to produce signal. TaqMan probes10 consist of a single oligonucleotide with a labeled reporter fluor on the 5′ end and a labeled quencher on the 3′end. Signal production in this system relies on 5′ exonuclease activity of Taq polymerase causing a diminution of signal quenching in parallel with amplicon production. Dual FRET probes produce signal as they anneal adjacent to one another on amplicons.11 Fluorophores placed on adjacent probes (one at the 3′end and the other at the 5′ end) interact when hybridized to amplified target to produce a FRET signal. Alternatively, labeled primer-based reporter systems can be used.12 Such systems also incorporate FRET chemistries, usually with a reporter-quencher fluorophore pair on one primer. When amplification occurs, the fluorophores are physically separated, quenching is relieved, and signal is produced. One variation on this method has recently been marketed as the MultiCode-RTx system (Eragen Biosciences, Inc., Madison, WI).12,13,14 The latter system is based on the use of two modified nucleotides, 2′-deoxy-5methylisocytidine and 2′-deoxy-isoguanosine (diC and diG), and two primers, one labeled with fluorescent reporter adjacent to diC and the other unlabeled. The quencher molecule is covalently attached to diGTP and incorporated opposite the diCTP, quenching the fluorescent dye accordingly.15 As target accumulates, fluorescence decreases.
Published comparative data between FRET-based methods is limited. Most studies have focused primarily on endpoint methods such as Southern blot or PCR-enzyme-linked immunosorbent assay.16,17 More recent publications have examined TaqMan methods; either compared to endpoint methods,18,19 or to one-another.4,18 The latter quantitative real-time PCR assays have been shown to provide accurate and reproducible results. While prior studies have primarily focused on TaqMan chemistries, the availability of other FRET-based methods offers an increasingly complex choice for clinical laboratories. Here, three different commercially produced ASRs were examined, including MultiCode-RTx CMV PCR, artus CMV TM PCR (QIAGEN, Valencia, CA), and CMV UL54 Primer/Hybridization Probes (Roche Diagnostics, Indianapolis, IN), each based on a different method of FRET signal production.
Materials and Methods
Clinical Samples and Control Viral Strains
Samples included 149 clinical whole blood nucleic acid extracts, remaining after routine clinical testing for CMV DNA by quantitative PCR, using a “conventional” endpoint detection PCR assay (data not shown). Samples were selected based on the results of that assay to assure a mix of both positive and negative samples. These samples were subsequently de-identified and only results of the three new methods used for comparative purposes. Whole blood specimens were anticoagulated with EDTA and DNA extracted using EZ1 or M48 instrumentation with the EZ1 tissue extraction kit (QIAGEN, Valencia, CA). 200 μl of each sample was processed, DNA eluted in a final volume of 200 μl of buffer, and stored at −70°C for 1 to 3 years (July 2004-September 2006). All samples were de-identified before use in this study. Approval for the use of this material was obtained from the St. Jude Children's Research Hospital Institutional Review Board. Quantified human CMV DNA (1.7 × 104 copies/μl), electron-microscope counted whole CMV particles (2.3 × 1010 viral particles/ml), and other control viruses including EBV, HHV6, HHV8, Adv5, HSV1, SV40, and VZV1/2 (used for testing assay specificity) were purchased from Advanced Biotechnologies, Inc (Columbia, MD).
CMV PCR Methods
TaqMan Method
To create a working master mix, 12.5 μl CMV TM Master (containing primers to a 105-bp region of the CMV major immediate early gene and packaged separately as artus CMV TM PCR ASR, QIAGEN, Valencia, CA), 2.5 μl CMV LC/RG/TM MG-Sol, and 1 μl CMV LC/RG/TM internal control (Supplement to artus CMV TM PCR) were combined. Ten μl of sample were added to 15 μl working master mix in a chilled 96-well optical plate, sealed with an optical adhesive film, briefly centrifuged, and amplified using the 7500 Real-Time PCR system (Applied Biosystems, Inc, Foster City, CA). Cycling parameters were 95°C for 10 minutes, 45 cycles of 95°C for 15 seconds, and 55°C for 1 minute. Results were analyzed using 7500 System Sequence Detection Software version 1.2.3. The amplification threshold was set at 25,000 ΔRn, corresponding to the lower end of the geometric phase of amplification. The formula used for calculating the amplification efficiency was: efficiency = 10 (−1/slope) − 1.
