

Abstract
UVB light promotes survival of initiated keratinocytes, in part, by the direct activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Novel chemopreventative agents targeting UVB-induced signaling pathways are needed to reduce the incidence of nonmelanoma skin cancer. Quercetin (Qu) is a dietary flavonoid and a known inhibitor of PI3K. We determined that Qu degrades rapidly when diluted in DMEM and incubated under normal cell culture conditions. Degradation was delayed by supplementing the medium with 1 mmol/L ascorbic acid (AA), and as expected, stabilization actually increased the effectiveness of Qu as a PI3K inhibitor because basal and UVB-induced Akt phosphorylation were reduced compared with Qu treatment in the absence of AA. Although AA stabilization increased Qu-induced apoptosis in mock-irradiated HaCaT cells, consistent with it acting as a PI3K inhibitor (13.4% Annexin V–positive cells for AA-stabilized Qu versus 6.3% for Qu), AA stabilization of Qu actually reduced the ability of the compound to induce apoptosis of UVB-irradiated HaCaTs (29.7% of Qu-treated cells versus 15.5% of AA + Qu–treated cells). Similar trends were seen in the analysis of caspase-3 and poly(ADP-ribose) polymerase cleavage. Qu is known to oxidize to form reactive products, and we found that dihydroethidium is oxidized by Qu regardless of whether or not it was stabilized. Although redox cycling occurs even in the presence of AA, stabilization reduces the accumulation of reactive Qu products that contribute to the proapoptotic effect of the compound, and thus reduces the ability of the compound to induce apoptosis of UVB-irradiated HaCaT cells.
Nonmelanoma skin cancers, consisting of squamous cell carcinomas and basal cell carcinomas, are the most common of all types of cancer in the United States (1). Squamous cell carcinomas occur primarily on the sun-exposed areas of the body, primarily the face, head, and arms, and there is a well-established link between chronic sun exposure and the development of squamous cell carcinoma (2). UVB light, which composes 1% to 10% of the UV light that reaches the Earth's surface, is classified as a complete carcinogen and is known to activate several signal transduction pathways, including the phosphatidylinositol 3-kinase (PI3K) pathway. This pathway has been shown by our laboratory and others to be activated by UVB in cultured cells as well as in both mouse and human skin (3–6). In addition, several UVB-induced intracellular signaling cascades are involved in numerous cellular functions, such as growth and proliferation, differentiation, protein expression, and apoptosis, and are often deregulated during tumorigenesis (3, 7–10).
The PI3K signaling pathway represents an important target for the prevention of nonmelanoma skin cancer because it activates Akt and mediates a cell survival response to UVB. In sun-damaged skin, activation of PI3K by UVB increases the survival of initiated keratinocytes and facilitates the progression of precancerous actinic keratoses to squamous cell carcinomas. Our laboratory has also previously determined that PI3K mediates the UVB-induced expression of cyclooxygenase-2 and c-Fos and the activation of activator protein-1 in cultured human keratinocytes and in mouse skin, all of which have been implicated in the development of squamous cell carcinoma (3, 11, 12).
Quercetin (Qu; 3,3′,4′,5,7-pentahydroxyflavone), a naturally produced flavonoid enriched in red wine as well as in numerous fruits, vegetables, and nuts, has well-established activity against intracellular targets known to mediate UVB signaling, including PI3K and mitogen-activated protein kinase/extracellular signal–regulated kinase kinase/extracellular signal–regulated kinase (13–15). Qu and other flavanoid compounds have been reported to induce apoptosis as a result of PI3K inhibition and through other mechanisms including reactive oxygen species (ROS) generation (16). However, the exact relationship between the formation of reactive species and Qu are still not fully understood. In fact, it has been reported that Qu degrades rapidly in aqueous medium, including DMEM (17), and the generation of ROS or reactive Qu intermediates may contribute to the proapoptotic effect. Inclusion of 1 mmol/L ascorbic acid (AA) in the cell culture medium actually delays the degradation of Qu and serves to stabilize the compound over several hours. However, regardless of the mechanism, the chemopreventative potential of Qu, like other PI3K inhibitors, is commonly believed to lie in its ability to induce apoptosis of initiated keratinocytes and therefore reduce the possibility of progression of actinic keratoses to squamous cell carcinomas.
