

Abstract
Synonymous codon usage varies both between organisms and among genes within a genome, and arises due to differences in G + C content, replication strand skew, or gene expression levels. Correspondence analysis (CA) is widely used to identify major sources of variation in synonymous codon usage among genes and provides a way to identify horizontally transferred or highly expressed genes. Four methods of CA have been developed based on three kinds of input data: absolute codon frequency, relative codon frequency, and relative synonymous codon usage (RSCU) as well as within-group CA (WCA). Although different CA methods have been used in the past, no comprehensive comparative study has been performed to evaluate their effectiveness. Here, the four CA methods were evaluated by applying them to 241 bacterial genome sequences. The results indicate that WCA is more effective than the other three methods in generating axes that reflect variations in synonymous codon usage. Furthermore, WCA reveals sources that were previously unnoticed in some genomes; e.g. synonymous codon usage related to replication strand skew was detected in Rickettsia prowazekii. Though CA based on RSCU is widely used, our evaluation indicates that this method does not perform as well as WCA.
Key words: correspondence analysis, synonymous codon usage, horizontal gene transfer, strand-specific mutational bias, translational selection
1. Introduction
Most amino acids are encoded by more than one codon, and these synonymous codons usually differ by one nucleotide in the third position. Generally, alternative synonymous codons are not used with equal frequency; their usage varies among different species, and often among genes within the same genome.1 Three principal factors have been proposed to account for the intragenomic variation in synonymous codon usage. First, intragenomic variation in G + C content is mostly related to the existence of regions with unusual base composition, so-called genomic islands, that may be the result of recent horizontal DNA transfer.2–4 Secondly, the excess of G over C in the leading strand of DNA replication relative to the lagging strand is observed in many bacteria, and this is thought to reflect strand-specific mutational bias.5,6 Thirdly, genes expressed at high levels in fast-growing bacteria tend to preferentially use translationally optimal codons that are recognized by the most abundant tRNAs. This presumably reflects natural selection for synonymous codons that are translated more efficiently and accurately.7,8 Thus, the use of synonymous codons in any gene can be the result of a mixture of these different evolutionary factors, and their relative contributions may vary among different species depending on their life history.9–11 It follows that information on synonymous codon usage can be used to identify certain kinds of genes, e.g. those that have been horizontally transferred12–14 or are highly expressed.15–18
To reliably detect and quantify synonymous codon usage patterns, it is necessary to employ appropriate statistical methods. One such method is correspondence analysis (CA), a multivariate statistical method that can be used to summarize high dimensional data, such as codon counts, by reducing them to a limited number of variables, called axes.19,20 The axes retain much of the information about the variability in codon usage among the genes, but in a way that makes those differences easier to understand. This method is widely used to identify major sources of variation in synonymous codon usage among genes.
A common issue in synonymous codon usage analysis is that variation in amino acid composition among proteins is a confounding factor in assessing variation in synonymous codon usage among nucleotide sequences. Different approaches have been taken to remove such amino acid composition effects. Most commonly, CA is performed on modified codon usage data that have been adjusted for the frequency of the amino acids they encode. The resulting relative codon frequency (RF) and relative synonymous codon usage (RSCU) are used instead of the original codon count data, which is also referred to as the absolute codon frequency (AF). However, previous studies showed that for some genomes the use of RF and RSCU to remove amino acid composition effects introduced a bias associated with the low frequency of cysteine in proteins.21,22 To validate findings, some researchers compared the results of CA using different input data (termed here CA-AF, CA-RF, and CA-RSCU).21,23,24 The within-group CA (WCA) has been proposed as an alternative method to dissociate the effects of different amino acid compositions from the effects directly related to synonymous codon usage.25 This method adjusts the value for each codon by the average value of all the codons encoding for the same amino acid using a different method than CA-RF or CA-RSCU. These four different CA methods have all been used for studying synonymous codon usage, but it remains unclear which one is the most effective. In spite of the lack of rigorous testing, CA-RSCU remains the most popular method.26–37
In this paper, we have evaluated and compared four CA methods for the analysis of synonymous codon usage (CA-AF, CA-RF, CA-RSCU and WCA) by applying them to 241 bacterial genomes for which complete genome sequences were available. Our results indicate that WCA is more effective than the other three methods in generating axes corresponding to variation in synonymous codon usage.