Dual Probe Method
Master mix included 2.0 μl 10× PCR reaction mix (LightCycler FastStart DNA Master HybProbe kit, Roche Diagnostics, Indianapolis, IN), 2.0 μl primer and probe mix (CMV UL54 Primer/Hybridization Probes, Roche), 0.4 μl internal control (recovery template, 5 × 102 copies/μl, LightCycler CMV UL54 Template Set, Roche Diagnostics), and 5.6 μl nuclease-free water. Ten μl of CMV master mix and 10 μl of sample were added to each reaction tube, centrifuged, and amplified (LightCycler 2.0, Roche) with the following parameters: denaturation at 95°C for 10 minutes; amplification at 95°C for 10 seconds, 55°C for 15 seconds, 72°C for 15 seconds; melt curve at 95°C for 0 seconds, 40°C for 1 minute, 85°C for 0 seconds, and cooling at 40°C for 10 minutes. LightCycler software version 4.05 was used to analyze results. Crossing point (threshold) of each sample was set automatically by the system software, followed by absolute quantification analysis and calculation of amplification efficiency.
Labeled Primer Method
Master mix included 0.5 μl 50× Titanium taq (Clontech Lab, Inc. Mountain View, CA), 1 μl 25× Primer mix (purchased pre-mixed as MultiCode-RTx CMV PCR and targeting the UL54 DNA Polymerase gene), 5 μl 5× ISOlution, 1 μl 25× DNA UR (internal control), and 7.5 μl nuclease-free water (packaged together as MultiCode-RTx System DNA Reagent Set). All reagents (unless otherwise specified) were manufactured by Eragen Biosciences, Madison, WI. Ten μl of sample were added to 15 μl master mix in a chilled 96-well optical plate, sealed with an optical adhesive film, briefly centrifuged, and amplified using the 7500 Real-Time PCR system with parameters of 95°C for 2 minutes; 50 cycles of 95°C for 15 seconds, 58°C for 10 seconds, 72°C for 35 seconds; and 1 cycle of 95°C for 15 seconds, 60°C for 1 minute, 95°C for 15 seconds. Results were analyzed using MultiCode-RTx Analysis Software version 1.5.4.8 (EraGen Biosciences). Threshold of amplification was automatically identified and efficiency of amplification was automatically calculated by the analysis software.
Research Design and Data Analysis
Analytical Evaluation
A CMV DNA concentration gradient from 1 to 1 × 105 copies per reaction (corresponding to 1 × 102 to 1 × 107copies/ml) was assayed to evaluate analytic sensitivity and linear dynamic range of each method. Conversion from copies per reaction to copies per ml was based on the volume of the original extract and sample used in the reaction, resulting in a dilution factor of 100. This can be seen in the following formula:
The specificity of each system was tested against control strains of other human viruses including HHV6, HHV8, VZV1/2, HSV1, Adv5, EBV, and SV40. Commercially prepared and quantified viral DNA solutions were used for each test, with a concentration of 5 × 103 copies per reaction.
Clinical Evaluation
The same 10-fold serial dilution series used to establish linearity was subsequently assayed in duplicate in parallel with each clinical sample run and used as a common quantitative calibrator for all three amplification methods. A linear regression curve was constructed based on these results and used to determine quantitative values for patient samples. A run-to-run comparison was performed including 74 clinical sample extracts assayed twice with each ASR method. The remaining 75 clinical sample extracts were tested in duplicate (single run) for an intrarun comparison of sensitivity. The first of two results determined for each sample was used for qualitative and quantitative comparisons of the three methods.
Statistics
Method performance using the three reagents was compared in a pairwise manner, including data from 149 clinical samples tested by all three systems. Concordance was examined using a cutoff of 100 with McNemar's test20 and regression lines were fit between any two assays and any two replicates. The cutoff value of 100 was based on the theoretical limit of the PCR reactions, corresponding to a lower limit of one copy per reaction.