The goal of the current study was to evaluate the ability of Qu to induce apoptosis in UVB-irradiated HaCaT cells, a human keratinocyte line representing precancerous initiated cells, and to gain insight into its mechanism of action. We initially hypothesized that the primary proapoptotic mechanism was through the inhibition of UVB-induced PI3K and Akt activation. However, we determined that Qu stabilization with 1 mmol/L AA actually reduced the proapoptotic effect compared with Qu treatment in the absence of AA although efficacy for PI3K inhibition was increased by stabilization. In addition, by using dihydroethidium to measure the generation of reactive products, we found that stabilization of Qu does not prevent it from undergoing redox cycling because HaCaTs treated with Qu in AA-supplemented DMEM still displayed increased ethidium fluorescence. The data suggest that degradation of Qu results in the formation of a reactive product, which is partially responsible for the apoptotic effect. Although Qu is stabilized by AA, redox cycling still occurs. However, it is possible that reactive Qu intermediates do not significantly accumulate in the presence of AA, which therefore results in reduced apoptosis in response to AA-stabilized Qu. Overall, our study suggests that reactive products are partially responsible for the chemopreventative properties of Qu and that stabilization actually reduces the ability of Qu to eliminate initiated cells.
Materials and Methods
Materials
Quercetin dihydrate, ascorbic acid, and propidium iodide were purchased from Sigma-Aldrich. Caspase-3, poly(ADP- ribose) polymerase (PARP), and phospho-specific antibodies and total antibodies for Akt were from Cell Signaling Technology. Annexin V-Alexa Fluor 488 and dihydroethidium were obtained from Invitrogen/Molecular Probes. All other reagents were reagent grade and purchased from Sigma.
Cells
The human keratinocyte cell line HaCaT was established from cells that were obtained from adult sun-damaged skin (18). HaCaT cells have two mutated p53 alleles caused by C→T and CC→TT mutations, which are known to be initiating mutations induced by exposure to UV light (19, 20). HaCaT cells were cultured in DMEM with 10% fetal bovine serum and 100 units/mL penicillin/streptomycin at 37°C and in 5% CO2. The cells were cultured to 90% to 95% confluence and then maintained in serum-free DMEM for 24 h before UVB exposure.
Quercetin stability
DMEM (10 mL), either not supplemented or supplemented with 1 mmol/L AA, was placed in a 100-mm tissue culture dish in the absence of cells and maintained under normal cell culture conditions (humidified incubator at 37°C and 5% CO2). A 1,000× Qu stock solution was added to the dishes at a final concentration of 50 μmol/L. At the indicated time points, 100-μL aliquots were removed from the dish, diluted 1:1 with 1 mmol/L AA to prevent further degradation, and snap frozen in liquid nitrogen. Samples were then analyzed by high-performance liquid chromatography against a DMEM reference sample and normalized to time 0 to determine relative amounts of Qu remaining.
UVB irradiation of HaCaT cells
HaCaT cells maintained in serum-free DMEM for 24 h were pretreated with Qu and/or ascorbate for 1 h before UVB irradiation. After incubation, HaCaTs were washed once in PBS, which was then removed. Cells were irradiated with a dose of 250 J/m2 using a bank of two SF20 UVB lamps (National Biological Corp.) providing a peak emission of 313 nm. Control cells were treated in the same manner and mock irradiated. Following irradiation, HaCaT cells were again washed with PBS and returned to DMEM containing appropriate drug treatments.
Western blotting
Cells were lysed in radioimmunoprecipitation assay buffer containing 50 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 1% Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 3 mmol/L β-glycerophosphate, 1 mmol/L NaVO4, 10 mmol/L NaF, and 1 mmol/L phenylmethylsulfonyl fluoride. Protein concentration was determined using Bio-Rad detergent-compatible protein assay reagent (Bio-Rad Laboratories). For Western blot analysis, 40-μg protein was resolved by SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. Membranes were blocked in TBS containing 0.1% Tween 20 and either 5% nonfat dry milk or 5% bovine serum albumin (for phospho-specific antibodies). After washing in TBS-0.1% Tween 20, membranes were incubated with appropriate horseradish peroxidase–conjugated secondary antibodies and then washed extensively in TBS-0.1% Tween 20. Antigen-antibody complexes were detected with Amersham enhanced chemiluminescence detection reagent (GE Healthcare).
Measurement of quercetin-induced oxidation
Following irradiation with 250 J/m2 UVB, cells were incubated in the presence of 10 μmol/L dihydroethidium for 1 h. Cells treated with Qu were treated for 1 h before irradiation and 1 h after UVB during the dihydroethidium loading period. At the end of the treatment, cells were washed once with PBS and trypsinized from tissue culture plates. Cells were then pelleted and resuspended in PBS containing 0.5 mol/L EDTA, at which point samples were analyzed by flow cytometry for ethidium fluorescence using a total of 10,000 events per treatment. One-color flow cyotmetric analysis was done using a FACScan flow cytometer (BD Biosciences) equipped with an air-cooled 15-mW argon ion laser tuned to 488 nm. List mode data files were acquired and analyzed using CellQuest PRO software (BD Biosciences). Qu-treated cells that were not loaded with dihydroethidium did not display any background fluorescence compared with untreated cells.
