

Abstract
We sequenced and analyzed the genome of a commensal Escherichia coli (E. coli) strain SE11 (O152:H28) recently isolated from feces of a healthy adult and classified into E. coli phylogenetic group B1. SE11 harbored a 4.8 Mb chromosome encoding 4679 protein-coding genes and six plasmids encoding 323 protein-coding genes. None of the SE11 genes had sequence similarity to known genes encoding phage- and plasmid-borne virulence factors found in pathogenic E. coli strains. The comparative genome analysis with the laboratory strain K-12 MG1655 identified 62 poorly conserved genes between these two non-pathogenic strains and 1186 genes absent in MG1655. These genes in SE11 were mostly encoded in large insertion regions on the chromosome or in the plasmids, and were notably abundant in genes of fimbriae and autotransporters, which are cell surface appendages that largely contribute to the adherence ability of bacteria to host cells and bacterial conjugation. These data suggest that SE11 may have evolved to acquire and accumulate the functions advantageous for stable colonization of intestinal cells, and that the adhesion-associated functions are important for the commensality of E. coli in human gut habitat.
Key words: Escherichia coli, commensal, human gut, genome sequencing
1. Introduction
Microbial communities (microbiota) inhabiting the human body sites have long been recognized to play critical roles in human health and disease. Collective genomes (microbiome) of the human microbiota have now become important targets to be studied in both microbiology and human biology.1 Among the human microbiota, the gut microbiota are most abundant in number of microbial species accounting for ≥1000 species, which shape a very complex and dynamic microbial community with high interindividual variations.2 The large-scale bacterial 16S ribosomal RNA sequence and metagenomic analyses of the gut microbiome have provided a great progress for a better understanding of the ecological and biological natures of the human gut microbiota.3–7 However, genomic sequences of individual members constituting the microbiota are also needed, and important to more precisely interpret the enumerative data that will be accumulated in future studies including the International Human Microbiome Project.8
Escherichia coli (E. coli) is one of the common members in the human gut microbiota. Over the past decades, there have been many reports on the phylogenetic and genomic analyses of E. coli strains isolated from various sources including humans, animals and various environments.9–19 Among isolated E. coli strains, the whole-genome sequencing analysis has been extensively performed for pathogenic strains to explore the pathogenicity and identify virulence-associated genes in these strains.20–26 In contrast, the whole-genomic sequencing of non-pathogenic E. coli strains has been limited for several E. coli K-12 strains that have long been used in genetic studies and recombinant DNA technologies.27–30 Since E. coli K-12 strain was originally isolated from the stool of a convalescent diphtheria patient in 1922, these sequenced K-12-derived strains, MG1655, W3110 and DH10B, may have undergone spontaneous genetic changes during preservation and successive passages at the laboratory, resulting in the accumulation of mutations in genes and loss of many features representing the wild-type commensal E. coli.31–33 Nevertheless, the genomic sequencing analysis of human commensal E. coli strains is quite scarce. Only two human commensal strains HS and Nissle 1917 have been completely or partially sequenced.26,34,35 This is surprising because the wild-type commensal strain is a good reference genome in the human gut microbiota research and useful to explore the genetic and functional features adapted to human gut habitat, and the comparison with the laboratory strain K-12 or pathogenic strains will provide new insights into the structural and evolutionary aspect of commensal E. coli strains.
In this study, we sequenced the genome of commensal E. coli strain-designated SE11 isolated from feces of a healthy adult and performed the comparative analysis with other sequenced E. coli genomes. This paper may be the first report demonstrating the complete genome sequence analysis of the wild-type commensal E. coli strain belonging to phylogenetic group B1, distant from strains K-12 and HS belonging to phylogenetic group A in E. coli reference (ECOR) collection.36
2. Materials and methods
2.1. Isolation of E. coli strains from fresh feces of a healthy adult human
One gram of feces collected from a healthy adult human was suspended in 9.0 mL of phosphate-buffered saline (pH 7.0). Serially diluted solutions were inoculated on deoxycholate hydrogen sulfide lactose (DHL) agar (Eiken Chemical Co. Ltd.) and incubated at 37°C for 24 h. Eight red colonies on the DHL agar plates were picked up and subjected to single colony isolation twice on Luria-Burtani (LB) agar plates. The eight isolates were identified as E. coli on the basis of the following characteristics: gram negative, rod shape, growth under the aerobic and anaerobic conditions, spore formation negative, motile, production of gas/lactic acid from glucose/lactose. Each isolate was grown in LB broth at 37°C for 24 h and stored in the LB medium containing 10% glycerol at −85°C until used for further analysis.
2.2. Random amplification of polymorphic DNA fingerprinting
Eight E. coli isolates were analyzed by random amplification of polymorphic DNA (RAPD) fingerprinting method using three primers (1247, 5′-AAGAGCCCGT-3′; 1254, 5′-CCGCAGCCAA-3′; 1290, 5′-GTGGATGCGA-3′).37 A fresh colony grown on LB agar plate was transferred to a 1.5 mL microtube. The cells were disrupted using microwave (500 W for 1 min) and suspended in 5.0 µL of double-distilled water (ddH2O). After spindown, the supernatant was used as template DNA in RAPD analysis. The 50.0 µL polymerase chain reaction (PCR) mixture contained 5.0 µL of template DNA, 4.0 µL of each primer (10 µM), 5.0 µL of 10× PCR buffer, 4.0 µL of dNTP mixture, 0.25 µL of Ex Taq polymerase (Takara Bio Inc.), and 27.75 µL of ddH2O. PCR amplification was performed in the iCycler Thermal Cycler (Bio-Rad) according to the following protocol: 1 cycle of 10 min at 94°C; 30 cycles of 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C; and 1 cycle of 10 min at 72°C. Amplified DNA fragments were separated on 1.0% agarose gels (100 V for 30 min) and stained with ethidium bromide (0.2 µg/mL) for 30 min.