2. Materials and methods
2.1. Sequences
Complete genome sequences of bacterial species in GenBank format38 were retrieved from the NCBI FTP site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria). In the case of species for which multiple strains have been sequenced, only one representative was randomly selected. An exception was made for the genomes of the following 10 strains, which were specifically selected as species representatives because they have been previously analyzed by CA: Borrelia burgdorferi B31 (B. burgdorferi B31),21,39,40 Chlamydia trachomatis D/UW-3/CX (C. trachomatis D/UW-3/CX),41 Clostridium perfringens 13 (C. perfringens 13),42 Escherichia coli K12 MG1655 (E. coli K12 MG1655),21,23,43 Haemophilus influenzae Rd KW20 (H. influenzae Rd KW20),44 Helicobacter pylori 26695 (H. pylori 26695),45 Mycoplasma genitalium G37 (M. genitalium G37),21,46 Rickettsia prowazekii Madrid E (R. prowazekii Madrid E),47 Thermotoga maritima MSB8 (T. maritima MSB8)22 and Treponema pallidum Nichols (T. pallidum Nichols).39 Moreover, genomes were excluded when genes used in the analysis (Section 2.4) were missing. The final data set included 241 genomes (see Supplementary Table S1 or S2 for a comprehensive list). All protein-coding sequences, except those containing letters other than A, C, G, or T were included in the analysis. Because methionine and tryptophan are generally encoded by only a single codon, the codons for methionine and tryptophan were excluded. Start and stop codons were also eliminated.
2.2. Definitions of codon usage data
We computed original codon count data, i.e. the AF, and two kinds of modified codon usage data that have been normalized for each individual amino acid. The latter included the RF, which is defined as the ratio of the number of occurrences of a codon to the sum of all synonymous codons21,48 and the RSCU, which is defined as the ratio of the observed number of occurrences of a codon to the number expected if all synonymous codons were used with equal frequency.49 The values of AF, RF and RSCU of the cth codon for the ath amino acid (AFac, RFac, and RSCUac, respectively) were calculated as follows:
| 1 |
| 2 |
| 3 |
where nac is the number of occurrences of the cth codon for the ath amino acid, and da the degree of codon degeneracy for the ath amino acid. RFac equals 1/da (e.g. 1/2 for cysteine and 1/6 for arginine) when alternative synonymous codons are used with equal frequency, and reaches the maximum value of 1 when only one of synonymous codons is used and all others are not present with value of 0. RSCUac equals 1 when alternative synonymous codons are used with equal frequency, and attains its maximum value of da (e.g. 2 for cysteine and 6 for arginine) when only one of synonymous codons is used for the amino acid.
2.3. Implementation of CA
CA was implemented using the ‘dudi.coa’ and ‘within’ functions in the ‘ade4’50 library of R.51 CA takes multivariate data and combines them into a small number of variables (axes) that explains most of the variation among the original variables.19,21,25 In our study our variables are the 59 codons for each gene in a genome, and the result of the CA yields the coordinates of each gene on each new axis. A matrix is created in which the rows correspond to the genes on one bacterial genome and the columns to the 59 codons, such that each row has the codon usage information for a specific gene. For the different CA methods, CA-AF, CA-RF, CA-RSCU, or WCA, the cells contain AF, RF, RSCU, or AF values, respectively, for each gene and codon.
We provide a brief explanation of our implementation of CA for analyzing synonymous codon usage. For each genome, the matrix X = [xij] is an input data table with N genes (rows) and 59 codons (columns). We denote the sum of values for the ith gene of X as xi+ and the jth codon as x+j. We denote the sum of all of the data in X as x++. The weight of the ith gene is defined as pi+ = xi+/x++, that of the jth codon is defined as p+j = x+j/x++. The matrix Y has elements 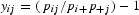 where pij is the weight of each cell pij = xij/x++. The matrix Y for WCA is obtained by replacing the elements yij in the matrix Y for CA-AF by
where pij is the weight of each cell pij = xij/x++. The matrix Y for WCA is obtained by replacing the elements yij in the matrix Y for CA-AF by 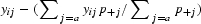 , where the sum extends over all codons j encoding amino acid a. This subtraction centers the data in each cell based upon the value of the codons that encode a particular amino acid. In other words, the yij values for WCA become the difference between the yij values for CA-AF and their adjusted average.
, where the sum extends over all codons j encoding amino acid a. This subtraction centers the data in each cell based upon the value of the codons that encode a particular amino acid. In other words, the yij values for WCA become the difference between the yij values for CA-AF and their adjusted average.
The matrix Z with elements 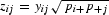 is submitted to singular value decomposition, producing three matrices: Z = USVt. S is a diagonal matrix whose diagonal elements sk are singular values, the matrices U and V have elements uik and vjk, respectively (the superscript t is the transposition operator). The coordinates for the ith gene or the jth codon in the kth axis (gik and cjk, respectively) are calculated as follows:
is submitted to singular value decomposition, producing three matrices: Z = USVt. S is a diagonal matrix whose diagonal elements sk are singular values, the matrices U and V have elements uik and vjk, respectively (the superscript t is the transposition operator). The coordinates for the ith gene or the jth codon in the kth axis (gik and cjk, respectively) are calculated as follows:
| 4 |
| 5 |
The gik scores are the values that are correlated with other gene features in the subsequent analyses (see Section 2.4).