Annexin V/propidium iodide labeling of cells
After incubation for 12 h after UVB irradiation, DMEM containing floating cells was collected. Cells were then washed once with PBS and detached using trypsin supplemented with EDTA. Once detached, trypsin was inactivated with DMEM containing 10% fetal bovine serum. The cells were then pooled with the floaters, at which point the entire sample was pelleted by centrifugation and DMEM was removed. The pellet was resuspended in Annexin binding buffer [containing 10 mmol/L HEPES (pH 7.4), 140 mmol/L NaCl, and 2.5 mmol/L CaCl2] at a concentration of 106 cells/mL and then transferred to 1.5-mL microcentrifuge tubes. Cells were incubated for 30 min with Annexin V-Alexa Fluor 488 and propidium iodide according to the manufacturer's instructions. Cells were then diluted up to a total volume of 0.5 mL in Annexin binding buffer and analyzed by two-color flow cytometry using a total of 10,000 events. The emission fluorescence of the Annexin V conjugate was detected and recorded through a 530/30 bandpass filter in the FL1 detector. Propidium iodide was detected in the FL2 detector through a 585/42 bandpass filter. List mode data files gated on forward scatter versus side scatter were acquired and analyzed using CellQuest PRO software. Appropriate electronic compensation was adjusted by acquiring cell populations stained with each dye or fluorophore individually, as well as an unstained control. Apoptotic cells were only those which stained positive for Annexin V and negative for propidium iodide, located in the bottom right quadrant.
Results
Stabilization of quercetin in DMEM by inclusion of ascorbic acid increases Its efficacy for PI3K inhibition
We were first interested in determining how long Qu remains intact when dissolved in aqueous medium and when incubated under our exact cell culture conditions because this may affect its efficacy as a PI3K inhibitor. Qu is known to undergo oxidation in DMEM and other cell culture medium to form an ortho-semiquinone and an ortho-quinone (21, 22). We determined that Qu degrades rapidly in DMEM, with 50% of the compound being lost before 30 minutes of incubation and almost complete loss by 4 hours (Fig. 1). Qu could be stabilized in DMEM by supplementing the medium with 1 mmol/L AA. AA supplementation led to nearly 100% Qu stabilization for 4 hours, followed by a 10% to 15% per hour decrease in Qu until the 12-hour time point at which ∼95% of the compound had been degraded.
Fig. 1.
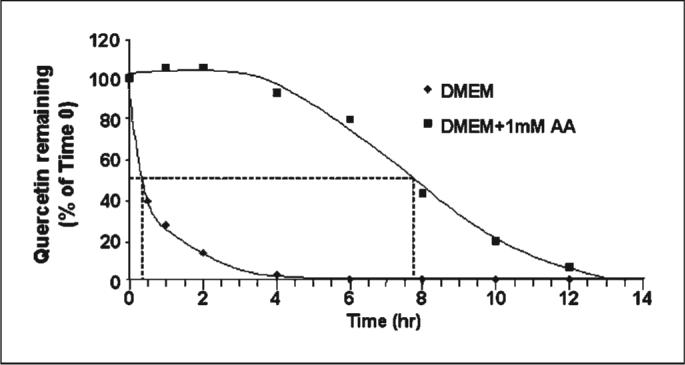
Ascorbic acid stabilizes Qu in DMEM under cell culture conditions. Qu (50 μmol/L) was added to 10-mL DMEM, which either contained or did not contain 1 mmol/L AA, and maintained at 37°C with 5% CO2 in a humidified incubator. Aliquots were collected at the indicated time points and subjected to high-performance liquid chromatography analysis to determine the remaining amount of Qu relative to time 0. Dashed line, the approximate times at which 50% of the Qu measured at time 0 had been degraded. AA stabilized Qu for several hours before any degradation had begun and also slowed the rate at which Qu degraded. Representative of two independent experiments.
We were next interested in determining what effect stabilizing Qu had on PI3K inhibition. Figure 2 indicates that Qu alone, without AA stabilization, significantly inhibited Akt phosphorylation in both mock-irradiated and UVB-irradiated HaCaTs, consistent with the compound acting as a PI3K inhibitor. As expected, stabilization of Qu with AA increased the efficacy of Qu as a PI3K inhibitor, indicated by a further reduction in phospho-Akt levels in both mock-irradiated and irradiated HaCaTs compared with Qu treatment alone.
Fig. 2.
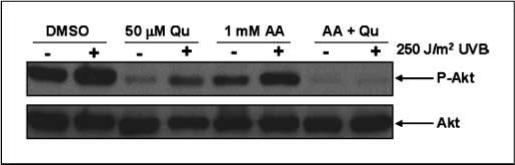
Qu inhibits Akt phosphorylation in mock-irradiated and UVB-irradiated HaCaT cells. HaCaT keratinocytes were pretreated with 50 μmol/L Qu for 1 h in the presence or absence of 1 mmol/L AA before irradiation with 250 J/m2 UVB. After irradiation, cells were placed back in DMEM containing appropriate treatments for 1 h before harvesting proteins. UVB increased phospho-Akt levels as expected, and Qu significantly decreased phospho-Akt in both irradiated and mock-irradiated cells, indicating that this compound is effectively inhibiting PI3K activity. AA slightly reduced phospho-Akt levels but had no effect on UVB induction of Akt phosphorylation. Qu inhibition of Akt phosphorylation was significantly increased when HaCaTs were treated in the presence of AA because basal levels were reduced compared with Qu alone and there was no increase in phospho-Akt levels with UVB irradiation. Representative of three independent experiments.