2.3. Genome sequencing
The genome sequence of SE11 was determined by a whole-genome shotgun strategy. We constructed small-insert [2 kilobases (kb)], large-insert (10 kb) and fosmid (40 kb) genomic libraries, and generated 55 296 sequences using ABI 3730xl sequencers (Applied Biosystems), giving eightfold coverage from both ends of the genomic clones. Sequence reads were assembled with the Phred–Phrap–Consed program38 and gaps were closed by direct sequencing of clones that spanned the gaps or of PCR products amplified with oligonucleotide primers designed to anneal to each end of neighboring contigs. The overall accuracy of the finished sequence was estimated to have an error rate of <1 per 10 000 bases (Phrap score of ≥40).
2.4. Informatics
An initial set of predicted protein-coding genes was identified using Glimmer 2.0.39 Genes consisting <120 base pairs (bp) and those containing overlaps were eliminated. All predicted proteins were searched against a non-redundant protein database (nr, NCBI) using BLASTP with a bit-score cutoff of 60. The start codon of each protein-coding gene was manually refined from BLASTP alignments. The tRNA genes were predicted by the tRNAscan-SE40 and the rRNA genes were detected by BLASTN search using known E. coli rRNA sequences as queries. Protein domains were identified using the Pfam database. Orthology across whole-genomes has been determined using BLASTP reciprocal best hits with a bit-score cutoff of 60 in all-against-all comparisons of amino acid sequences. Two sequences were identified as poorly conserved orthologs if their BLAST score ratio is <0.8.41 Sequences of seven housekeeping genes of the ECOR strains were obtained from the multilocus sequence typing (MLST) website (http://web.mpiib-berlin.mpg.de) in the Max Planck Institute.42 These sequences were concatenated, and aligned by the neighbor-joining method with 1000 bootstrap iterations using ClustalW. The sequence data of the SE11 genome have been deposited in DDBJ/GenBank/EMBL and the accession numbers are as follows: AP009240 (chromosome), AP009241 (pSE11-1), AP009242 (pSE11-2), AP009243 (pSE11-3), AP009244 (pSE11-4), AP009245 (pSE11-5) and AP009246 (pSE11-6).
3. Results
3.1. Isolation and phylogenetic analysis of SE11
We isolated eight E. coli strains from feces of a healthy adult as described in Materials and methods, and examined them by the RAPD method. Seven strains exhibited the same RAPD patterns in respective experiments using three different primer sets, thus revealing their structural identity of genomes. We therefore selected one E. coli strain-designated SE11 for further analysis and sequencing. The 16S rRNA sequence of SE11 showed the highest similarity (98.8% identity) to that of E. coli ATCC 11775T (accession no. X80725). From the MLST analysis based on the nucleotide sequences of seven housekeeping genes,42 SE11 was found to belong to phylogenetic group B1 whose members predominate in the human gut microbiota,9 and is phylogenetically distinct from K-12 and HS strains in group A and more from human pathogenic strains mostly belonging to group B2 or E in ECOR collection (Supplementary Fig. S1). Most of commensal E. coli strains belonging to groups A and B1 were shown to be avirulent in mice.43 SE11 has an O152:H28 serotype, which is less frequently found in enteroinvasive E. coli.44,45
3.2. General features and gene content in SE11
The genome of E. coli SE11 consists of a circular chromosome of 4 887 515 bp and six plasmids (100.0, 91.2, 60.6, 6.9, 5.4 and 4.1 kb) (Figs 1 and 2). General features of the SE11 genome were shown in Table 1. The chromosome size of SE11 is larger than those of the laboratory K-12 strains, and smaller than those of pathogenic strains sequenced to date (Supplementary Table S1). The SE11 chromosome contained 4679 predicted protein-coding genes, 86 tRNA genes, and 22 rRNA genes, and the six plasmids contained a total of 323 predicted protein-coding genes. Of all protein-coding genes predicted in SE11, we could assign 2944 (59%) protein-coding genes to known functions, 1895 (38%) to genes of unknown function conserved in many bacterial genomes, and 163 (3%) to novel hypothetical genes. We identified 52 copies of insertion sequence (IS) elements in the SE11 genome (Supplementary Table S2). These IS elements are classified into 27 families, and the IS677 family (10 copies as intact forms) was most predominant in SE11.
Figure 1.

Circular representation of the SE11 chromosome. From the outside in: circles 1 and 2 of the chromosome show the positions of protein-coding genes on the positive and negative strands, respectively. Circles 3 and 4 show the positions of protein-coding genes that have orthologs in E. coli strains E24377A and K-12 MG1655, respectively. Circle 5 shows the positions of the prophages PP_SE11 (blue), integrative elements IE_SE11 (orange), and large segment near the aspV (green). Circle 6 shows the positions of tRNA genes (purple) and rRNA genes (brown). Circle 7 shows a plot of GC skew [(G – C)/(G + C); khaki indicates values > 0; purple indicates values < 0]. Circle 8 shows a plot of G + C content (higher values outward).
Figure 2.

Circular representations of three larger plasmids of SE11. The outer and inner circles of each plasmid represent genes on the positive and negative strands, respectively.
Table 1.