The contribution of the jth codon to the kth axis is given by 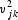 . The sum of the contributions of all 59 codons to each axis is one; that is,
. The sum of the contributions of all 59 codons to each axis is one; that is, 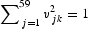 . We compared the sum of the contributions of 18 codons with twofold degeneracy (those coding for asparagine, aspartic acid, cysteine, glutamic acid, glutamine, histidine, lysine, phenylalanine, and tyrosine) and the sum of the contributions of 18 codons with sixfold degeneracy (those coding arginine, leucine, and serine).
. We compared the sum of the contributions of 18 codons with twofold degeneracy (those coding for asparagine, aspartic acid, cysteine, glutamic acid, glutamine, histidine, lysine, phenylalanine, and tyrosine) and the sum of the contributions of 18 codons with sixfold degeneracy (those coding arginine, leucine, and serine).
Supplementary Table S1 shows the percentage of total variance explained by the first 10 axes, as generated by these four CA methods for 241 bacterial genomes. Because the percentage of variance explained by axes >3 was small overall, our subsequent analyses were focused on the first three axes.
2.4. Interpretation of axes generated by CA
To identify major sources of variation among genes on the axes generated by CA of codon usage data, we conducted two analyses that considered four commonly used features of protein-coding genes: GRAVY, GC3content, GC3skew, and Expression.22,52 First, we tested for the correlation between scores of each of three axes [Equation (4)] and values of GRAVY, GC3content, or GC3skew. GRAVY is the mean of the sum of the hydropathic index of each amino acid in the protein, and thus reflects amino acid composition.53 GC3content is the relative frequency of guanine and cytosine, (G + C)/(A + T + G + C), at the third codon position in the nucleotide sequence, and GC3skew is the deviation from equal amounts of guanine and cytosine, (G − C)/(G + C), at the third codon position in the nucleotide sequence. Pearson’s product moment correlation coefficient (r) between the axis scores and gene feature values was calculated. The square of r measures the percentage of variance; e.g. the square of 0.70 indicates that 49% of the variance in the axis scores is explained by the variance in the gene feature values. For each axis, the gene feature with an absolute r value (|r|) >0.70 was identified as the main source of variation among genes on the axis. At lower threshold |r| values, different gene features were detected on the same axis and/or the same gene feature was detected on more than one axis, and thus the interpretation of the axes becomes quite difficult. Additionally, low |r| values may be statistically significantly different from zero due to very large sample sizes, but weak correlations may have no biological meaning.
Secondly, to analyze the correlation between scores of each of the three axes [Equation (4)] and levels of gene expression (Expression), we tested for the distribution of the axis scores for 40 genes expected to be expressed constitutively at high levels.10 This set included the genes encoding translation elongation factors Tu (tuf), Ts (tsf) and G (fus), and 37 of the larger ribosomal proteins (encoded by genes rplA-rplF, rplI-rplT, and rpsB-rpsT). In each axis, the score for each gene was standardized by subtracting the mean and dividing by the standard deviation of scores for all protein genes. For each axis, Expression was detected as the main source of variation among genes on the axis when the mean absolute standard score for the 40 highly expressed genes was >1.644854 (an interval in which theoretically only 5% of all protein genes are included).
3. Results and discussion
3.1. Performance of different CA methods
CA summarizes high dimensional data, such as codon counts, by reducing them to a limited number of variables (axes). We tested the ability of the four CA methods, CA-AF, CA-RF, CA-RSCU, and WCA, to generate axes that correspond to variation in synonymous codon usage. We considered two commonly used gene features: GC3content is the G + C content at the third codon position, and GC3skew that reflects the bias in G over C content at the third codon position. We investigated how often these two gene features were correlated with one of the first three axes in 241 bacterial genomes (Table 1). To illustrate our method, Fig. 1 shows scatter plots of axis 1 scores obtained by the four methods, plotted against GC3skew for R. prowazekii Madrid E genes. At the threshold |r| value of 0.70, GC3skew values were significantly correlated with axis 1 scores of WCA (|r| = 0.84), but not with those of CA-AF (|r| = 0.46), CA-RF (|r| = 0.32), and CA-RSCU (|r| = 0.04). Thus, in R. prowazekii Madrid E, GC3skew was detected on axis 1 of WCA, but not on axis 1 of CA-AF, CA-RF, and CA-RSCU. GC3content was detected in 191 genomes when the WCA method was used, which was more than when CA-AF (150), CA-RF (143), or CA-RSCU (145) were used (Table 1A). Likewise, the total number of genomes where GC3skew was detected (108) was also greater when WCA was used than when CA-AF (46), CA-RF (30), and CA-RSCU (53) were used (Table 1B). Thus, WCA detected GC3content and GC3skew more often than CA-AF, CA-RF, and CA-RSCU.