Quercetin forms reactive products in cell culture medium
Qu is known to undergo oxidation and form radical Qu intermediates in aqueous medium. In addition, Qu has been proposed to generate ROS. We tested the oxidative potential of Qu by loading HaCaTs with dihydroethidium, a compound that fluoresces when oxidized, and then treating with Qu. We found that Qu treatment increased ethidium fluorescence significantly over control. The dose of UVB used in these experiments was not sufficient to induce dihydroethidium oxidation alone (Fig. 3). Qu treatment increased dihydroethidium oxidation in both irradiated and mock-irradiated HaCaTs, and the difference between the two groups was not statistically significant (Fig. 3A and B). Interestingly, when the assay was done using DMEM supplemented with AA, Qu treatment still oxidized dihydroethidium significantly in both UVB-irradiated and mock-irradiated HaCaTs compared with the respective controls (Fig. 3C and D). In addition, we found no significant reduction in the oxidative potential of Qu because there was no statistical difference between Qu treatment in DMEM compared with Qu treatment in AA-supplemented DMEM.
Fig. 3.
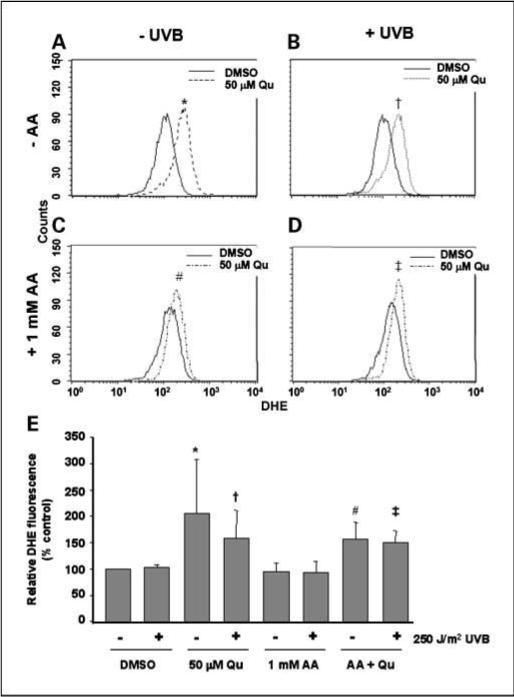
Qu acts as an oxidant by generating reactive species. HaCaT cells were treated with 50 μmol/L Qu in the presence or absence of 1 μmol/L AA for 1 h before UVB irradiation. After exposure to UVB, cells were placed back in DMEM containing appropriate treatments for 1 h. During this time, 10 μmol/L dihydroethidium (DHE) was added to the medium to measure the generation of reactive species. Generation of ROS was then determined by flow cytometry (A–D). Treatment with Qu caused significant oxidation of dihydroethidium in both mock-irradiated and UVB-irradiated cells regardless of the presence of AA. E, the data were pooled from five independent experiments each measuring a total of 10,000 events. Columns, mean; bars, SD. Statistical significance was set at P < 0.05 using one-tailed t test: *, versus DMSO-UVB; †, versus DMSO + UVB; #, versus AA-UVB; ‡, versus AA + UVB. No statistical significance was found between any Qu-treated condition regardless of the presence of AA.
Stabilization of quercetin reduces its ability to induce apoptosis of UVB-irradiated HaCaTs
The enhanced PI3K inhibition that occurs when Qu is stabilized by AA suggests that the apoptotic response of UVB-irradiated HaCaT cells to Qu + AA would be greater than the response to Qu alone. However, we determined that Qu treatment in the absence of AA actually had a greater effect on UVB-induced apoptosis. To quantitate the apoptotic response of HaCaT cells to Qu, Annexin V staining was done to identify changes in the apoptotic cell population. The mean percentage of Annexin V–positive/propidium iodide–negative cells in untreated HaCaTs was 1.9 ± 0.9% (Fig. 4A and I), and this population increased significantly to 10.9 ± 0.4% of UVB-irradiated HaCaTs (Fig. 4B). Qu treatment also significantly increased the Annexin V–positive cell population to 6.3 ± 3.1% (Fig. 4C). We again determined that Qu potentiated the UVB effect because irradiated cells treated with Qu displayed the greatest percentage of apoptotic cells, 29.7 ± 7.3% (Fig. 4D).
Fig. 4.