Results
Analytical Performance
Analytical sensitivity and the lower limit of linearity reached 10 copies/reaction (103 copies/ml, if no CMV DNA were lost during extraction) for the TaqMan and Labeled Primer systems and 100 copies/reaction for the Dual Probe system (104 copies/ml). Sensitivity for each system was determined after completion of a minimum of 14 individual determinations at each target concentration and defined by detection of target 95% of the time at a given concentration. Linearity extended to 105 copies/reaction (107 copies/ml of sample) for all three methods (Figure 1). The amplification efficiencies for all three methods reached greater than 95% (99.25%, 95.81%, and 98.20% for TaqMan method, Labeled Primer method, and Dual Probe method, respectively). No cross-reactivity was seen in any assay when control strains of HHV6, HHV8, VZV1/2, HSV1, Adv5, EBV, and SV40 were tested (100% analytical specificity).
Figure 1.

Ten-fold serial dilutions of CMV DNA from 10 to 1 × 105 copies/reaction demonstrating comparative linearity of optimized TaqMan, Labeled Primer, and Dual Probe methods.
Clinical Performance
Pairwise analysis of all three methods showed comparable qualitative results, suggesting no significant difference in sensitivities (Tables 1, A–C, all P values >1.00). PCR inhibition, based on absence of internal control amplification in both replicates of negative samples, was noted in a total of five specimens (one in dual probe, one in Taqman, and three in labeled primer). All negative samples without amplified internal control were excluded from comparative analyses. When detected in negative samples, the internal controls generated consistent results, with cycle threshold coefficients of variation of 2 to 7%. Linear regression analysis with both values above the cutoff produced intercepts and slopes approximating 0 and 1 in all cases (Figure 2, A–C), indicating general agreement between quantitative results achieved by the different systems. Concordance was variable, with an R2 = 0.82 when results from TaqMan and Labeled Primer methods were compared, but with lower R2 values seen for the other assay pairs (R2 = 0.55 and R2 = 0.60 Figure 2, A–C). Although not statistically significant, a slight shift in regression lines indicated that the Dual Probe assay tended to produce somewhat larger viral load results than did the other techniques. This shift in quantitative results is also seen in discrepant values (Table 1, A–C), which tended to be higher for samples detected only by the Dual Probe method.
Table 1.
Qualitative Comparison of Methods
| A. Concordance between TaqMan and Labeled Primer Methods*# | |||
|---|---|---|---|
| Labeled primer − | Labeled primer + | Total | |
| TaqMan − | 99 | 5 (130, 170, 790, 120, 240) | 104 |
| TaqMan+ | 5 (108, 227, 197, 139, 322) | 33 | 38 |
| Total | 104 | 38 | 142 |
| B. Concordance between Dual Probe and Labeled Primer*# | |||
|---|---|---|---|
| Labeled primer − | Labeled primer + | Total | |
| Dual probe − | 100 | 6 (130, 150, 150, 180, 120, 240) | 106 |
| Dual probe + | 5 (9050, 14000, 155, 30100, 2840) | 32 | 37 |
| Total | 105 | 38 | 143 |
| C. Concordance between Dual Probe and TaqMan*# | |||
|---|---|---|---|
| TaqMan − | TaqMan + | Total | |
| Dual probe − | 104 | 6 (119, 295, 227, 123, 197, 139) | 110 |
| Dual probe + | 5 (14000, 155, 22100, 36600, 30100) | 32 | 37 |
| Total | 109 | 38 | 147 |
McNemar's Test for Equal Sensitivity, P = 1.00.
Positive values of discrepant pairs listed parenthetically.
Figure 2.