General features of the SE11 genome
| Chromosome | Plasmids |
||||||
|---|---|---|---|---|---|---|---|
| pSE11-1 | pSE11-2 | pSE11-3 | pSE11-4 | pSE11-5 | pSE11-6 | ||
| Size (bp) | 4 887 515 | 100 021 | 91 158 | 60 555 | 6929 | 5366 | 4082 |
| GC content (%) | 50.8 | 50.5 | 50.2 | 48.6 | 48.0 | 46.2 | 49.4 |
| Protein-coding gene | 4679 | 124 | 112 | 67 | 10 | 7 | 3 |
| Assigned function | 2772 | 70 | 49 | 46 | 4 | 2 | 1 |
| Conserved hypothetical | 1474 | 48 | 38 | 11 | 2 | 3 | 2 |
| Unknown function | 118 | 6 | 23 | 10 | 4 | 2 | 0 |
| Phage related | 315 | 0 | 2 | 0 | 0 | 0 | 0 |
| IS element | 33 | 2 | 5 | 12 | 0 | 0 | 0 |
| rRNA gene | 86 | 0 | 0 | 0 | 0 | 0 | 0 |
| tRNA gene | 22 | 0 | 0 | 0 | 0 | 0 | 0 |
Comparison of all 5002 protein-coding genes in SE11 with those in the strain K-12 MG1655 identified 1186 genes absent in MG1655, 62 poorly conserved genes and 3754 highly conserved genes. Classification of the 5002 protein-coding genes in SE11 was summarized in Fig. 3. Of the 3754 highly conserved genes, 2802 were also conserved in all 14 sequenced E. coli genomes. Of the 1186 genes, 170 were unique to SE11 among the 14 E. coli genomes. The 1186 genes absent in MG1655 comprised 438 mobile elements-related, 356 conserved function-unknown, 108 hypothetical and 284 genes with assigned functions including metabolic genes of oligosaccharides such as sugar, cellobiose, mannose and N-acetylgalactosamine, a gene for bile salt hydrolase, tetracycline-resistant genes and genes associated with fimbriae on the bacterial cell surface (discussed later). The 356 conserved hypothetical genes and 284 genes with assigned functions in SE11 were listed in Supplementary Table S3. On the other hand, the 317 genes that were present in MG1655 but absent in SE11 comprised 186 (59%) mobile elements-related and 131 unique genes including those involved in the restriction/modification system and acetoacetate metabolism (Supplementary Table S4). The 62 poorly conserved genes between SE11 and MG1655 may have the higher mutation rate than other conserved E. coli genes and included some of genes of the lipopolysaccharide (LPS) biosynthesis (Supplementary Table S5). Outer core oligosaccharide in LPS is highly variable in structure and five distinct outer core types in E. coli are known.46 The outer core oligosaccharide of SE11 was found to be of R3 type in this study. The comparative analysis with the phylogenetically closest E24377A, an enterotoxigenic E. coli (ETEC) isolate, also revealed that both strains shared the highest number of 4112 orthologs, of which only 41 protein-coding genes were not found in other sequenced E. coli strains and may be regarded as group B1-specific genes (Supplementary Table S3). From these comparative analyses of E. coli genes, it was found that SE11 lacked genes homologous to known or suspected toxins and extracellular enzymes such as Shiga toxins, alpha-hemolysin and enterohemolysin that are involved in the virulence of pathogenic E. coli strains as well as genes for heat-labile and heat-stable enterotoxins encoded on the plasmids in ETEC E24377A.26
Figure 3.
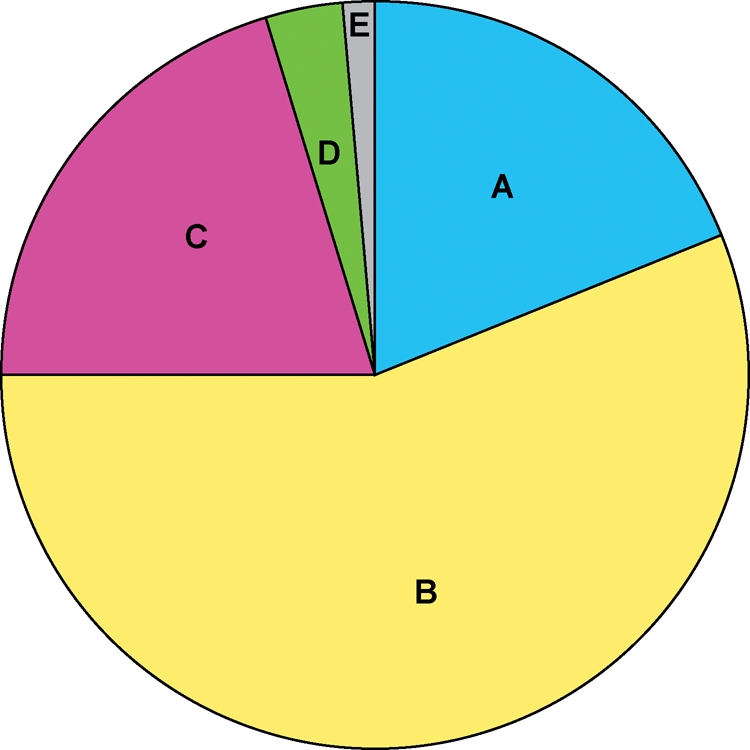
Classification of all 5002 protein-coding genes in SE11 based on comparison with those in MG1655 and 12 other E. coli strains. The 5002 protein-coding genes annotated in SE11 were compared with those in 13 other sequenced E. coli strains and classified into given categories with the percentage ratio. A: highly conserved genes with MG1655 (952); B: highly conserved genes in all 14 strains (2802); C: SE11 genes absent in MG1655 (1016); D: SE11-specific genes in all 14 E. coli strains (170); E: poorly conserved genes with MG1655 (62); A + B: total highly conserved genes with MG1655 (3754); C + D: total SE11 genes absent in MG1655 (1186). Number of classified genes are given in parentheses.
3.3. Prophages and integrative elements
In the SE11 chromosome, there are seven prophage regions (PP_SE11-1 to -7; 36–53 kb in length) and three integrative elements (IE_SE11-1 to -3; 7–33 kb in length) that contained an integrase gene but no genes for apparent phages, transposons and integrative conjugative elements. Many of these integrated regions were flanked by short sequence duplications that are hallmarks of the lateral transfer event (Table 2). Comparative analysis with MG1655 showed that the SE11 chromosome contained 47 regions (>5 kb) that are absent in MG1655 (Fig. 4). Of these additional regions in the SE11 chromosome, we identified nine large segments (>30 kb), eight of which overlapped with all seven prophage regions (PP_SE11-1 to -7) and one integrative element (IE_SE11-1) described earlier (Table 2). Only a large segment (ECSE_0213–0239) near the aspV tRNA gene contained no apparent integrase gene, phage-related gene, transposase gene, and direct repeat, and mostly encoded proteins with unknown function. Integrative elements corresponding to this large segment were also retained at the same loci in the chromosomes of E24377A and phylogenetically distant strain EHEC O157 and UPEC 536, suggesting that MG1655 might have lost this locus during evolution (Supplementary Fig. S2).
