

Abstract
The chemistry of 1,2,3,4-tetrahydro-1,5-naphthyridines and 2,3,4,5-tetrahydro-1H-pyrido[3,2-b]azepines has been explored with the goal of discovering reactions at N1 suitable for library development. Epoxide openings, palladium-catalyzed N-arylations, DEPBT-promoted acylations, and urea formation through the reaction with isocyanates were all successful. The epoxide opening chemistry using homochiral epichlorohydrin, with epoxide reclosure and a second nucleophilic opening led to the preparation of a small 24-membered library.
1. Introduction
Recently we reported the details of a new procedure for the efficient synthesis of 1,2,3,4-tetrahydro-1,5-naphthyridines 1 and 2,3,4,5-tetrahydro-1H-pyrido[3,2-b]azepines 2 using an intramolecular inverse electron demand Diels-Alder reaction between an imidazole dienophile and a 1,2,4-triazine linked from the imidazole N1 position to the triazinyl C3 with a tri- or tetramethylene tether, respectively (Scheme 1).1 Using this chemistry, a series of heterocycles was prepared, which demonstrated variation in ring size in the saturated ring as well as substitution diversity in both the saturated and pyridine rings (Chart 1). As small nitrogen heterocycles with variable substitution in the aromatized pyridine ring, this series presents an ideal ”drug-like” structure type for library development in the search for new biologically active compounds. Thus, the next goal was to examine the chemistry of the N1 position to determine the suitability of these scaffolds for library synthesis. There were no previous reports in the literature addressing this issue, and thus the degree of nucleophilicity of this nitrogen, how this nucleophilicity would be affected by pyridine ring substitution, and which reactions could be exploited for library synthesis were uncertain. We herein report the results of this investigation, which concludes with the preparation of a small, twenty-four membered library, setting the stage for larger library syntheses.
Scheme 1.

Chart 1.

2. Results and Discussion
Our probe into the chemistry of the tetrahydronaphthyridines with a few select models focused on derivatizations at N-1. Five basic reactions (Scheme 2) were initially examined: (1) epoxide opening to secondary alcohols 3 (Route A), (2) Pd-catalyzed aryl aminations to 4 (Route B),2 (3) Petasis-Mannich chemistry3 to 6 (Route D), (4) DEPBT-mediated acylation4 to amides 7 (Route E), and (5) urea formation with isocyanates to 8 (Route F). In addition, N-tosylated 1h could also serve as the aryl halide in Pd-catalyzed aryl aminations to produce 5 (Route C), as anticipated. While all of these reactions worked well, the homoenamine products from the Petasis-Mannich chemistry (e.g. 6) formed with β–styrylboronic acid5 proved to be very unstable and this chemistry was no longer pursued. All attempts at Mitsunobu couplings, even with ADDA, TMAD, and TIPA, and the more acidic of the naphthyridines 1g with the two electron withdrawing ester groups failed.6 The products from the other exploratory reactions (A – C, and E and F) proved to be sufficiently stable for further exploration.
Scheme 2.

Aryl amination of 5,6-diphenyltetrahydronaphthyridine 1a proceeded in excellent yield with bromobenzene under Hartwig conditions7 to give the 1,5,6-triphenyltetrahydronaphthyridine 4 (89%, Route B), thereby establishing that these tetrahydronaphthyridine N1 nitrogens are sufficiently nucleophilic to participate in palladium catalyzed aryl aminations. Lower yields (65%) of 4 were obtained using the Pd(dba)3/BINAP/NaOtBu system.8 The 6-p-bromophenyltetrahydronaphthyridine 1h, protected as the N-tosylate, also participated in the aryl amination with morpholine using the same conditions, demonstrating that appropriately substituted naphthyridines can serve as the oxidative addition substrates in these cross-couplings. Morpholine derivative 5 was produced in 56% unoptimized yield (Route C).
Direct acylation of the tetrahydronaphthyridines with carboxylic acids was then explored using 1d, 1e, 1h as a models (Scheme 2, Route E). While EDCI/HOBt proved ineffective in promoting the acylation, Goodman’s 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4-one (DEPBT) reagent4 was successful and amides 7d, 7e, and 7h were isolated in 78 – 95% yield from the couplings with 4-pentynoic acid. The alkynyl functionality in 7d,e,h can serve as a diversification point for further chemistry such as dipolar cycloadditions.9 In the 1H NMR spectra of all the amides, H-8 suffered severe broadening, as did C-8 in the 13C NMR, due to slow rotation about the amide bond. Heating the samples to 50 °C sharpened the resonances considerably.