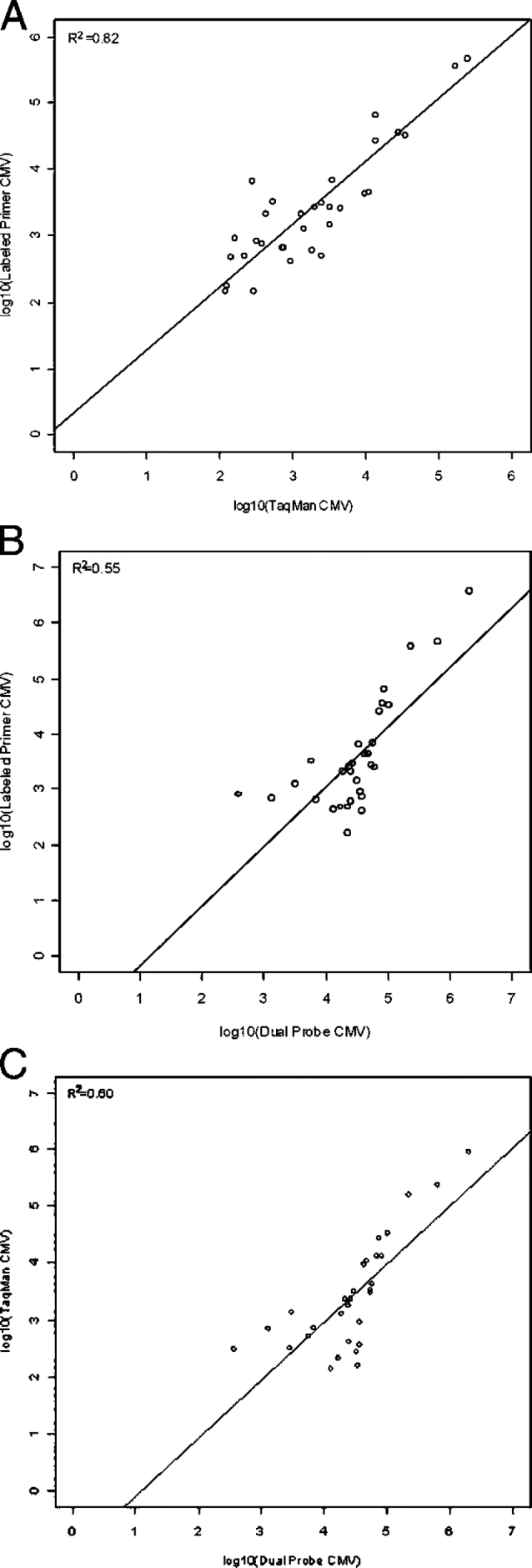
A: Comparison of quantitative results between Labeled Primer and TaqMan methods. B: Comparison of quantitative results between Labeled Primer and Dual Probe methods. C: Comparison of quantitative results between TaqMan and Dual Probe methods.
Comparison of replicate assay results demonstrated excellent between and within run reproducibility without significant discordance in TaqMan and Labeled Primer systems (within run R2 = 0.97, between run R2 = 0.73 and 0.95 respectively). The Dual Probe assay was less precise (within run R2 = 0.4, between run R2 = 0.45).
Technical Considerations
All three assays were easy to use and reagents were conveniently packaged. PCR set up and run time for the TaqMan and Labeled Primer assays were similar. The Dual Probe assay set-up and run time was considerably shorter; however, maximum run capacity was approximately one-third that of the other assays. Analysis using the 7500 System Sequence Detection Software and the LightCycler Software was straightforward and took a short amount of time to complete; the MultiCode-RTx Analysis Software required exporting data from the 7500 system slightly prolonging the time to completion (see Table 2).
Table 2.
Comparison of Assay Performance Times
| Time (minutes) |
|||||
|---|---|---|---|---|---|
| Assay | # of Samples assayed | PCR set-up | PCR run | Analysis | Time to completion (minutes) |
| TaqMan | 94 | 90 | 120 | 15 | 225 |
| Dual probe | 32 | 45 | 70 | 15 | 130 |
| Labeled primer | 94 | 90 | 120 | 25 | 235 |
Discussion
As real-time PCR becomes the primary method in use for quantitative detection of blood-borne viruses, an increasing variety of FRET-based chemistries have become available. The findings above suggest that three of these chemistries, TaqMan, Dual Probe, and Labeled Primer can provide comparable results when testing whole blood nucleic acid extracts for CMV. Sensitivity and linear dynamic range extended somewhat lower with the Labeled Primer and TaqMan methods than with the Dual Probe chemistry, although it is uncertain if this difference was a clinically significant one. The latter point appears borne out by results achieved during testing of patient samples, as qualitative results were without significant difference between the three methods. Viral load results also correlated among all three assay pairs, with the tightest correlation seen between the Labeled Primer and TaqMan assays. Minor differences between the methods were seen related to quantitative reproducibility, with the Labeled Primer and TaqMan-based assays showing a higher degree of correlation between replicates than did the Dual Probe test. Again, the clinical significance of this variability, which was seen primarily in samples with lower viral loads (less than 500 copies/ml), is uncertain.