Table 2.
Prophages and integrative elements in the SE11 chromosome
| Start | End | Size (kb) | Integration site | Phage type | Sequence duplication (nt) | |
|---|---|---|---|---|---|---|
| Prophage | ||||||
| PP_SE11-1 | 608 272 | 658 623 | 50.4 | argU | Lambda-like | 47 |
| PP_SE11-2 | 1 475 327 | 1 527 950 | 52.6 | ttcA | lambda-like | 43 |
| PP_SE11-3 | 1 739 450 | 1 786 245 | 46.8 | between ydfJ and rspB | lambda-like | – |
| PP_SE11-4 | 1 928 706 | 1 964 507 | 35.8 | between btuC and ihfA | P2-like | 25 |
| PP_SE11-5 | 2 122 554 | 2 167 374 | 44.8 | yecE | Lambda-like | 11 |
| PP_SE11-6 | 2 261 390 | 2 309 029 | 47.6 | serU | Unclear | 77 |
| PP_SE11-7 | 2 660 266 | 2 696 947 | 36.7 | yfcI | Mu-like | 5 |
| Integrative element | ||||||
| IE_SE11-1 | 295 509 | 328 178 | 32.7 | thrW | – | 19 |
| IE_SE11-2 | 3 014 785 | 3 021 558 | 6.8 | ssrA | – | – |
| IE_SE11-3 | 4 769 885 | 4 786 593 | 16.7 | leuX | – | – |
Figure 4.

Locations and lengths of the strain-specific segments. Horizontal axis represents the MG1655 chromosome location and vertical axis shows lengths of the strain-specific segments (>5 kb) compared with the MG1655 chromosome. The positions of PP_SE11 and IE_SE11 are indicated in SE11. Prophages at the same locus as the large SE11-specific segments are indicated in O157. Positions of 10 prophages in MG1655 are shown at the top of the figure. The total length of the strain-specific segment (>5 kb) is indicated under each strain name.
The genomes of pathogenic E. coli strains contain many prophages and other genetic elements that are the major sources for genes encoding virulence factors, such as toxins, type III secretion systems (TTSS), and effector proteins secreted by the TTSS.47,48 We compared the highly conserved prophages in SE11, MG1655 and O157 Sakai to analyze differences in structure and gene contents. PP_SE11-1 exhibits structural features similar to those of lambda-like prophage Sp8 in O157 Sakai and lambda-like prophage DLP12 in MG1655 at the same integration loci, but both PP_SE11-1 and DLP12 lacked the virulence-related catalase gene in Sp8 (Fig. 5A). PP_SE11-1 and DLP12 share 19 genes including the nmpC gene encoding a porin protein that allows small metabolites such as sugars, ions and amino acids to permeate. In addition, PP_SE11-1 and PP_SE11-5 shared a 23 kb almost identical segment that contains the additional nmpC homolog, suggesting that a very recent duplication of these regions may have occurred in SE11 (data not shown). The integrated locus of PP_SE11-2 is the same as those of lambda-like prophage Sp10 in O157 Sakai and lambda-like prophage Rac in MG1655 (Fig. 5B). These three prophages share many conserved genes but genes for three TTSS effectors and a Cu/Zn-superoxide dismutase encoded by Sp10 were missing in PP_SE11-2 and Rac. PP_SE11-3 was also found to integrate at the same locus as those of lambda-like prophages Sp11–Sp12 in O157 Sakai and lambda-like prophage Qin in MG1655, but the genes for TTSS effectors and a transcriptional regulator (PchB) encoded in Sp11–Sp12 were missing in PP_SE11-3 and Qin (Fig. 5C). Taken together with the results obtained from the analysis of genes in SE11, these data further indicate that SE11 and MG1655 do not possess prophage-borne virulence-associated genes found in O157 Sakai, despite the high conservation of these integrated elements among the three evolutionarily distant strains (Supplementary Fig. S1). At present, it is unknown whether the ancestral E. coli acquired virulence genes by prophage integration and thereafter they have been retained in O157 Sakai and lost in SE11 or it acquired non-virulent prophages and thereafter O157 Sakai has independently acquired the virulence genes but SE11 has not.
Figure 5.

Comparisons of the genomic location of three SE11 prophages with the corresponding location of the related prophages of K-12 and O157 strains. Genomic organizations of PP_SE11-1 (A); PP_SE11-2 (B) and PP_SE11-3 (C). Genes and their orientations are depicted with arrows using the following colors: red, integrase genes; orange, phage-related genes; yellow, transposase genes; green, genes outside PP_SE11; blue, virulence-associated genes in O157; gray, genes in MG1655 and O157 conserved in SE11. Light blue bars indicate orthologous regions.