Table 1.
Numbers of genomes where the gene feature GC3content, GC3skew, or GRAVY was significantly correlated with one of three axes generated by different CA methods, CA-AF, CA-RF, CA-RSCU, and WCA, in 241 bacterial genomes
| Method | Axis 1 | Axis 2 | Axis 3 |
|---|---|---|---|
| A. GC3content | |||
| CA-AF | 121 | 17 | 12 |
| CA-RF | 129 | 9 | 5 |
| CA-RSCU | 134 | 11 | 0 |
| WCA | 150 | 34 | 7 |
| B. GC3skew | |||
| CA-AF | 26 | 7 | 13 |
| CA-RF | 26 | 4 | 0 |
| CA-RSCU | 25 | 25 | 3 |
| WCA | 38 | 57 | 13 |
| C. GRAVY | |||
| CA-AF | 20 | 69 | 55 |
| CA-RF | 0 | 0 | 0 |
| CA-RSCU | 0 | 0 | 0 |
| WCA | 0 | 0 | 0 |
Figure 1.

Scatter plot showing axis 1 scores obtained by different CA methods, CA-AF (A), CA-RF (B), CA-RSCU (C), and WCA (D), plotted against GC3skew for R. prowazekii Madrid E genes. Each point represents a gene.
It is important to note that these results remained similar when all complete bacterial genomic sequences available from the NCBI repository on August 2008 were included (data not shown). Similar results were obtained when only long sequences with >300 codons were used (data not shown). We also verified the consistency of the results when using detection thresholds below |r| = 0.70 (data not shown). Thus we conclude that WCA is more effective than the other three methods in generating axes that correspond to variation in synonymous codon usage, regardless of the data sets and statistical criteria used.
WCA may have performed best because it does not mask variation in synonymous codon usage caused by amino acid composition and codon degeneracy. CA-AF may have performed worse because it is confounded by amino acid composition. CA-RF and CA-RSCU did not perform as well as WCA possibly because their input data depend on the degree of codon degeneracy, which differs among amino acids [da in Equations (2) and (3) in Section 2.2].54 Later, we demonstrate these effects on the four CA methods.
3.2. Effect of amino acid composition and codon degeneracy in different CA methods
To determine the effect of amino acid composition, we tested the ability of the four CA methods, CA-AF, CA-RF, CA-RSCU, and WCA, to generate axes that correspond to variation in amino acid composition. The protein feature GRAVY, which represents the global hydrophobicity of proteins, can be used to measure the variation in amino acid composition among proteins.55 We investigated how often GRAVY was correlated with one of the first three axes in 241 bacterial genomes. CA-AF detected the correlation between GRAVY and one of the first three axes in 144 genomes, whereas CA-RF, CA-RSCU, and WCA did not detect it (Table 1C). This result suggests that CA-AF can generate axes corresponding to variation in amino acid composition as well as synonymous codon usage, whereas CA-RF, CA-RSCU, and WCA never generate such axes because they compensate for differences in amino acid composition.
The use of RF and RSCU to remove the confounding effects of amino acid composition introduces other effects associated with the degree of codon degeneracy, which may be pronounced for rare amino acids. To determine the effect of the difference in the degree of codon degeneracy between amino acids, we compared the contributions to axis 1 of nine amino acids with low (twofold) degeneracy and three amino acids with high (sixfold) degeneracy, totaling 18 codons each. This was done for the four CA methods, CA-AF, CA-RF, CA-RSCU, and WCA. Fig. 2 shows scatter plots of the contribution of twofold degenerate codons (y-axis) plotted against that of sixfold degenerate codons (x-axis) for 241 bacterial genomes. The scatter plots for CA-AF and WCA (Fig. 2A and D) displayed genome distributions less biased toward twofold or sixfold degenerate codons than the scatter plots for CA-RF and CA-RSCU (Fig. 2B and C). For CA-RF, 208 (86%) of the 241 genomes fell above the line y = x, indicating that twofold degenerate codons contributed more to the axis than sixfold degenerate codons in most genomes (Fig. 2B). For CA-RSCU, 238 (99%) of the 241 genomes were below the line y = x, indicating that sixfold degenerate codons contributed more to the axis than twofold degenerate codons in most genomes (Fig. 2C). Thus, CA-RF and CA-RSCU tend to generate axes corresponding to variation in low (twofold) and high (sixfold) degenerate codons, respectively. This observation can be explained by the dependence of their input data on the degree of codon degeneracy [da in Equations (2) and (3) in Section 2.2]. Thus, the use of RF and RSCU to remove effects of amino acid usage introduces other effects associated with the degree of codon degeneracy, whereas WCA does not. In spite of these shortcomings, these methods, in particular CA-RSCU, are still frequently used.26–37 We recommend using WCA for analyzing synonymous codon usage.