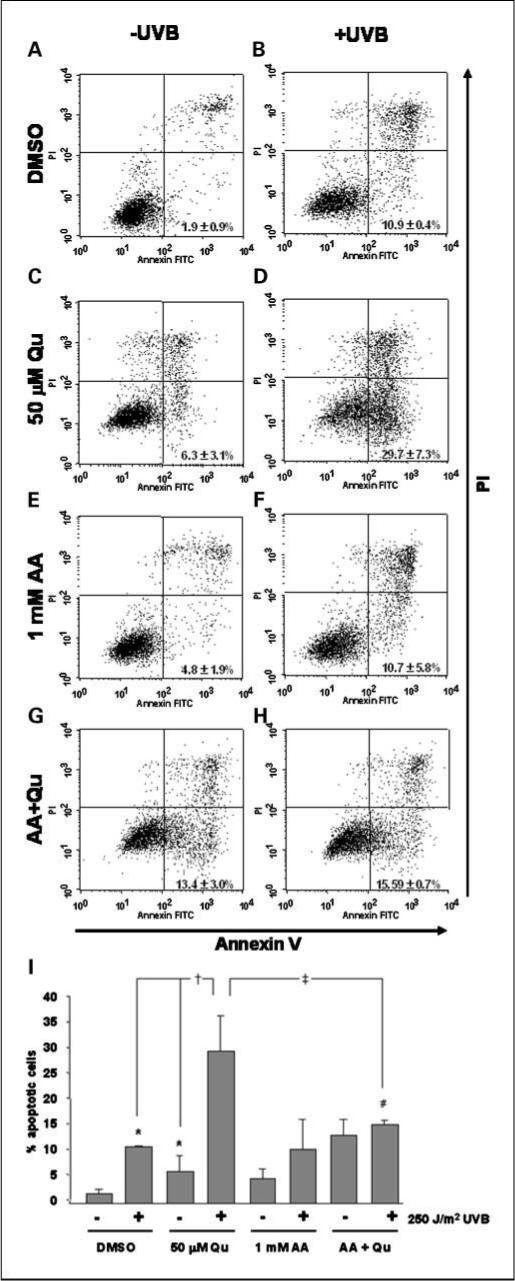
Degradation of Qu is partially responsible for its proapoptotic effect on HaCaT keratinocytes. HaCaT cells were treated with 50 μmol/L Qu in the presence and absence of 1 mmol/L AA for 1 h before UVB irradiation and for 12 h after 250 J/m2 UVB. A to H, UVB and Qu each independently induced apoptosis of HaCaT cells as expected. HaCaTs treated with both Qu and UVB displayed higher Annexin V staining than either Qu-treated cells or UVB-irradiated cells alone. Stabilization of Qu with 1 mmol/L AA resulted in a significant reduction of Qu + UVB–induced Annexin V staining. Interestingly, UVB did not have any further significant effect on Annexin V staining in AA + Qu–treated HaCaTs. Each figure is representative of three independent experiments analyzing 10,000 events per condition. I, all data from independent experiments were pooled and statistical significance was set at P < 0.05 using one-tailed t test: *, versus DMSO-UVB; †, versus DMSO + UVB and Qu-UVB; #, versus AA-UVB; ‡, versus Qu + UVB.
Caspase-3 and PARP cleavage were both increased in a manner similar to Annexin V staining. Again, both UVB irradiation and Qu treatment alone induced cleavage of caspase-3 and PARP (Fig. 5). Cleavage of caspase-3 and PARP were both increased in cells treated with Qu in combination with UVB irradiation. Interestingly, when Qu was stabilized with AA and the efficacy for PI3K inhibition increased (Fig. 2), the enhancement of UVB-induced apoptosis was lost. The increase in the percentage of Annexin V–positive cells caused by AA compared with DMSO-treated controls (both mock irradiated and UVB irradiated) was not statistically significant (Fig. 4E and F), and there was no apparent effect of AA on the cleavage of either caspase-3 or PARP. The slight reduction in Akt phosphorylation caused by AA treatment alone (Fig. 2) therefore did not have a significant effect on apoptosis of HaCaT keratinocytes. However, inclusion of 1 mmol/L AA in DMEM during Qu treatment resulted in a significant reduction in the ability of Qu to enhance UVB-induced apoptosis. The Annexin V–positive cell population was reduced from 29.7% to 15.5% (Fig. 4H), and we observed a similar effect on caspase-3 and PARP, where AA reduced the degree of cleavage in Qu- and UVB-treated HaCaTs compared with Qu and UVB in the absence of AA (Fig. 5).
Fig. 5.