3.4. Plasmids
The six plasmids in SE11 encoded a total of 323 protein-coding genes (Table 1 and Fig. 2). Copy numbers of each plasmid in SE11 were estimated to be one copy for pSE11-1, pSE11-2, pSE11-3, and pSE11-4, and ∼2 copies for pSE11-5 and pSE11-6 by the number of sequence reads assembled in respective plasmids. Three small plasmids (pSE11-4, pSE11-5 and pSE11-6) were found to be cryptic. Four plasmids (pSE11-1, pSE11-2, pSE11-3 and pSE11-6) had the genes encoding replication protein. The replication proteins of pSE11-1, pSE11-2 and pSE11-3 showed the high sequence similarity to those of IncFII, ColV and F plasmids, respectively, and pSE11-6 had the replication protein 100% identical to that of the plasmid pSMS35_130 of E. coli SMS-3-5. Two plasmids (pSE11-4 and pSE11-5) have the genes for mobilization protein. Thus, the six plasmids found in SE11 are compatible in a cell. The pSE11-1 (100 021 bp) contained almost identical gene sets to those in the conjugate plasmid ColIb-P9 (93 399 bp, accession no. AB021078) except for several genes including tetracycline resistance genes tetR (ECSE_P1-0010) and tetA (ECSE_P1-0011). Both pSE11-1 and ColIb-P9 encoded the same set of genes for conjugational transfer (tra and trb genes), biogenesis of type IV pili (pil genes), and colicin Ib production and immunity. It has been reported that type IV pili encoded by IncI1 group plasmids of enteric bacteria (e.g. ColIb-P9) are required both for plasmid conjugation and adherence to host epithelial cells.49 The pSE11-2 (91 158 bp) is a conjugative plasmid containing the fimbrial operon (ECSE_P2-0001-0005) homologous to that of the F1 (Caf1) pili biogenesis whose genes are encoded on the virulence plasmid pMT1 in Yersinia pestis.50 The gene products encoded by the fimbrial operon in pSE11-2 and the caf1 operon in pMT1 showed 31–70% amino acid sequence identities. The pSE11-3 is a non-conjugative plasmid of 60 555 bp, and contained two chaperone-usher fimbrial operons. One is the fae operon encoding F4 (or K88) fimbriae (ECSE_P3-0031-0037), which was flanked by transposase genes. F4 fimbriae are the major colonization factors in some ETEC strains associated with porcine neonatal and postweaning diarrhea.51 The other fimbrial operon (ECSE_P3-0060-0066) showed no strong similarity to entries in public databases.
3.5. Genes for fimbriae and autotransporter in SE11
Three loci of chaperone-usher pathways and one operon of type IV pilus encoded on the SE11 plasmids were almost completely missing in other sequenced E. coli strains. Certain E. coli strains were shown to be fimbriated and conferred the ability to adhere to host intestinal cells by the presence of a plasmid encoding fimbrial genes.52 SE11 also contains at least 13 loci for the fimbrial biosynthesis on the chromosome, accounting for a total of 17 loci, many of which were missing or present as truncated forms in other sequenced E. coli genomes (Table 3). MG1655 lacked two of 13 chromosomal loci encoding the fimbrial biosynthesis in SE11. One of these two loci is the lpf operon (ECSE_4015–4018) for the synthesis of long polar fimbriae that are known to mediate bacterial cell adhesion to host epithelial cells.53 Of sequenced E. coli strains, E24377A, SMS-3-5 and O157 Sakai contained the lpf operon at the position between glmS and pstS. The lpf operon in SE11 is almost identical with 99–100% amino acid sequence identity of that in E24377A and divergent from those of SMS-3–5 (83–98%) and O157 (34–63%). Another fimbrial operon locus (ECSE_3375–3378) is similar to CS1 fimbriae that are a major colonization factor of some ETEC strains.54 The CS1-like fimbrial operon in SE11 is also conserved in E24377A, HS, ATCC 8739, UPEC strain 536 and SMS-3-5 with the sequence similarity of 71–100% amino acid sequence identities. Of the fimbrial operons conserved between SE11 and MG1655, only three genes (ECSE_2643–2645) in the yfc operon showed low similarities of 52–59% amino acid sequence identities between them, while the three genes showed 98–99% amino acid sequence identities with those of Shigella flexneri.
Table 3.
Genes for fimbriae in SE11
| Locus | Presence in:a |
|||||||
|---|---|---|---|---|---|---|---|---|
| MG1655 | E24377A | HS | O157 Sakai | CFT073 | UTI89 | 536 | ||
| Chromosome | ||||||||
| ECSE_0135–ECSE_0141 | yad | + | + | + | + | + | + | (+) |
| ECSE_0555–ECSE_0560 | sfm | + | + | + | + | − | − | − |
| ECSE_0775–ECSE_0778 | ybg | + | + | + | + | − | − | − |
| ECSE_0999–ECSE_1005 | ycb | + | + | + | + | − | − | − |
| ECSE_1099–ECSE_1106 | Curli | + | + | + | + | + | + | (+) |
| ECSE_1592–ECSE_1597 | yde | (+) | (+) | (+) | + | + | (+) | (+) |
| ECSE_2377–ECSE_2380 | yeh | + | + | + | (+) | + | + | + |
| ECSE_2643–ECSE_2648 | yfc | + | + | + | + | + | + | + |
| ECSE_3324–ECSE_3326 | yqi | + | + | + | − | + | + | − |
| ECSE_3375–ECSE_3378 | CS1-like | − | + | + | − | − | − | + |
| ECSE_3428–ECSE_3431 | yra | + | + | + | (+) | − | − | − |
| ECSE_4015–ECSE_4018 | lpf | − | + | − | (+) | − | − | − |
| ECSE_4585–ECSE_4593 | type 1 | + | (+) | + | + | + | + | + |
| Plasmid | ||||||||
| ECSE_P1-0108–ECSE_P1-0120 | type IV pili | − | (+) | − | − | − | − | − |
| ECSE_P2-0001–ECSE_P2-0005 | Caf-like | − | − | − | − | − | − | − |
| ECSE_P3-0031–ECSE_P3-0037 | F4-like | − | − | − | − | − | − | − |
| ECSE_P3-0061–ECSE_P3-0066 | − | − | − | − | − | − | − | |
a‘+’ indicates a locus where all genes are present; ‘−’ indicates a locus where all genes are absent; and ‘(+)’ indicates a locus where one or more genes, but not all, are absent or disrupted.