Figure 2.

Contributions of twofold and sixfold degenerate codons to axis 1, obtained by different CA methods, CA-AF (A), CA-RF (B), CA-RSCU (C), and WCA (D), for 241 bacterial genomes. Each point represents a genome.
3.3. Sources of intragenomic variation in synonymous codon usage among genes
We applied WCA to the genomes of 241 bacterial species to identify major sources of intragenomic variation in synonymous codon usage among genes. In addition to the two gene features described earlier (GC3content and GC3skew), gene expression level (Expression) was also considered. In 57 genomes, WCA detected one of the three gene features, GC3content, GC3skew, and Expression on axis 1 but none of the features on axes 2 and 3 (Supplementary Table S2). In 97 other genomes, WCA detected two of the three gene features on axes 1 and 2 but none of the gene features on axis 3. All three features were detected on the first three axes of 40 genomes, and only in nine genomes were no gene features detected on the first three axes. The results demonstrate that the three gene features can contribute to intragenomic variations in synonymous codon usage among genes, and that their relative contributions vary among different genomes.
CA of codon usage data generated axes on which no gene feature was detected. There are three possible explanations for this observation. First, in some cases, the axis was moderately correlated with one of the gene features considered here, but the correlation was not strong enough to reach the detection threshold. For example in Shewanella putrefaciens CN-32, the |r| value between axis 1 of WCA and GC3content (0.68) was below the threshold |r| value of 0.70. Secondly, although the axis was not correlated with any of the gene features considered here, it may be correlated with other relevant gene features that can be determined computationally or experimentally; e.g. protein abundance56 and mRNA half-life.57 Thirdly, variation among genes on the axis, even if the axis accounts for the largest fractions of the total variation among genes, may have no biological meaning. These possibilities should be kept in mind when interpreting the axes generated by CA of codon usage data.
For 10 genomes in our study that were previously analyzed by CA (Table 2), we compared our findings with previous conclusions. First, GC3content was detected as a primary source of synonymous codon usage variation among genes in E. coli K12 MG1655, M. genitalium G37, T. maritima MSB8, and H. pylori 26695. G + C content was previously detected in these first three genomes (previous analysis for H. pylori is not directly comparable). Intragenomic variation in G + C content mostly reflects the existence of regions with anomalous nucleotide composition, putatively acquired by horizontal transfer.2 The exception to this is M. genitalium, in which intragenomic G + C variation is continuous along the genome.58 Thus if the WCA axis clearly separates anomalous gene clusters from other genes, the axis scores can be used to predict genes that have recently transferred.
Table 2.
Gene features that are significantly correlated with one of three axes generated by WCA in 10 bacterial genomes
| Bacterial strain | Axis 1 | Axis 2 | Axis 3 | Referencesa |
|---|---|---|---|---|
| B. burgdorferi B31 | GC3skew | ndb | nd | 21,39,40 |
| C. trachomatis D/UW-3/CX | GC3skew | Expression | nd | 41 |
| C. perfringens 13 | Expression | nd | nd | 42 |
| E. coli K12 MG1655 | GC3content | Expression | nd | 21,23,43 |
| H. influenzae Rd KW20 | Expression | GC3content | nd | 44 |
| H. pylori 26695 | GC3content | GC3skew | nd | 45 |
| M. genitalium G37 | GC3content | nd | nd | 21,46 |
| R. prowazekii Madrid E | GC3skew | GC3content | nd | 47 |
| T. maritima MSB8 | GC3content | nd | nd | 22 |
| T. pallidum Nichols | GC3skew | GC3content | nd | 39 |
aPrevious studies, whose results do not necessarily agree with those shown here. See Section 3.3 for conflicts.
bnd, none of the gene features considered here were detected.
The second feature, GC3skew was detected as a primary source of synonymous codon usage variation among genes in B. burgdorferi B31, C. trachomatis D/UW-3/CX, R. prowazekii Madrid E, and T. pallidum Nichols (Table 2 and Fig. 1). Intragenomic variation in GC3skew presumably reflects differences in mutational bias between the leading and lagging strands of replication.5,6 This mutational bias was previously detected in each of these genomes, except R. prowazekii.47 Thus in genomes where GC3skew is detected on axis 1 of WCA, the axis scores can be used to predict whether the gene is located on the leading or lagging strands.