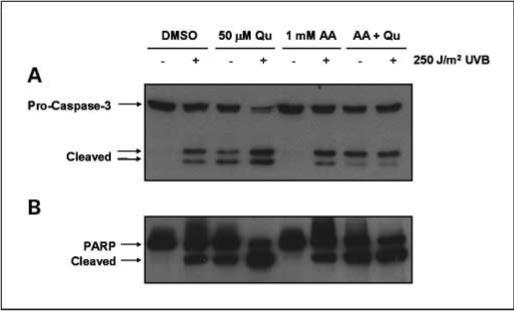
Qu degradation increases caspase-3 and PARP cleavage. HaCaT cells were treated with 50 μmol/L Qu in the presence and absence of 1 mmol/L AA for 1 h before UVB irradiation and for 16 h after 250 J/m2 UVB. Cleavage of caspase-3 (A) and PARP (B) were induced by both UVB irradiation and Qu treatment alone. UVB irradiation followed by Qu treatment resulted in an increase in caspase-3 cleavage compared with either UVB or Qu alone. AA had no effect alone on caspase-3 or PARP cleavage; however, stabilization of Qu with AA resulted in a significant reduction in the cleavage of both proteins compared with Qu alone. Representative of three independent experiments.
Discussion
Quercetin is an abundantly available dietary compound present in a wide variety of foods, with well-established pharmacologic activity on intracellular signaling pathways involved in the development of cancers. We were initially interested in this compound due to its ability to inhibit PI3K activation because our laboratory and others have established PI3K as an important signaling protein involved in UVB-induced signaling (3–6). Activation of PI3K by UVB has been linked to the development and progression of nonmelanoma skin cancer, and compounds with inhibitory activity against PI3K are viewed as potential chemopreventative agents.
In the current study, we investigated the chemopreventative potential of Qu by analyzing its ability to induce apoptosis of HaCaT cells, an initiated human keratinocyte cell line. We showed that there is a significant increase in the apoptotic cell population after irradiation with 250 J/m2 UVB (10.9%). Treatment of the cells with Qu was also sufficient to induce apoptosis to a small but significant degree (6.3%). Interestingly, the combination of UVB irradiation and Qu treatment resulted in a potentiated apoptotic response, with nearly 30% of the cells found to be in the early phase of apoptosis. Qu is a well-established inhibitor of PI3K, and we showed its ability to inhibit PI3K in both irradiated and mock-irradiated HaCaT cells by showing reduced Akt phosphorylation under either condition. PI3K and Akt are activated in response to UVB irradiation, indicating that this pathway represents an important survival response to limit the amount of apoptotic cell death after UVB. Clearly, because Qu inhibits PI3K and Akt activation, and the apoptotic response to UVB is greatly increased, this represents an important mechanism by which Qu induces cell death.
Further investigation led us to conclude that the PI3K/Akt signaling pathway only partially explains the mechanism behind Qu-induced cell death. Dihal et al. (17) reported that Qu is unstable in aqueous cell culture medium supplemented with 10% FCS. We investigated this phenomenon in DMEM under serum-free conditions and found a similar effect but with a much faster rate of degradation. We found that 50% of Qu degraded in DMEM before 30 minutes of incubation. We also confirmed that Qu could be stabilized by supplementing DMEM with 1 mmol/L AA, and we found that Qu was nearly 100% stabilized for a period of 4 hours before any significant reduction of intact Qu was seen. We were interested in howstabilization of Qu would affect its proapoptotic properties. Our first finding was that stabilization of Qu increased its efficacy of PI3K inhibition resulting in reduced basal levels of p-Akt compared with Qu treatment in the absence of AA. In addition, AA-stabilized Qu had a greater inhibitory effect on UVB-induced Akt phosphorylation, whereas activation was only partially blocked by Qu treatment in the absence of AA, suggesting that stabilized Qu would be more effective at inducing apoptosis. However, we determined that stabilizing Qu completely prevented any further increase in apoptosis by UVB. Qu treatment of UVB-irradiated HaCaT cells increased the apoptotic cell population by 172% compared with UVB alone. When cells were incubated in AA-supplemented DMEM, Qu treatment of irradiated HaCaTs resulted only in a 45% increase in the number of apoptotic cells, and this increase was not statistically significant.