Several autotransporters such as E. coli AIDA-I and Ag43 are also known to have the function as fimbrial adhesions.55 Autotransporters are a large and diverse superfamily of proteins that are composed of an N-terminal variable passenger domain translocated across the membrane and a C-terminal beta domain. SE11 possesses at least eight genes encoding intact autotransporters (Table 4), of which five autotransporters (ECSE_1215, ECSE_1251, ECSE_1600, ECSE_2459 and ECSE_2494) contained the pertactin motif (Pfam PF03212) and thus may function as adhesins like pertactins of Bordetella.56 Other sequenced E. coli strains also possess these homologous genes in various combinations but encode many of them as pseudogenes or completely lacked (Table 4). For instance, MG1655 has six orthologous genes of the eight autotransporter genes encoded in SE11, but three of the six genes were fragmented and seemed to no longer function. O157 Sakai and three UPEC strains (CTF073, UTI89 and 536) possess only four autotransporter genes homologous to four intact genes (ECSE_0327, ECSE_0393, ECSE_2494 and ECSE_3884) in SE11. Absence of orthologs of three genes (ECSE_1215, ECSE_1251 and ECSE_1600) is common in the three strains (CTF073, UTI89 and 536) belonging to phylogenetic group B2, suggesting that these orthologous genes may have been lost only in the lineage to the B2 group after divergence of the ancestor of the B2 group from the common ancestral E. coli (see Supplementary Fig. S1). Relative abundance of autotransporter homologs was observed in ETEC E24377A and a commensal HS belonging to groups B1 and A, respectively, both of which possess six intact autotransporter genes. The orthologs of ECSE_3884 are widely conserved and distributed throughout E. coli and Shigella, and its passenger domain contains the short repeats (PF05658) and motifs (PF05662) found in hemagglutinins, suggesting that the autotransporter encoded by ECSE_3884 may also be involved in the mechanism of bacterial attachment to host cells.57 It is also noteworthy that SE11 contained no autotransporter that exports host-damaging proteins with the serine protease activity such as Sat and Pic produced by UPEC strains.58,59
Table 4.
Genes for autotransporter in SE11
| Locus | Length (aa) | Presence in:a |
||||||
|---|---|---|---|---|---|---|---|---|
| MG1655 | E24377A | HS | O157 Sakai | CFT073 | UTI89 | 536 | ||
| ECSE_0327 | 765 | − | −+ | (+) | + | + | + | |
| ECSE_0393 | 968 | (+) | + | (+) | + | + | + | + |
| ECSE_1215 | 773 | (+) | + | (+) | − | − | − | − |
| ECSE_1251 | 961 | + | + | + | (+) | − | − | − |
| ECSE_1600 | 1806 | (+) | (+) | + | (+) | − | − | − |
| ECSE_2459 | 761 | + | + | + | + | (+) | (+) | (+) |
| ECSE_2494 | 1244 | + | + | + | + | + | + | + |
| ECSE_3884 | 1616 | − | + | + | + | + | + | + |
a‘+’ indicates presence; ‘−’ indicates absence; and ‘(+)’ indicates presence of a truncated gene.
4. Discussion
From the detailed analysis of the genome sequence of the wild-type commensal strain SE11, we found that SE11 is notably abundant in the adhesion functions such as fimbriae and autotransporters that have been originally identified as virulence-associated functions in pathogenic E. coli strains.60,61 Although many of these adhesion-associated genes are also conserved in pathogenic E. coli strains, our data indicated that SE11 does not accompany other known virulence-associated genes found in the pathogenic strains. Furthermore, many of these adhesion-associated genes were encoded in the integrated regions on the chromosome and in the transmittable plasmids in SE11, indicating that they have been horizontally acquired in SE11. Lack of known virulence-associated genes in SE11 was also evident from the structural comparisons of several conserved prophages in SE11, MG1655 and O157 Sakai, showing that virulence-associated genes present in the prophages of O157 were completely missing in those of SE11 and MG1655, while other genes were retained. The SE11 plasmids also encoded many genes associated with bacterial conjugation. This feature may be advantageous for the efficient distribution of plasmids through cell–cell contacts in the gut environment with the high microbial density.5 These data suggest that the adhesion-associated genes are transferable genetic elements between E. coli and rather serve as a versatile function enhancing the ability of E. coli to colonize the gut. This notion is consistent with the recent finding that commensal and pathogenic E. coli strains use a common pilus adherence factor for the colonization.62
The comparison of SE11 with the laboratory-adapted strain MG1655 revealed that SE11 possessed more genes involved in the metabolism of carbohydrates as well as the genes for the adhesion than MG1655. These genes are associated with uptake of available nutrients, allowing E. coli to survive in the intestinal tract rich in oligo- and polysaccharides.63 The genomic features of SE11 shown here may indicate the consequence of adaptation of the commensal E. coli strain to human gut habitat.
Supplementary Data
Supplementary data are available online at www.dnaresearch.oxfordjournals.org.
Funding
This research was supported by Grants-in-Aid for Scientific Research on Priority Areas ‘Comprehensive Genomics’ (M.H.) and ‘Applied Genomics’ (T.H.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Supplementary Material
Acknowledgements
We thank K. Furuya, C. Yoshino, H. Inaba, K. Motomura, and Y. Hattori (University of Tokyo), A. Yamashita, A. Tamura, and N. Itoh (Kitasato University) and K. Hasebe (Azabu University) for technical assistance. We also thank the Institute for Genomic Research (TIGR) for their policy of making preliminary sequence data publicly available.