Somewhat more interesting was the potential use of the tetrahydronaphthyridines as scaffolds for preparing libraries via urea formation in the reaction with isocyanates, or via epoxide ring opening reactions (Scheme 2, Routes F and A, respectively). Urea formation (Scheme 2, Route F) proceeded in good to excellent yields and could be accomplished with commercially available isocyanates such as phenylisocyanate, producing 8a in 99+% yield, or by adding the naphthyridine to a solution of the isocyanate generated in situ from a carboxylic acid and DPPA/Et3N (Scheme 3).10 One advantage of this latter approach beginning with the carboxylic acids is the ability to use a greater variety of reagents for the isocyanate domain, as was done in the preparation of 8b (60%) and 8c (73%), rather than rely upon the more structurally limited, commercially available isocyanates.
Scheme 3.

Our initial library work, however, focused on epoxide opening since this chemistry has the potential for adding a stereochemically encoded appendage to the naphthyridine scaffold (Scheme 2, Route A). The use of enantiomerically pure epoxides, or a kinetic resolution in the course of opening a racemic mixture of epoxides would therefore be required in order to obtain enantiomerically pure individual library members. We briefly examined the application of Jacobsen’s (R,R)-oligosalen-Co(III) catalyst11 in the kinetically resolving epoxide opening of rac-propylene oxide (Scheme 4). By the use of a large excess of epoxide (10–15 eq) at −78 °C, warming to −40 °C overnight, in the presence of 4 mol% catalyst and 8 mol% 2,6-lutidinium p-toluenesulfonate (LPTS) an excellent yield (95%) and ee (99+%) of secondary alcohol 3a was obtained after passing through a solid-liquid extraction plug (SLE-Celite preconditioned with saturated aqueous NaHCO3 solution), selecting for the (S)-epoxide. Only a single enantiomer was detected by chiral HPLC, which also allowed assignment of the enantiomer upon comparison with that prepared from enantiomerically pure (S)-propylene oxide. At higher temperatures (0 °C), the ee was only 58%. Given the need for the large amount of epoxide, and the ready availability of epoxides with high to complete enantioenrichment,12 opening of enantiomerically pure epoxides was deemed a more suitable strategy for library development.
Scheme 4.

Preliminary work to optimize epoxide openings with enantiomerically pure epoxides used 1h with epichlorohydrin (Scheme 5, Table 1). A significant part of this effort optimized procedures for compound purification applicable to parallel synthesis. While several Lewis acid catalysts were examined in the epoxide opening, adaptation of Crotti’s Yb(OTf)313,14 (0.2 eq) protocol proved the most effective. Other Lewis acids examined, LiClO4,15 B(C6F5)3,16 Sc(OTf)3,14b and ZrCl4,17 gave lower yields or were completely ineffective. Purification of the secondary alcohols was readily achieved by passage of the reaction mixture through an SLE-Celite cartridge as described above, then scavenging the small amount of unreacted tetrahydronaphthyridine from the eluant with MP-NCO resin.18 From the available naphthyridines and pyridoazepines (Chart 1), 1c, 1h, 1i, and 2c were then selected for conversion to the corresponding enantiomerically pure chlorohydrins 3/9 using these optimized conditions and purification protocols with (+)-(S)-epichlorohydrin, (Scheme 5/Table 1). The intent was to then use these chlorohydrins as building blocks for library development. Tetrahydronaphthyridine 1j with the ester substituent was also reacted with rac-epichlorohydrin under these conditions to form the chlorohydrin (3j), though in this case the reaction was considerably more sluggish, requiring 10 h to produce only a 53% yield of 3j (Table 1, Entry 4). Presumably the electron withdrawing ester substituent significantly reduces the nucleophilic ity in comparison to the other naphthyridines. Given this reduced reactivity, 1j was not further applied to library synthesis exploiting N1 nucleophilicity.
Scheme 5.