The findings here are generally consistent with those of previous investigators, who have shown that the development of real-time PCR chemistries provides a practical option for CMV testing that compares well with end-point methods.4,7,19,21 Those studies focused mainly on TaqMan chemistry-based techniques, describing consistent performance characteristics appropriate for clinical use. The TaqMan assay used here resulted in a linear range and limit of detection (1 × 103 to 1 × 107copies/ml) similar to that of these earlier studies.4,8,22 Likewise, other investigators have examined the use of various real-time chemistries for the quantitative detection of other blood-borne viruses.23,24,25,26 Most of these have involved the use of TaqMan or Dual Probe methods, and no direct comparison of all three chemistries discussed here has been previously published.
Just as the use of real-time PCR has been applied to a variety of pathogens, the clinical implications of comparing different FRET chemistries finds significance broadly, as laboratories attempt to find the optimal method in their respective individual practice settings. The data presented here suggest that such decisions may in some cases be made primarily based on factors such as ease-of-use, cost-effectiveness, throughput, and instrumentation, as analytical performance characteristics may appear comparable between some chemistries or reagents. All three tests included in this study were subjectively easy to perform. There was variability in packaging and number of reagents required. Hands-on time for a full PCR run was similar for both the TaqMan and Labeled Primer methods but shorter for the Dual Probe method due to the 32 tube capacity of the LightCycler. Throughput was primarily affected by the platform used, with microtiter plate-based instrumentation having a longer run-time than the LightCycler, but offering advantages in turnaround time and time spent per specimen when large numbers of samples are anticipated. This information points to the importance of considering the needs of a given institution or laboratory when selecting among commercially available reagents or methods.
Although this study suggests that performance may sometimes not be the primary differentiating factor when comparing methods, this statement must be taken in the context described. Only three chemistries were evaluated, each using only a single procedure. Certainly, variability in performance may be seen depending on whether different chemistries, reagents, target sites, or cycling conditions are used.27,28 Further differences in assay performance may also become apparent when larger numbers of samples are tested, if a different sample type is tested,22,25,29,30 or if another virus is targeted. The blood compartment used for CMV testing has varied widely in the literature, depending on the patient population at hand and historical clinical practice. Whole blood was used in this study based on current accepted practice at our center; determination of assay performance with other specimen types would certainly require further evaluation. Other factors that may play into method performance, accuracy, and precision include steps of specimen preparation and quantitative calibration materials31,32 and methods. The latter factors were performed in common between all three methods in this study, removing them as sources of variability. However, these issues must also be thoroughly assessed before deciding which method to introduce in a given laboratory setting.
Although such issues will continue to be the subject of ongoing study, the results presented here demonstrated that any one of several commercial reagents now on the market may be useful for the routine clinical detection and quantification of CMV in peripheral blood specimens. The growing availability and variety of such high quality reagents offers increasing choices for the clinical microbiology laboratory, and with those choices, an increasing ability to tailor assay selection to individual patient care settings, institutional, and laboratory needs.
Acknowledgements
We are grateful to Deo Kumar Srivastava (Department of Biostatistics, St. Jude Children's Research Hospital) for assistance with statistical analysis.
Footnotes
Supported in part by the American Lebanese Syrian Associated Charities (ALSAC).
R.T.H. has received an honorarium from Eragen Biosciences for a previous speaking engagement.
Reagents were supplied by QIAGEN, Roche Diagnostics, and EraGen Biosciences, Inc.