References
- 1.Dethlefsen L., McFall-Ngai M., Relman D. A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooper L. V., Gordon J. I. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 3.Eckburg P. B., Bik E. M., Bernstein C. N., et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gill S. R., Pop M., Deboy R. T., et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley R. E., Turnbaugh P. J., Klein S., Gordon J. I. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 6.Frank D. N., St Amand A. L., Feldman R. A., Boedeker E. C., Harpaz N., Pace N. R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurokawa K., Itoh T., Kuwahara T., et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnbaugh P. J., Ley R. E., Hamady M., Fraser-Liggett C. M., Knight R., Gordon J. I. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duriez P., Clermont O., Bonacorsi S., et al. Commensal Escherichia coli isolates are phylogenetically distributed among geographically distinct human populations. Microbiology. 2001;147:1671–1676. doi: 10.1099/00221287-147-6-1671. [DOI] [PubMed] [Google Scholar]
- 10.Oelschlaeger T. A., Dobrindt U., Hacker J. Pathogenicity islands of uropathogenic E. coli and the evolution of virulence. Int. J. Antimicrob. Agents. 2002;19:517–521. doi: 10.1016/s0924-8579(02)00092-4. [DOI] [PubMed] [Google Scholar]
- 11.Dobrindt U., Agerer F., Michaelis K., et al. Analysis of genome plasticity in pathogenic and commensal Escherichia coli isolates by use of DNA arrays. J. Bacteriol. 2003;185:1831–1840. doi: 10.1128/JB.185.6.1831-1840.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escobar-Paramo P., Clermont O., Blanc-Potard A. B., Bui H., Le Bouguenec C., Denamur E. A specific genetic background is required for acquisition and expression of virulence factors in Escherichia coli. Mol. Biol. Evol. 2004;21:1085–1094. doi: 10.1093/molbev/msh118. [DOI] [PubMed] [Google Scholar]
- 13.Fukiya S., Mizoguchi H., Tobe T., Mori H. Extensive genomic diversity in pathogenic Escherichia coli and Shigella strains revealed by comparative genomic hybridization microarray. J. Bacteriol. 2004;186:3911–3921. doi: 10.1128/JB.186.12.3911-3921.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrindt U. (Patho-)Genomics of Escherichia coli. Int. J. Med. Microbiol. 2005;295:357–371. doi: 10.1016/j.ijmm.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Hejnova J., Dobrindt U., Nemcova R., et al. Characterization of the flexible genome complement of the commensal Escherichia coli strain A0 34/86 (O83:K24:H31) Microbiology. 2005;151:385–398. doi: 10.1099/mic.0.27469-0. [DOI] [PubMed] [Google Scholar]
- 16.Chen Q., Savarino S. J., Venkatesan M. M. Subtractive hybridization and optical mapping of the enterotoxigenic Escherichia coli H10407 chromosome: isolation of unique sequences and demonstration of significant similarity to the chromosome of E. coli K-12. Microbiology. 2006;152:1041–1054. doi: 10.1099/mic.0.28648-0. [DOI] [PubMed] [Google Scholar]
- 17.Sabate M., Moreno E., Perez T., Andreu A., Prats G. Pathogenicity island markers in commensal and uropathogenic Escherichia coli isolates. Clin. Microbiol. Infect. 2006;12:880–886. doi: 10.1111/j.1469-0691.2006.01461.x. [DOI] [PubMed] [Google Scholar]
- 18.Ihssen J., Grasselli E., Bassin C., et al. Comparative genomic hybridization and physiological characterization of environmental isolates indicate that significant (eco-)physiological properties are highly conserved in the species Escherichia coli. Microbiology. 2007;153:2052–2066. doi: 10.1099/mic.0.2006/002006-0. [DOI] [PubMed] [Google Scholar]
- 19.Clermont O., Lescat M., O’Brien C. L., Gordon D. M., Tenaillon O., Denamur E. Evidence for a human-specific Escherichia coli clone. Environ. Microbiol. 2008;10:1000–1006. doi: 10.1111/j.1462-2920.2007.01520.x. [DOI] [PubMed] [Google Scholar]
- 20.Perna N. T., Plunkett G., III, Burland V., et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi T., Makino K., Ohnishi M., et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22. doi: 10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- 22.Welch R. A., Burland V., Plunkett G., III, et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl Acad. Sci. USA. 2002;99:17020–17024. doi: 10.1073/pnas.252529799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brzuszkiewicz E., Bruggemann H., Liesegang H., et al. How to become a uropathogen: comparative genomic analysis of extraintestinal pathogenic Escherichia coli strains. Proc. Natl Acad. Sci. USA. 2006;103:12879–12884. doi: 10.1073/pnas.0603038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S. L., Hung C. S., Xu J., et al. Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc. Natl Acad. Sci. USA. 2006;103:5977–5982. doi: 10.1073/pnas.0600938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson T. J., Kariyawasam S., Wannemuehler Y., et al. The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J. Bacteriol. 2007;189:3228–3236. doi: 10.1128/JB.01726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rasko D. A., Rosovitz M. J., Myers G. S., et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blattner F. R., Plunkett G., III, Bloch C. A., et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi K., Morooka N., Yamamoto Y., et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100049. 2006.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herring C. D., Palsson B. Ø. An evaluation of comparative genome sequencing (CGS) by comparing two previously-sequenced bacterial genomes. BMC Genomics. 2007;8:274. doi: 10.1186/1471-2164-8-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durfee T., Nelson R., Baldwin S., et al. The complete genome sequence of Escherichia coli DH10B: insights into the biology of a laboratory workhorse. J. Bacteriol. 2008;190:2597–2606. doi: 10.1128/JB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith H. W. Survival of orally administered E. coli K-12 in alimentary tract of man. Nature. 1975;255:500–502. doi: 10.1038/255500a0. [DOI] [PubMed] [Google Scholar]
- 32.Lerat E., Ochman H. Psi-Phi: exploring the outer limits of bacterial pseudogenes. Genome Res. 2004;14:2273–2278. doi: 10.1101/gr.2925604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobman J. L., Penn C. W., Pallen M. J. Laboratory strains of Escherichia coli: model citizens or deceitful delinquents growing old disgracefully? Mol. Microbiol. 2007;64:881–885. doi: 10.1111/j.1365-2958.2007.05710.x. [DOI] [PubMed] [Google Scholar]