Table 1.
| Item | 1/2 | Chlorohydrin 3/9 | Epoxide 10/11 |
|---|---|---|---|
| 1 | 1c |

3c (85%) |

10c (98%) |
| 2 | 1h |

3h (95%) |

10h (99%) |
| 3 | 1i |

3i (86%) |

10i (98%) |
| 4 | 1j |

3j (53%) |
- |
| 5 | 2c |

9c(70%) |

11c |
Isolated yields.
With epichlorohydrins 3c, 3h, 3i, and 9c in-hand, two separate reactivities were then tested (Scheme 5): closure to epoxides 10/11 for a second nucleophilic epoxide opening to 12/13, and conversion to the oxazolidinone 15, the latter process adapting Qian’s procedure (Scheme 5 above).19 Qian’s original procedure called for the reaction of epoxides with YbCl3, generating a chlorohydrin in situ, which then condenses with an isocyanate to form the target oxazolidinone, regenerating the YbCl3 catalyst. Attempts to use these protocols on epoxide 10i prepared from 1i, was not successful and only starting epoxide was recovered. Beginning with chlorohydrin 3i (R = H, R′ = p-MeOPh), however, reaction with phenylisocyanate in the presence of Yb(OTf)3 produced carbamate 14. Furthermore, a one-pot procedure optimized the carbamate preparation wherein 1i was treated with (S)-epichlorohydrin in the presence of Yb(OTf)3 (0.2 eq), excess epichlorohydrin was removed under reduced pressure, then the isocyanate was added to produce 14 in 91% yield from 1i. Treatment of 14 with NaH then yielded the oxazolidinone 15 (99%). As with the amides 7, the 1H NMR spectrum of 14 at rt also showed slight broadening for the H8 due to hindered rotation, which sharpened upon heating to 50 °C.
Alternatively, chlorohydrins 3/9 could be efficiently closed with NaH to the corresponding epoxides 10/11 (98 – 99%, Table 1), then these epoxides subjected to a nucleophilic opening either with a second naphthyridine or another nucleophile under Yb(OTf)3 catalysis (Scheme 5, above) using a small excess (1.2 – 2 eq) of nucleophile. Work-up was accomplished by passage through an SLE-Celite cartridge, pre-conditioned with saturated aqueous NaHCO3 solution, to remove the Lewis acid, followed by scavenging of the excess nucleophile with MP-NCO from the eluant. Some of the secondary alcohols 12/13 produced upon epoxide opening with amines were unstable to storage, some decomposing within 24 hours of isolation. Stable products from the epoxide openings could be obtained following removal of the unreacted nucleophile by treatment with HCl/ether, forming the more stable HCl salts of 12/13. Attempts to purify the product secondary alcohols by resin capture with PS-SiEt2Cl20 or PS-DHP21 were not successful. Other efforts to remove excess nucleophile (naphthyridine) with PS-TsCl,22 and PL-MIA23, or to remove excess epoxide 10/11, with the second nucleophile as the limiting reagent, using PS-thiophenol,24 PS-Trisamine,18c,18d or PS-NH218b were also incomplete, though MP-MIA res in could be used to remove secondary amines other than the naphthyridines. The enantiomeric purity of epoxide 10h was confirmed by chiral HPLC.
A small, 24-membered library (Table 2) was then prepared in a block reactor from the four epoxides 10c, 10h, 10i, and 11c by reacting with six nucleophiles using the protocols described above, preparing a minimum of 10 mg per sample with an avergae yield of 80%. The yields were noticeably higher with morpholine and diethylamine as the nucleophile (90 – 96%, Table 2, columns 6 and 7) in comparison to a second naphthyridine as the nucleophile (66 – 81%, Table 2, columns 2 – 5). Purity analysis was accomplished by LC/MS and NMR, which determined that each library member was ≥ 65 % pure with an average purity of 92.8%; only three members of the library were < 80% pure. Prior to library preparation, model reactions to establish block reactor protocols were run, and the secondary alcohol products 12g, 12i, and 12j (see Chart 2) were purified by passing through the SLE cartridge followed by flash chromatography for accurate assessment of yields based on 100% purity, and full characterization (13C NMR and HRMS spectra). Included in this library are three naphthyridine dimers 12a, 12i, and 12p, and thirteen pseudodimers (12b–d, 12g,h, 12j, 12m–o, 13a–d). In addition, by reversing the naphthyridinyl identities of the epoxide electrophile and amine nucleophile, three sets of enantiomers were also produced: 12c/12g, 12d/12m, and 12j/12o. The remaining eight members of the library were prepared from the epoxide openings using diethylamine and morpholine, respectively.
Table 2.
Twenty-four Membered Library Prepared by Nucleophilic Opening of Naphthyridine and Pyridoazepine Epoxides (Scheme 5).a
| Reagents |
 1c |
 1d |
 1h |
 1i |
 |
Et2NH |
|---|---|---|---|---|---|---|