References
- 1.Razonable RR, Emery VC. Management of CMV infection and disease in transplant patients. Herpes. 2004;11:77–86. [PubMed] [Google Scholar]
- 2.Baldanti F, Lilleri D, Gerna G. Monitoring human cytomegalovirus infection in transplant recipients. J Clin Virol. 2008;41:237–241. doi: 10.1016/j.jcv.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Vernet G. Molecular diagnostics in virology. J Clin Virol. 2004;31:239–247. doi: 10.1016/j.jcv.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gault E, Michel Y, Dehée A, Belabani C, Nicolas JC, Garbarg-Chenon A. Quantification of human cytomegalovirus DNA by real-time PCR. J Clin Microbiol. 2001;39:772–775. doi: 10.1128/JCM.39.2.772-775.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Res. 2002;30:1292–1305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen X, Zehnbauer B, Gnirke A, Kwok PY. Fluorescence energy transfer detection as a homogeneous DNA diagnostic method. Proc Natl Acad Sci USA. 1997;94:10756–10761. doi: 10.1073/pnas.94.20.10756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griscelli F, Barrois M, Chauvin S, Lastere S, Bellet D, Bourhis JH. Quantification of human cytomegalovirus DNA in bone marrow transplant recipients by real-time PCR. J Clin Microbiol. 2001;39:4362–4369. doi: 10.1128/JCM.39.12.4362-4369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nitsche A, Streuer N, Schmidt CA, Landt O, Ellerbrok H, Pauli G, Siegert W. Detection of human cytomegalovirus DNA by real-time quantitative PCR. J Clin Microbiol. 2000;38:2734–2737. doi: 10.1128/jcm.38.7.2734-2737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernando S, Folgueira L, Lumbreras C, San JR, Maldonado S, Prieto C, Babiano MJ, Delgado J, Andres A, Moreno E, Aguado JM, Otero JR. Comparison of cytomegalovirus viral load measure by real-time PCR with pp65 antigenemia for the diagnosis of cytomegalovirus disease in solid organ transplant patients. Transplant Proc. 2005;37:4094–4096. doi: 10.1016/j.transproceed.2005.10.087. [DOI] [PubMed] [Google Scholar]
- 10.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 11.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 12.Moser MJ, Ruckstuhl M, Larsen CA, Swearingen AJ, Kozlowski M, Bassit L, Sharma PL, Schinazi RF, Prudent JR. Quantifying mixed populations of drug-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49:3334–3340. doi: 10.1128/AAC.49.8.3334-3340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Svarovskaia ES, Moser MJ, Bae AS, Prudent JR, Miller MD, Borroto-Esoda K. MultiCode-RTx real-time PCR system for detection of subpopulations of K65R human immunodeficiency virus type 1 reverse transcriptase mutant viruses in clinical samples. J Clin Microbiol. 2006;44:4237–4241. doi: 10.1128/JCM.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moser MJ, Christensen DR, Norwood D, Prudent JR. Multiplexed detection of anthrax-related toxin genes. J Mol Diagn. 2006;8:89–96. doi: 10.2353/jmoldx.2006.050049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherrill CB, Marshall DJ, Moser MJ, Larsen CA, ude-Snow L, Jurczyk S, Shapiro G, Prudent JR. Nucleic acid analysis using an expanded genetic alphabet to quench fluorescence. J Am Chem Soc. 2004;126:4550–4556. doi: 10.1021/ja0315558. [DOI] [PubMed] [Google Scholar]
- 16.Razonable RR, Brown RA, Epsy MJ, Riverom A, Kremers W, Wilson J, Groettum C, Smith TF, Paya CV. Comparative quantitation of cytomegalovirus (CMV) DNA in solid organ transplant recipients with CMV infection by using two high-throughput automated systems. J Clin Microbiol. 2001;39:4472–4476. doi: 10.1128/JCM.39.12.4472-4476.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Preiser W, Rabenau HF, Vogel JU, Brixner V, Doerr HW. Performance characteristics of an automated PCR assay for the quantification of cytomegalovirus DNA in plasma. J Virol Methods. 2002;101:149–157. doi: 10.1016/s0166-0934(01)00438-4. [DOI] [PubMed] [Google Scholar]