- 34.Grozdanov L., Raasch C., Schulze J., et al. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J. Bacteriol. 2004;186:5432–5441. doi: 10.1128/JB.186.16.5432-5441.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun J., Gunzer F., Westendorf A. M., et al. Genomic peculiarity of coding sequences and metabolic potential of probiotic Escherichia coli strain Nissle 1917 inferred from raw genome data. J. Biotechnol. 2005;117:147–161. doi: 10.1016/j.jbiotec.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 36.Ochman H., Selander R. K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984;157:690–693. doi: 10.1128/jb.157.2.690-693.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Venieri D., Vantarakis A., Komninou G., Papapetropoulou M. Differentiation of faecal Escherichia coli from human and animal sources by random amplified polymorphic DNA-PCR (RAPD-PCR) Water Sci. Technol. 2004;50:193–198. [PubMed] [Google Scholar]
- 38.Gordon D., Desmarais C., Green P. Automated finishing with autofinish. Genome Res. 2001;11:614–625. doi: 10.1101/gr.171401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delcher A. L., Harmon D., Kasif S., White O., Salzberg S. L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowe T. M., Eddy S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasko D. A., Myers G. S., Ravel J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics. 2005;6:2. doi: 10.1186/1471-2105-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wirth T., Falush D., Lan R., et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 2006;60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Picard B., Garcia J. S., Gouriou S., et al. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 1999;67:546–553. doi: 10.1128/iai.67.2.546-553.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olsson U., Lycknert K., Stenutz R., Weintraub A., Widmalm G. Structural analysis of the O-antigen polysaccharide from Escherichia coli O152. Carbohydr. Res. 2005;340:167–171. doi: 10.1016/j.carres.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 45.Stenutz R., Weintraub A., Widmalm G. The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol. Rev. 2006;30:382–403. doi: 10.1111/j.1574-6976.2006.00016.x. [DOI] [PubMed] [Google Scholar]
- 46.Raetz C. R., Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tobe T., Beatson S. A., Taniguchi H., et al. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl Acad. Sci. USA. 2006;103:14941–14946. doi: 10.1073/pnas.0604891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayashi T., Ooka T., Ogura Y., Asadulghani Md. Evolutionary Biology of Bacterial and Fungal Pathogens. ASM press; 2008. pp. 407–419. [Google Scholar]
- 49.Dudley E. G., Abe C., Ghigo J. M., Latour-Lambert P., Hormazabal J. C., Nataro J. P. An IncI1 plasmid contributes to the adherence of the atypical enteroaggregative Escherichia coli strain C1096 to cultured cells and abiotic surfaces. Infect. Immun. 2006;74:2102–2114. doi: 10.1128/IAI.74.4.2102-2114.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu P., Elliott J., McCready P., et al. Structural organization of virulence-associated plasmids of Yersinia pestis. J. Bacteriol. 1998;180:5192–5202. doi: 10.1128/jb.180.19.5192-5202.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaper J. B., Nataro J. P., Mobley H. L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 52.Karch H., Heesemann J., Laufs R., O’Brien A. D., Tacket C., Levine M. M. A plasmid of enterohemorrhagic Escherichia coli O157:H7 is required for expression of a new fimbrial antigen and for adhesion to epithelial cells. Infect. Immun. 1987;55:455–461. doi: 10.1128/iai.55.2.455-461.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ideses D., Biran D., Gophna U., Levy-Nissenbaum O., Ron E. Z. The lpf operon of invasive Escherichia coli. Int. J. Med. Microbiol. 2005;295:227–236. doi: 10.1016/j.ijmm.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 54.Perez-Casal J., Swartley J. S., Scott J. R. Gene encoding the major subunit of CS1 pili of human enterotoxigenic Escherichia coli. Infect. Immun. 1990;58:3594–3600. doi: 10.1128/iai.58.11.3594-3600.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henderson I. R., Nataro J. P. Virulence functions of autotransporter proteins. Infect. Immun. 2001;69:1231–1243. doi: 10.1128/IAI.69.3.1231-1243.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J., Fairweather N. F., Novotny P., Dougan G., Charles I. G. Cloning, nucleotide sequence and heterologous expression of the protective outer-membrane protein P.68 pertactin from Bordetella bronchiseptica. J. Gen. Microbiol. 1992;138:1697–1705. doi: 10.1099/00221287-138-8-1697. [DOI] [PubMed] [Google Scholar]
- 57.Johnson J. R. Virulence factors in Escherichia coli urinary tract infection. Clin. Microbiol. Rev. 1991;4:80–128. doi: 10.1128/cmr.4.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guyer D. M., Henderson I. R., Nataro J. P., Mobley H. L. Identification of Sat, an autotransporter toxin produced by uropathogenic Escherichia coli. Mol. Microbiol. 2000;38:53–66. doi: 10.1046/j.1365-2958.2000.02110.x. [DOI] [PubMed] [Google Scholar]
- 59.Heimer S. R., Rasko D. A., Lockatell C. V., Johnson D. E., Mobley H. L. Autotransporter genes pic and tsh are associated with Escherichia coli strains that cause acute pyelonephritis and are expressed during urinary tract infection. Infect. Immun. 2004;72:593–597. doi: 10.1128/IAI.72.1.593-597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Connell I., Agace W., Klemm P., Schembri M., Mărild S., Svanborg C. Type 1 fimbrial expression enhances Escherichia coli virulence for the urinary tract. Proc. Natl Acad. Sci. USA. 1996;93:9827–9832. doi: 10.1073/pnas.93.18.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pizarro-Cerdá J., Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124:715–727. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 62.Rendón M. A., Saldaña Z., Erdem A. L., et al. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proc. Natl Acad. Sci. USA. 2007;104:10637–10642. doi: 10.1073/pnas.0704104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flint H. J., Bayer E. A., Rincon M. T., Lamed R., White B. A. Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nature Rev. Microbiol. 2008;6:121–131. doi: 10.1038/nrmicro1817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.