10c |

12a (75%/100%) |

12b (66%/85%) |
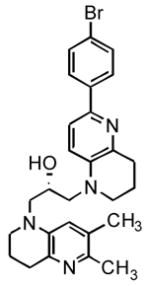
12c(73%/89%) |

12d (68%/65%) |

12e (92%/100%) |

12f(91%/97%) |

10h |

12g (74%/100%) |

12h (79%/89%) |

12i (81%/98%) |

12j (78%/73%) |

12k (94%/92%) |

12l (92%/97%) |

10i |

12m (68%/100%) |

12n (72%/100%) |

12o (73%/100%) |

12p (80%/80%) |

12q (90%/100%) |

12r (96%/100%) |

11c |

13a (74%/100%) |

13b (70%/100%) |

13c(80%/83%) |

13d(72%/74%) |

13e (93%/95%) |

13f (95%/100%) |
(% yield/% purity as determined by ELSD-MS).
3. Conclusions
The chemistry of 1,2,3,4-tetrahydro-1,5-naphthyridines has been explored to delineate the potential for the use of these heterocycles as scaffolds in library development, focusing primarily upon the reactivity of the N1 nitrogen as a nucleophile. The nucleophilicity proved quite sensitive to the electronic effects of the substituents in the B-ring. Those with electron withdrawing groups such as esters (e.g. 1j) were not as reactive as other naphthyridines and were not included in libraries requiring N1 nucleophiles, such as epoxide openings. Ultimately, Yb(OTf)3–catalyzed opening of the epoxides derived from epichlorohydrin was selected for library development leading to the preparation of a small series of linked naphthyridine dimers and related heterocycles 16. The successful conversion of the intermediate chlorohydrins 3 to oxazolidinones was also intriguing and this chemistry is currently being pursued for a third library generation. Future libraries will also exploit the naphthyridine scaffolds as amines in Pd-catalyzed arylation chemistry.
4. Experimental Procedures
General Procedure A (Scheme 5): Preparation of 1-(3-chloro-2-hydroxypropyl)-1,2,3,4-tetrahydro-1,5-naphthyridines 3c, 3h, 3i, 3j, and 1-(3-chloro-2-hydroxypropyl)-2,3,4,5-tetrahydro-1H-pyrido[3,2-b]]azepine (9c)
(a) Large scale
To a solution of tetrahydro-1,5-naphthyridine 1 or tetrahydropyridoazepine 2 and Yb(OTf)3 (typically 0.2 eq) in CH2Cl2 was added epichlorohydrin (3 – 5 eq) in a pressure tube, and the reaction mixture was then heated to 60 °C for 3 – 5 h. After cooling to rt, the mixture was diluted with saturated aqueous NaHCO3 and CH2Cl2, then the organic layer was separated and washed with saturated brine, then dried over Na2SO4. After filtration, the organic layer was concentrated in vacuo, and the residue was purified by flash chromatography on a silica gel column.
(b) Small scale
To a solution of tetrahydro-1,5-naphthyridine 1 or tetrahydropyridoazepine 2 and Yb(OTf)3 (typically 0.2 eq) in CH2Cl2 was added epichlorohydrin (3–5 eq) in a pressure tube, and the reaction mixture was heated to 60 °C for 5 h. After cooling to rt, the mixture was passed through an SLE-Celite cartridge, pre-conditioned with saturated aqueous NaHCO3, eluting with CH2Cl2. The organic eluant was concentrated in vacuo, and the residue was purified by flash chromatography on a silica gel column.