The relationship between Qu and ROS is not fully understood. For example, investigators working with glioma cells have reported that ROS is not involved in Qu-induced apoptosis (23), or that Qu acts as an antioxidant because it inhibits ROS-induced apoptosis (24). Still other investigators have found both prooxidant and antioxidant effects of Qu depending on the concentrations used for experimentation (25). The findings from these studies indicate that there are likely cell type–specific effects of Qu and that there are effects of Qu that are dependent on the experimental design. Based on the published data and our findings on Qu-induced apoptosis, we hypothesized that Qu in the absence of AA generated more reactive species than AA-stabilized Qu. We tested this by analyzing dihydroethidium fluorescence in response to both treatments. However, we determined that Qu was capable of inducing dihydroethidium fluorescence regardless of whether or not it was stabilized, and although it seemed that there was reduced reactive species generation in the AA-stabilized conditions, this reduction was not significantly different from Qu alone. Qu has been determined to form reactive intermediate species, including an ortho-semiquinone radical and an ortho-quinone, which can cycle back to Qu using intracellular glutathione (22). In addition, Boots et al. (21) reported that Qu oxidized by tyrosinase could be converted from an ortho-quinone back to Qu by AA, resulting in sustained levels of Qu composed of a fraction that did not undergo tyrosinase-induced oxidation and a fraction that was regenerated after oxidation. Our data also indicate that redox cycling still occurs in the presence of AA, but because Qu levels remain stable for several hours, the reactive products that mediate part of the proapoptotic effect do not accumulate (Fig. 6). The current study therefore suggests that there is a combined effect of reactive Qu products and UVB that does not occur when Qu is stabilized, resulting in a reduced apoptotic response to Qu in the presence of AA. Ultimately, the maximum chemopreventative potential of Qu might lie both in the pharmacologic inhibition of PI3K and in its ability to form reactive products. Both of these mechanisms seem to be critical in promoting apoptosis and could therefore contribute to the removal of initiated keratinocytes from the epidermis and limit the development of squamous cell carcinoma.
Fig. 6.
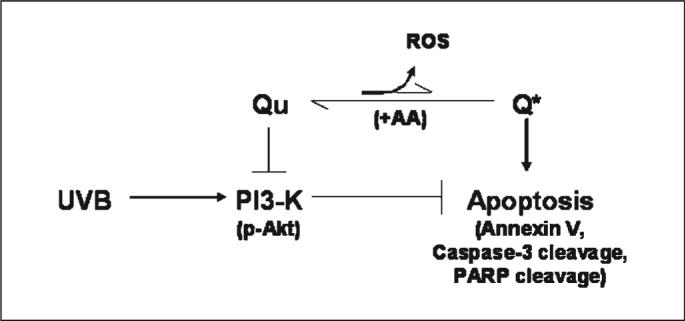
Proposed mechanisms of action of Qu. Qu-induced apoptosis is proposed to occur through the combined effect of PI3K inhibition and generation of reactive intermediates, including o-semiquinone and o-quinone Qu products (Q*) and potentially ROS. Stabilization of Qu with AA reduces apoptosis, although the efficacy for PI3K inhibition is increased. Redox cycling still occurs in the presence of AA, but because Qu remains intact for several hours, Q*, which mediate part of the proapoptotic effect, do not accumulate and the apoptotic response to Qu is therefore reduced.
Acknowledgments
We thank Dr. Sherry Chow, Dr. Steven Stratton, and Cathy Cordova in the Analytical Core Service at the Arizona Cancer Center for technical assistance with high-performance liquid chromatography analysis of quercetin degradation; Barb Carolus and Paula Campbell in the Arizona Research Labs-Division of Biotechnology Cytometry Core Facility at the Arizona Cancer Center for the collection of flow cytometry data; and Dr. Sally Dickinson for critical review of the manuscript and Anne Cione for administrative support.
Grant support: NIH grants: Arizona Cancer Center Core Grant CA23074, Chemoprevention of Skin Cancer Program Project Grant CA27502, R25T Cancer Prevention and Control Postdoctoral Fellowship CA78447, and Southwest Environmental Health Sciences Center Support Grant ES06694.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Kwa RE, Campana K, Moy RL. Biology of cutaneous squamous cell carcinoma. J Am Acad Dermatol. 1992;26:1–26. doi: 10.1016/0190-9622(92)70001-v. [DOI] [PubMed] [Google Scholar]
- 3.Bachelor MA, Cooper SJ, Sikorski ET, Bowden GT. Inhibition of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model. Mol Cancer Res. 2005;3:90–9. doi: 10.1158/1541-7786.MCR-04-0065. [DOI] [PubMed] [Google Scholar]
- 4.Einspahr JG, Bowden GT, Alberts DS, et al. Cross-validation of murine UV signal transduction pathways in human skin. Photochem Photobiol. 2008;84:463–76. doi: 10.1111/j.1751-1097.2007.00287.x. [DOI] [PubMed] [Google Scholar]
- 5.Umeda J, Sano S, Kogawa K, et al. In vivo cooperation between Bcl-xL and the phosphoinositide 3-kinase-Akt signaling pathway for the protection of epidermal keratinocytes from apoptosis. FASEB J. 2003;17:610–20. doi: 10.1096/fj.02-0597com. [DOI] [PubMed] [Google Scholar]