The third feature, Expression, was detected as a major source of synonymous codon usage variation among genes in C. trachomatis D/UW-3/CX, C. perfringens 13, E. coli K12 MG1655 and H. influenzae Rd KW20, which is consistent with previous findings (Table 2). The relative contribution of Expression varies among different genomes; e.g. Expression is a primary source in H. influenzae, while it is a secondary source in E. coli. The anomalous codon usage of highly expressed genes presumably reflects natural selection for optimal codons that are translated more efficiently and accurately; so-called translational selection.7,8 In B. burgdorferi and M. genitalium, conflicting conclusions regarding the presence or absence of translational selection on synonymous codon usage have been reported.21 In the present analysis, Expression was not detected in these two genomes, suggesting there is no evidence for translational selection. This is in agreement with conclusions drawn using a different statistical method.10 Thus in genomes where Expression is detected by WCA, the axis scores can be used to predict gene expression level and compared with experimental expression data obtained by DNA microarray (transcriptomes) and 2D gel electrophoresis (proteomes).
3.4. Conclusion
Of the four CA methods, WCA was found to be most useful for the analysis of synonymous codon usage. Using WCA, it may be possible to find new factors that can explain variation in synonymous codon usage among genes, and improve the accuracy of identifying genes that have been horizontally transferred or are highly expressed.
4. Availability
All analyses are implemented using G-language Genome Analysis Environment version 1.8.3,59,60 available at http://www.g-language.org/.
Supplementary Data
Supplementary data are available online at www.dnaresearch.oxfordjournals.org.
Funding
This project was supported by the Microbial Genome Sequencing Program of the National Science Foundation (EF-0627988), and by the National Institutes of Health grant R01 GM073821 from the National Institute of General Medical Sciences, and COBRE and INBRE grants P20RR016454 and P20RR16448 from the National Center for Research Resources, National Institutes of Health.
Supplementary Material
Acknowledgements
We thank Kazuharu Arakawa (Institute for Advanced Biosciences, Keio University) for his technical advice on the G-language Genome Analysis Environment and Christopher J. Williams (Department of Statistics, University of Idaho) for statistical advice.
References
- 1.Sharp P. M., Cowe E., Higgins D. G., Shields D. C., Wolfe K. H., Wright F. Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res. 1988;16:8207–8211. doi: 10.1093/nar/16.17.8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karlin S. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 2001;9:335–343. doi: 10.1016/s0966-842x(01)02079-0. [DOI] [PubMed] [Google Scholar]
- 3.Lawrence J. G., Ochman H. Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- 4.Ochman H., Lawrence J. G., Groisman E. A. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 5.Lobry J. R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- 6.McLean M. J., Wolfe K. H., Devine K. M. Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes. J. Mol. Evol. 1998;47:691–696. doi: 10.1007/pl00006428. [DOI] [PubMed] [Google Scholar]
- 7.Eyre-Walker A. Synonymous codon bias is related to gene length in Escherichia coli: selection for translational accuracy? Mol Biol Evol. 1996;13:864–872. doi: 10.1093/oxfordjournals.molbev.a025646. [DOI] [PubMed] [Google Scholar]
- 8.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 9.Carbone A., Kepes F., Zinovyev A. Codon bias signatures, organization of microorganisms in codon space, and lifestyle. Mol. Biol. Evol. 2005;22:547–561. doi: 10.1093/molbev/msi040. [DOI] [PubMed] [Google Scholar]
- 10.Sharp P. M., Bailes E., Grocock R. J., Peden J. F., Sockett R. E. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 2005;33:1141–1153. doi: 10.1093/nar/gki242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willenbrock H., Friis C., Friis A. S., Ussery D. W. An environmental signature for 323 microbial genomes based on codon adaptation indices. Genome Biol. 2006;7:R114. doi: 10.1186/gb-2006-7-12-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Vallve S., Guzman E., Montero M. A., Romeu A. HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003;31:187–189. doi: 10.1093/nar/gkg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koski L. B., Morton R. A., Golding G. B. Codon bias and base composition are poor indicators of horizontally transferred genes. Mol. Biol. Evol. 2001;18:404–412. doi: 10.1093/oxfordjournals.molbev.a003816. [DOI] [PubMed] [Google Scholar]
- 14.Mrazek J., Karlin S. Detecting alien genes in bacterial genomes. Ann. N Y Acad. Sci. 1999;870:314–329. doi: 10.1111/j.1749-6632.1999.tb08893.x. [DOI] [PubMed] [Google Scholar]