General Procedure B (Scheme 5): Preparation of 1-(2,3-epoxypropyl)-1,2,3,4-tetrahydro-1,5-naphthyridines 10c, 10h, 10i, and 1-(2,3-epoxypropyl)-2,3,4,5-tetrahydro-1H-pyrido[3,2-b]azepine 11c
A suspension of chlorohydrin 3 or 9 (0.10 mmol to 1.0 mmol) and NaH (60% in mineral oil, 2.0 eq) in THF (1 – 10 mL) was stirred at rt overnight. The reaction mixture was quenched with water, then extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and the eluant concentrated in vacuo. The residue was purified by flash chromatography on silica gel column.
General Procedure C (Scheme 5): Nucleophilic Opening of Epxoides 10 and 11
A solution of epoxynaphthyridine 10 or 11 (1 eq), nucleophilic amine (1.1 eq) and Yb(OTf)3 (1.0 eq) in CH2Cl2 was stirred for 30 h at 60 °C in a pressure tube. After cooling to rt, the reaction mixture was passed through an SLE-Celite cartridge, pre-conditioned with saturated aqueous NaHCO3, eluting with CH2Cl2. The organic eluant was concentrated in vacuo, and the crude product was placed in a pressure tube along with MP-NCO (3 eq) and CH2Cl2 (500 μL), and heated to 60 °C for 24 h. After cooling to rt, the resin was removed by filtration and washed with CH2Cl2, then the filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel column.
General Procedure D: Preparation of 24-Membered Library 12/13 by Nucleophilic Epoxide Opening (Chart 2)
Solutions of epoxides 10c, 10h, 10i, or 11c (0.05 mmol, 1 eq), 1,2,3,4-tetrahydro-1,5-naphthyridines 1d, 1h, 1i, or 11c (0.06 mmol, 1.2 eq each), or morpholine, or diethylamine (0.10 mmol, 2 eq each) and Yb(OTf)3 (0.05 mmo l, 1.0 eq) in CH2Cl2 (250 μL, 0.2 M) were stirred for 30 h at 60 °C in capped microwave reactor tubes (10 mL CEM microwave tubes with aluminum cap) in a Bhodan MiniBlock XT block reactor for 24 reactions. After cooling to rt, the reaction mixtures were passed through SLE-Celite cartridges, pre-conditioned with saturated aqueous NaHCO3, eluting with CH2Cl2 (3 × 1 mL), and collected in a CEM microwave tube (10 mL). The organic eluant was concentrated in vacuo using a Genevac centrifugal vacuum evaporator, and then nucleophile scavenger (MP-NCO, 0.15 mmol, 3 eq) and CH2Cl2 (500 μL) were added and heated to 60 °C for 24 h in the block reactor after recapping the tubes. After the cooling, the resin was removed by filtration, and washed with CH2Cl2 (3 × 1 mL). The filtrate was collected in a test tube, and then concentrated under reduced pressure using the Genevac centrifugal vacuum evaporator. The residue was treated with 2 M HCl in diethyl ether (500 μL), stirred for 15 minutes at rt, then the product was collected by filtration. Purity was established by LC/MS using ELSD as the primary detector and DAD as the secondary detector; column ACT ACE C3 3 × 50 mm, 3 μm; eluant: 5 mM formic acid in CH3CN/water gradient. Characterization was accomplished by 1H NMR.