- 6.Wang HQ, Quan T, He T, Franke TF, Voorhees JJ, Fisher GJ. Epidermal growth factor receptor-dependent, NF-κB-independent activation of the phosphatidylinositol 3-kinase/Akt pathway inhibits ultraviolet irradiation-induced caspases-3, -8, and -9 in human keratinocytes. J Biol Chem. 2003;278:45737–45. doi: 10.1074/jbc.M300574200. [DOI] [PubMed] [Google Scholar]
- 7.Balasubramanian S, Kim KH, Ahmad N, Mukhtar H. Activation of telomerase and its association with G1-phase of the cell cycle during UVB-induced skin tumorigenesis in SKH-1 hairless mouse. Oncogene. 1999;18:1297–302. doi: 10.1038/sj.onc.1202417. [DOI] [PubMed] [Google Scholar]
- 8.Cooper SJ, Bowden GT. Ultraviolet B regulation of transcription factor families: roles of nuclear factor-κB(NF-κB) and activator protein-1 (AP-1) in UVB-induced skin carcinogenesis. Curr Cancer Drug Targets. 2007;7:325–34. doi: 10.2174/156800907780809714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JH, An HT, Chung JH, Kim KH, Eun HC, Cho KH. Acute effects of UVB radiation on the proliferation and differentiation of keratinocytes. Photodermatol Photoimmunol Photomed. 2002;18:253–61. doi: 10.1034/j.1600-0781.2002.02755.x. [DOI] [PubMed] [Google Scholar]
- 10.Van Laethem A, Nys K, Van Kelst S, et al. Apoptosis signal regulating kinase-1 connects reactive oxygen species to p38 MAPK-induced mitochondrial apoptosis in UVB-irradiated human keratinocytes. Free Radic Biol Med. 2006;41:1361–71. doi: 10.1016/j.freeradbiomed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Gonzales M, Bowden GT. The role of PI 3-kinase in the UVB-induced expression of c-fos. Oncogene. 2002;21:2721–8. doi: 10.1038/sj.onc.1205366. [DOI] [PubMed] [Google Scholar]
- 12.Tang Q, Gonzales M, Inoue H, Bowden GT. Roles of Akt and glycogen synthase kinase 3β in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer Res. 2001;61:4329–32. [PubMed] [Google Scholar]
- 13.Granado-Serrano AB, Martin MA, Bravo L, Goya L, Ramos S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J Nutr. 2006;136:2715–21. doi: 10.1093/jn/136.11.2715. [DOI] [PubMed] [Google Scholar]
- 14.Gulati N, Laudet B, Zohrabian VM, Murali R, Jhanwar-Uniyal M. The antiproliferative effect of quercetin in cancer cells is mediated via inhibition of the PI3K-Akt/PKB pathway. Anticancer Res. 2006;26:1177–81. [PubMed] [Google Scholar]
- 15.Lee KW, Kang NJ, Heo YS, et al. Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 2008;68:946–55. doi: 10.1158/0008-5472.CAN-07-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang YF, Chi CW, Wang JJ. Reactive oxygen species production is involved in quercetin-induced apoptosis in human hepatoma cells. Nutr Cancer. 2006;55:201–9. doi: 10.1207/s15327914nc5502_12. [DOI] [PubMed] [Google Scholar]
- 17.Dihal AA, Woutersen RA, van Ommen B, Rietjens IM, Stierum RH. Modulatory effects of quercetin on proliferation and differentiation of the human colorectal cell line Caco-2. Cancer Lett. 2006;238:248–59. doi: 10.1016/j.canlet.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrhart JC, Gosselet FP, Culerrier RM, Sarasin A. UVB-induced mutations in human key gatekeeper genes governing signalling pathways and consequences for skin tumourigenesis. Photochem Photobiol Sci. 2003;2:825–34. doi: 10.1039/b302281a. [DOI] [PubMed] [Google Scholar]
- 20.Lehman TA, Modali R, Boukamp P, et al. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14:833–9. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- 21.Boots AW, Bast A, Haenen GR. No role of DT-diaphorase (NQO1) in the protection against oxidized quercetin. FEBS Lett. 2005;579:677–82. doi: 10.1016/j.febslet.2004.12.044. [DOI] [PubMed] [Google Scholar]
- 22.Metodiewa D, Jaiswal AK, Cenas N, Dickancaite E, Segura-Aguilar J. Quercetin may act as a cytotoxic prooxidant after its metabolic activation to semiquinone and quinoidal product. Free Radic Biol Med. 1999;26:107–16. doi: 10.1016/s0891-5849(98)00167-1. [DOI] [PubMed] [Google Scholar]
- 23.Kim EJ, Choi CH, Park JY, Kang SK, Kim YK. Underlying mechanism of quercetin-induced cell death in human glioma cells. Neurochem Res. 2008;33:971–9. doi: 10.1007/s11064-007-9416-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen TJ, Jeng JY, Lin CW, Wu CY, Chen YC. Quercetin inhibition of ROS-dependent and -independent apoptosis in rat glioma C6 cells. Toxicology. 2006;223:113–26. doi: 10.1016/j.tox.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Robaszkiewicz A, Balcerczyk A, Bartosz G. Antioxidative and prooxidative effects of quercetin on A549 cells. Cell Biol Int. 2007;31:1245–50. doi: 10.1016/j.cellbi.2007.04.009. [DOI] [PubMed] [Google Scholar]