- 15.Henry I., Sharp P. M. Predicting gene expression level from codon usage bias. Mol. Biol. Evol. 2007;24:10–12. doi: 10.1093/molbev/msl148. [DOI] [PubMed] [Google Scholar]
- 16.Karlin S., Mrazek J., Campbell A., Kaiser D. Characterizations of highly expressed genes of four fast-growing bacteria. J. Bacteriol. 2001;183:5025–5040. doi: 10.1128/JB.183.17.5025-5040.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puigbo P., Romeu A., Garcia-Vallve S. HEG-DB: a database of predicted highly expressed genes in prokaryotic complete genomes under translational selection. Nucleic Acids Res. 2008;36:D524–D527. doi: 10.1093/nar/gkm831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharp P. M., Li W. H. The codon adaptation index – a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fellenberg K., Hauser N. C., Brors B., Neutzner A., Hoheisel J. D., Vingron M. Correspondence analysis applied to microarray data. Proc. Natl Acad. Sci. USA. 2001;98:10781–10786. doi: 10.1073/pnas.181597298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grantham R., Gautier C., Gouy M., Mercier R., Pave A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980;8:r49–r62. doi: 10.1093/nar/8.1.197-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perriere G., Thioulouse J. Use and misuse of correspondence analysis in codon usage studies. Nucleic Acids Res. 2002;30:4548–4555. doi: 10.1093/nar/gkf565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zavala A., Naya H., Romero H., Musto H. Trends in codon and amino acid usage in Thermotoga maritima. J. Mol. Evol. 2002;54:563–568. doi: 10.1007/s00239-001-0040-y. [DOI] [PubMed] [Google Scholar]
- 23.dos Reis M., Wernisch L., Savva R. Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 2003;31:6976–6985. doi: 10.1093/nar/gkg897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lerat E., Biemont C., Capy P. Codon usage and the origin of P elements. Mol. Biol. Evol. 2000;17:467–468. doi: 10.1093/oxfordjournals.molbev.a026326. [DOI] [PubMed] [Google Scholar]
- 25.Charif D., Thioulouse J., Lobry J. R., Perriere G. Online synonymous codon usage analyses with the ade4 and seqinR packages. Bioinformatics. 2005;21:545–547. doi: 10.1093/bioinformatics/bti037. [DOI] [PubMed] [Google Scholar]
- 26.Das S., Paul S., Chatterjee S., Dutta C. Codon and amino acid usage in two major human pathogens of genus Bartonella – optimization between replicational–transcriptional selection, translational control and cost minimization. DNA Res. 2005;12:91–102. doi: 10.1093/dnares/12.2.91. [DOI] [PubMed] [Google Scholar]
- 27.Basak S., Ghosh T. C. Temperature adaptation of synonymous codon usage in different functional categories of genes: a comparative study between homologous genes of Methanococcus jannaschii and Methanococcus maripaludis. FEBS Lett. 2006;580:3895–3899. doi: 10.1016/j.febslet.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Das S., Paul S., Bag S. K., Dutta C. Analysis of Nanoarchaeum equitans genome and proteome composition: indications for hyperthermophilic and parasitic adaptation. BMC Genomics. 2006;7:186. doi: 10.1186/1471-2164-7-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das S., Paul S., Dutta C. Evolutionary constraints on codon and amino acid usage in two strains of human pathogenic actinobacteria Tropheryma whipplei. J. Mol. Evol. 2006;62:645–658. doi: 10.1007/s00239-005-0164-6. [DOI] [PubMed] [Google Scholar]
- 30.Das S., Paul S., Dutta C. Synonymous codon usage in adenoviruses: influence of mutation, selection and protein hydropathy. Virus Res. 2006;117:227–236. doi: 10.1016/j.virusres.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Ahn I., Son H. S. Comparative study of the hemagglutinin and neuraminidase genes of influenza A virus H3N2, H9N2, and H5N1 subtypes using bioinformatics techniques. Can. J. Microbiol. 2007;53:830–839. doi: 10.1139/W07-044. [DOI] [PubMed] [Google Scholar]
- 32.Ranjan A., Vidyarthi A. S., Poddar R. Evaluation of codon bias perspectives in phage therapy of Mycobacterium tuberculosis by multivariate analysis. In Silico Biol. 2007;7:0030. [PubMed] [Google Scholar]
- 33.Sau K., Gupta S. K., Sau S., Mandal S. C., Ghosh T. C. Studies on synonymous codon and amino acid usage biases in the broad-host range bacteriophage KVP40. J. Microbiol. 2007;45:58–63. [PubMed] [Google Scholar]
- 34.Sen G., Sur S., Bose D., et al. Analysis of codon usage patterns and predicted highly expressed genes for six phytopathogenic Xanthomonas genomes shows a high degree of conservation. In Silico Biol. 2007;7:547–558. [PubMed] [Google Scholar]