Supplementary Material
Supplementary Data
Reaction procedures and spectral characterization data for all compounds. Supplementary data associated with this article can be found in the on-line version.
Acknowledgments
We thank the NIH for financial support (P50 GM067041). We also thank Professor John A. Porco, Jr. for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lahue BR, Lo SM, Wan ZK, Woo GHC, Snyder JK. J Org Chem. 2004;69:7171–7182. doi: 10.1021/jo040193z. [DOI] [PubMed] [Google Scholar]
- 2.Reviews: Hartwig JF. Synlett. 1997:329–340.Yang BH, Buchwald SL. J Organomet Chem. 1999;576:125–146.Hartwig JF. Palladium-Cata,yzed Amination of Aryl Halides and Related Reactions. In: Negishi E, editor. Handbook of Organopalladium Chemistry for Organic Synthesis. Vol. 1. Wiley & Sons; Hoboken, NJ: 2002. pp. 1051–1096.
- 3.(a) Petasis NA, Akritopoulou I. Tetrahedron Lett. 1993;34:583–586. [Google Scholar]; (b) Petasis NA, Zavialov IA. J Am Chem Soc. 1998;120:11798–11799. and references therein. [Google Scholar]
- 4.Li H, Jiang X, Ye Y-H, Fan C, Romoff T, Goodman M. Org Lett. 1999;1:91–93. doi: 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]
- 5.(a) Brown HC, Gupta SK. J Am Chem Soc. 1972;94:4370–4371. [Google Scholar]; (b) Brown HC, Gupta SK. J Am Chem Soc. 1975;97:5249–5255. [Google Scholar]
- 6.(a) Tsunoda T, Yamamiya Y, Ito S. Tetrahedron Lett. 1993;34:1639–1642. [Google Scholar]; (b) Tsunoda T, Otsuka J, Yamamiya Y, Ito S. Chem Lett. 1994:539–642. [Google Scholar]; (c) Ito S, Tsunoda T. Pure Appld Chem. 1999;71:1053–1057. [Google Scholar]
- 7.Louie J, Driver MS, Hamann BC, Hartwig JF. J Org Chem. 1997;62:1268–1273. [Google Scholar]
- 8.(a) Wolfe JP, Wagaw S, Buchwald SL. J Am Chem Soc. 1996;118:7215–7216. [Google Scholar]; (b) Wagaw S, Buchwald SI. J Org Chem. 1996;61:7240–7241. doi: 10.1021/jo9612739. [DOI] [PubMed] [Google Scholar]; (c) Marcoux JF, Wagaw S, Buchwald SL. J Org Chem. 1997;62:1568–1569. [Google Scholar]
- 9.For example: Quan C, Kurth M. J Org Chem. 2004;69:1470–1474. doi: 10.1021/jo0352124.Tornoe CW, Sanderson SJ, Mottram JC, Coombs GH, Meldal M. J Comb Chem. 2004;6:312–324. doi: 10.1021/cc020085v.Khanetskyy B, Dallinger D, Kappe CO. J Comb Chem. 2004;6:884–892. doi: 10.1021/cc0498938.Coats SJ, Link JS, Gauthier D, Hlasta DJ. Org Lett. 2005;7:1469–1472. doi: 10.1021/ol047637y.
- 10.Procedure followed: Kedrowski BL. J Org Chem. 2003;63:5403–5406. doi: 10.1021/jo034170g.
- 11.Ready JM, Jacobsen EN. J Am Chem Soc. 2001;123:2687–2688. doi: 10.1021/ja005867b. [DOI] [PubMed] [Google Scholar]
- 12.For example: Johnson RA, Sharpless KB. Catalytic Asymmetric Epoxidation of Allylic Alcohols. In: Ojima I, editor. Catalytic Asymmetric Synthesis. VCH; New York: 1993. pp. 103–158.Jacobsen EN. Epoxidation of Alkenes Other Than Allylic Alcohols. In: Jacobsen EN, Pflatz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis I-III. Vol. 2. Springer-Verlag; Berlin: 1999. pp. 649–677.Denmark SE, Wu Z. Synlett. 1999:847–859.