- 18.Caliendo AM, Ingersoll J, Fox-Canale AM, Pargman S, Bythwood T, Hayden MK, Bremer JW, Lurain NS. Evaluation of real-time PCR laboratory-developed tests using analyte-specific reagents for cytomegalovirus quantification. J Clin Microbiol. 2007;45:1723–1727. doi: 10.1128/JCM.02558-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piiparinen H, Hockerstedt K, Gronhagen-Riska C, Lautenschlager I. Comparison of two quantitative CMV PCR tests. Cobas Amplicor CMV Monitor and TaqMan assay, and pp65-antigenemia assay in the determination of viral loads from peripheral blood of organ transplant patients. J Clin Virol. 2004;30:258–266. doi: 10.1016/j.jcv.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 20.Agresti A. Categorical Data Analysis. John Wiley & Sons; Hoboken, New Jersey: 2002. pp. 348–350. [Google Scholar]
- 21.Pang XL, Chui L, Fenton J, LeBlanc B, Preiksaitis JK. Comparison of LightCycler-based PCR. COBAS amplicor CMV monitor, and pp65 antigenemia assays for quantitative measurement of cytomegalovirus viral load in peripheral blood specimens from patients after solid organ transplantation. J Clin Microbiol. 2003;41:3167–3174. doi: 10.1128/JCM.41.7.3167-3174.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machida U, Kami M, Fukui T, Kazuyama Y, Kinoshita M, Tanaka Y, Kanda Y, Ogawa S, Honda H, Chiba S, Mitani K, Muto Y, Osumi K, Kimura S, Hirai H. Real-time automated PCR for early diagnosis and monitoring of cytomegalovirus infection after bone marrow transplantation. J Clin Microbiol. 2000;38:2536–2542. doi: 10.1128/jcm.38.7.2536-2542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ratge D, Scheiblhuber B, Nitsche M, Knabbe C. High-speed detection of blood-borne hepatitis C virus RNA by single-tube real-time fluorescence reverse transcription-PCR with the LightCycler. Clin Chem. 2000;46:1987–1989. [PubMed] [Google Scholar]
- 24.Brechtbuehl K, Whalley SA, Dusheiko GM, Saunders NA. A rapid real-time quantitative polymerase chain reaction for hepatitis B virus. J Virol Methods. 2001;93:105–113. doi: 10.1016/s0166-0934(01)00260-9. [DOI] [PubMed] [Google Scholar]
- 25.Gu Z, Belzer SW, Gibson CS, Bankowski MJ, Hayden RT. Multiplexed, real-time PCR for quantitative detection of human adenovirus. J Clin Microbiol. 2003;41:4636–4641. doi: 10.1128/JCM.41.10.4636-4641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevens SJ, Verschuuren EA, Verkuujlen SA, Van Den Brule AJ, Meijer CJ, Middeldorp JM. Role of Epstein-Barr virus DNA load monitoring in prevention and early detection of post-transplant lymphoproliferative disease. Leuk Lymphoma. 2002;43:831–840. doi: 10.1080/10428190290016971. [DOI] [PubMed] [Google Scholar]
- 27.Neumaier M, Braun A, Wagener C. Fundamentals of quality assessment of molecular amplification methods in clinical diagnostics. International Federation of Clin Chem Scientific Division Committee on Molecular Biology Techniques. Clin Chem. 1998;44:12–26. [PubMed] [Google Scholar]
- 28.Saunders GC, Parkes HC, Primrose SB. Analytical Molecular Biology Quality and Validation. The Royal Society of Chemistry; Cambridge, UK: 1999. pp. 58–80. [Google Scholar]
- 29.Razonable RR, Brown RA, Wilson J, Groettum C, Kremers W, Espy M, Smith TF, Paya CV. The clinical use of various blood compartments for cytomegalovirus (CMV) DNA quantitation in transplant recipients with CMV disease. Transplantation. 2002;73:968–973. doi: 10.1097/00007890-200203270-00025. [DOI] [PubMed] [Google Scholar]
- 30.Hakim H, Gibson C, Pan J, Srivastava K, Gu Z, Bankowski MJ, Hayden RT. Comparison of various blood compartments and reporting units for the detection and quantification of Epstein-Barr virus in peripheral blood. J Clin Microbiol. 2007;45:2151–2155. doi: 10.1128/JCM.02308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai KK, Cook L, Krantz EM, Corey L, Jerome KR. Calibration curves for real-time PCR. Clin Chem. 2005;51:1132–1136. doi: 10.1373/clinchem.2004.039909. [DOI] [PubMed] [Google Scholar]
- 32.Hayden RT, Hokanson KM, Pounds SB, Bankowski MJ, Belzer SW, Carr J, Diorio D, Forman MS, Joshi Y, Hillyard D, Hodinka RL, Nikiforova MN, Romain CA, Stevenson J, Valsamakis A, Balfour HH., Jr Multicenter comparison of different real-time PCR assays for quantitative detection of Epstein-Barr virus. J Clin Microbiol. 2008;46:157–163. doi: 10.1128/JCM.01252-07. [DOI] [PMC free article] [PubMed] [Google Scholar]