- 35.Wang H. C., Hickey D. A. Rapid divergence of codon usage patterns within the rice genome. BMC Evol. Biol. 2007;7(Suppl 1):S6. doi: 10.1186/1471-2148-7-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong J., Li Y., Zhao S., Liu S., Zhang Z. Mutation pressure shapes codon usage in the GC-rich genome of foot-and-mouth disease virus. Virus Genes. 2007;35:767–776. doi: 10.1007/s11262-007-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao S., Zhang Q., Liu X., et al. Analysis of synonymous codon usage in 11 Human Bocavirus isolates. Biosystems. 2008;92:207–214. doi: 10.1016/j.biosystems.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Wheeler D. L. GenBank. Nucleic Acids Res. 2008;36:D25–D30. doi: 10.1093/nar/gkm929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lafay B., Lloyd A. T., McLean M. J., Devine K. M., Sharp P. M., Wolfe K. H. Proteome composition and codon usage in spirochaetes: species-specific and DNA strand-specific mutational biases. Nucleic Acids Res. 1999;27:1642–1649. doi: 10.1093/nar/27.7.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McInerney J. O. Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc. Natl Acad. Sci. USA. 1998;95:10698–106703. doi: 10.1073/pnas.95.18.10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero H., Zavala A., Musto H. Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 2000;28:2084–2090. doi: 10.1093/nar/28.10.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musto H., Romero H., Zavala A. Translational selection is operative for synonymous codon usage in Clostridium perfringens and Clostridium acetobutylicum. Microbiology. 2003;149:855–863. doi: 10.1099/mic.0.26063-0. [DOI] [PubMed] [Google Scholar]
- 43.Medigue C., Rouxel T., Vigier P., Henaut A., Danchin A. Evidence for horizontal gene transfer in Escherichia coli speciation. J. Mol. Biol. 1991;222:851–856. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- 44.Perriere G., Thioulouse J. On-line tools for sequence retrieval and multivariate statistics in molecular biology. Comput. Appl. Biosci. 1996;12:63–69. doi: 10.1093/bioinformatics/12.1.63. [DOI] [PubMed] [Google Scholar]
- 45.Lafay B., Atherton J. C., Sharp P. M. Absence of translationally selected synonymous codon usage bias in Helicobacter pylori. Microbiology. 2000;146(Pt 4):851–860. doi: 10.1099/00221287-146-4-851. [DOI] [PubMed] [Google Scholar]
- 46.McInerney J. O. Prokaryotic genome evolution as assessed by multivariate analysis of codon usage patterns. Microbiol Comp. Genomics. 1997;2:89–97. [Google Scholar]
- 47.Andersson S. G., Sharp P. M. Codon usage and base composition in Rickettsia prowazekii. J. Mol. Evol. 1996;42:525–536. doi: 10.1007/BF02352282. [DOI] [PubMed] [Google Scholar]
- 48.Rispe C., Delmotte F., van Ham R. C., Moya A. Mutational and selective pressures on codon and amino acid usage in Buchnera, endosymbiotic bacteria of aphids. Genome Res. 2004;14:44–53. doi: 10.1101/gr.1358104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharp P. M., Tuohy T. M., Mosurski K. R. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thioulouse J., Chessel D., Dolédec S., Olivier J. M. ADE-4: a multivariate analysis and graphical display software. Stat. Comput. 1997;7:75–83. [Google Scholar]
- 51.R Development Core Team. Vienna, Austria: 2007. R: a language and environment for statistical computing, R Foundation for Statistical Computing. [Google Scholar]
- 52.Grocock R. J., Sharp P. M. Synonymous codon usage in Pseudomonas aeruginosa PA01. Gene. 2002;289:131–139. doi: 10.1016/s0378-1119(02)00503-6. [DOI] [PubMed] [Google Scholar]
- 53.Kyte J., Doolittle R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki H., Saito R., Tomita M. A problem in multivariate analysis of codon usage data and a possible solution. FEBS Lett. 2005;579:6499–504. doi: 10.1016/j.febslet.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 55.Lobry J. R., Gautier C. Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res. 1994;22:3174–3180. doi: 10.1093/nar/22.15.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghaemmaghami S., Huh W. K., Bower K., et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 57.Carlini D. B. Context-dependent codon bias and messenger RNA longevity in the yeast transcriptome. Mol. Biol. Evol. 2005;22:1403–1411. doi: 10.1093/molbev/msi135. [DOI] [PubMed] [Google Scholar]
- 58.Kerr A. R., Peden J. F., Sharp P. M. Systematic base composition variation around the genome of Mycoplasma genitalium, but not Mycoplasma pneumoniae. Mol. Microbiol. 1997;25:1177–1179. doi: 10.1046/j.1365-2958.1997.5461902.x. [DOI] [PubMed] [Google Scholar]
- 59.Arakawa K., Mori K., Ikeda K., Matsuzaki T., Kobayashi Y., Tomita M. G-language genome analysis environment: a workbench for nucleotide sequence data mining. Bioinformatics. 2003;19:305–306. doi: 10.1093/bioinformatics/19.2.305. [DOI] [PubMed] [Google Scholar]
- 60.Arakawa K., Tomita M. G-language system as a platform for large-scale analysis of high-throughput omics data. J. Pesticide Sci. 2006;31:282–288. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.