- 13.Chini M, Crotti P, Favero L, Macchia F, Pineschi M. Tetrahedron Lett. 1994;35:433–436. [Google Scholar]
- 14.(a) Meguro M, Asao N, Yamamoto Y. J Chem Soc, Perkin Trans I. 1994:2597–2601. [Google Scholar]; (b) Likhar PR, Kumar MP, Bandyopadhyay AK. Tetrahedron Lett. 2002;43:3333–3335. [Google Scholar]
- 15.(a) Chini M, Crotti P, Macchia F. Tetrahedron Lett. 1990;31:4661–4664. [Google Scholar]; (b) Chini M, Crotti P, Macchia F. J Org Chem. 1991;56:5939–5942. [Google Scholar]; (c) Chng BL, Ganesan A. Bioorg Med Chem Lett. 1997;7:1511–1514. [Google Scholar]
- 16.Chandrasekhar S, Reddy ChR, Babu BN, Chandrashekar G. Tetrahedron Lett. 2002;43:3801–3803. [Google Scholar]
- 17.Chakraborti AK, Kondaskar A. Tetrahedron Lett. 2003;44:8315–8319. [Google Scholar]
- 18.Argonaut Technologies Technical Note 503, “MP-Isocyanate”. Foster City, CA: 2002. Also, see: Kaldor SW, Siegel MG, Fritz JE, Dressman BA, Hahn PJ. Tetrahedron Lett. 1996;37:7193–7196.Booth RJ, Hodges JC. J Am Chem Soc. 1997;119:4882–4886.Creswell MW, Bolton GL, Hodges JC, Meppen M. Tetrahedron. 1998;54:3983–3998.Galaffu N, Bradley M. Tetrahedron Lett. 2005;46:859–861.
- 19.Qian C, Zhu D. Synlett. 1994:129–130. [Google Scholar]
- 20.Hu Y, Porco JA, Jr, Ladadie JW, Gooding OW, Trost BM. J Org Chem. 1998;63:4518–4521. [Google Scholar]
- 21.(a) Thompson LA, Ellman JA. Tetrahedron Lett. 1994;35:9333–9336. [Google Scholar]; (b) Pearson WH, Clark RB. Tetrahedron Lett. 1997;38:7669–7672. [Google Scholar]; (c) Ramaseshan M, Ellingboe JW, Dory YL, Deslongchamps P. Tetrahedron Lett. 2000;41:4743–4749. [Google Scholar]; (d) Ramaseshan Dory YL, Deslongchamps P. J Comb Chem. 2000;2:615–623. doi: 10.1021/cc000030y. [DOI] [PubMed] [Google Scholar]; (e) Ma S, Duan D, Wang Y. J Comb Chem. 2002;4:239–247. doi: 10.1021/cc010084n. [DOI] [PubMed] [Google Scholar]
- 22.(a) Zhang HM, Greco MN, Maryanoff BE. J Org Chem. 1997;62:9326–9330. [Google Scholar]; (b) Zhang HC, Ye H, Moretto AF, Brumfield KK, Maryanoff BE. Org Lett. 2000;2:89–92. doi: 10.1021/ol991255o. [DOI] [PubMed] [Google Scholar]; (c) MacCoss RN, Brennan PE, Ley SV. Org Bioorg Chem. 2003;1:2029–2031. doi: 10.1039/b303951j. [DOI] [PubMed] [Google Scholar]
- 23.(a) Coppola GM. Tetrahderon Lett. 1998;39:8233–8236. [Google Scholar]; (b) Lizarzaburu ME, Shuttleworth SJ. Tetrahedron Lett. 2003;44:4873–4876. [Google Scholar]
- 24.Ermann M, Simkovsky NM, Roberts SM, Parry DM, Baxter AD, Montana JG. Tetrahedron Lett. 2000;41:2483–2485. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Reaction procedures and spectral characterization data for all compounds. Supplementary data associated with this article can be found in the on-line version.