

Abstract
Sialidases, or neuraminidases, are enzymes that cleave terminal sialic acid residues from complex sialic acid-containing structures. They have been found in many animals and microorganisms and are important in various physiological and pathological processes. In order to understand the biological significance of diverse sialidases, it is important to study in detail the structural determinants of their natural substrates. Here, we report the synthesis of sialoside libraries containing para-nitrophenol-tagged sialosides with different naturally occurring sialic acid forms, different sialyl linkages, and different penultimate monosaccharides using a highly efficient one-pot three-enzyme chemoenzymatic approach. Using these compounds in a 96-well plate based colorimetric high-throughput screening platform, the diversity of substrate preference is shown for seven bacterial sialidases. The sialoside libraries and the screening method are convenient tools for unravelling the substrate specificity and the biological function of sialidases.
Keywords: carbohydrates, chemoenzymatic synthesis, high-throughput, sialic acids, sialidases
Introduction
Sialidases, or neuraminidases (EC 3.2.1.18), are sialic acid-releasing exoglycosidases that catalyze the removal of terminal sialic acids from sialosides and sialoglycoconjugates in nature. Sialidases have been found in animals, bacteria, viruses, fungi, and protozoa.[1,2] Human sialidases have been shown to play pivotal roles in sialic acid metabolism[3] and to relate to a number of disease states such as sialidosis (a disease caused by lysosomal storage disorder)[4] and cancer.[5] Sialidases are also believed to play significant roles on the pathogenesis and pathology of bacterial and viral infections.[2,6] In some pathogenic bacteria, they serve as important virulence factors associated with bacterial invasion and colonization within the host.[2] Viral sialidases such as influenza virus neuraminidases catalyze the removal of sialic acid from the surface of infected host cells to release the newly formed progeny virus.[7]
Sialic acids, the monosaccharide residues that are cleaved by sialidases, are a family of α-keto acids with a nine-carbon backbone. They have been predominantly found as the outermost carbohydrates in vertebrates or as cell surface components in certain types of bacteria. Currently, more than 50 different sialic acid structures have been found in nature, including Neu5Ac (N-acetylneuraminic acid), Neu5Gc (N-glycolylneuraminic acid), KDN (deaminoneuraminc acid), and their acetylated, methylated, lactylated, sulfated, and phosphorylated derivatives (Scheme 1).[8] Among them, more than fifteen structures (mainly Neu5Ac and its derivatives) have been found in humans.[9] Terminal sialic acid residues are usually linked to a galactose residue through an α2,3- or an α2,6- linkage, to an N-acetylgalactosamine residue through an α2,6-linkage, or to another sialic acid through an α2,8-linkage.
Scheme 1.

Three basic forms of naturally occurring sialic acids. Neu5Ac, N-acetylneuraminic acid; Neu5Gc, N-glycolylneuraminic acid; KDN, deaminoneuraminc acid.
Sialidases from different sources vary in their preference in recognizing different sialic acid containing structures. The linkage specificity of some sialidases, especially those from bacterial and viral sources, has been studied using synthetic and natural substrates, including natural glycoproteins, purified milk oligosaccharides, desialylated and resialylated glycoproteins and cells.[10] Studies using synthetic substrates have been restricted to a limited number of compounds. Natural glycoproteins and their desialylated and resialylated forms can provide general information about the specificity of sialidase recognition, but data analyses could be difficult due to heterogeneity of the substrates. In order to systematically study the substrate specificity of sialidases related to the sialic acid forms, the sialyl linkages, and the penultimate monosaccharide residues in sialosides, we report here the chemoenzymatic synthesis of sialoside libraries containing Siaα2,3Galβ-pNP and Siaα2,6Gal(NAc)β-pNP with different naturally occurring sialic acid forms. A 96-well plate based colorimetric method for high-throughput screening is also developed to determine the structure determinants of the substrates recognized by different bacterial sialidases. This method can be easily used for quickly screening the substrate specificity of other sialidases, including those from bacterial, viral, and mammalian sources.
Results and Discussion
Synthesis of sialoside libraries
In order to elucidate the relationship of the different structural determinants of naturally occurring sialosides and the hydrolytic activity of sialidases, sialoside libraries containing fifteen sialosides were synthesized using a highly efficient one-pot three-enzyme chemoenzymatic method (Scheme 2).[9,11] In this method, N-acetylmannosamine (ManNAc) derivatives such as 6-O-acetyl ManNAc (ManN2,6Ac2) 2, 6-O-lactyl ManNAc (ManNAc6Lt) 3, and N-glycolylmannosamine (ManNGc) 4, were chemically or enzymatically synthesized as described previously.[9] These compounds, together with ManNAc 1 and mannose 5, were used as sialic acid precursors. They were converted to naturally occurring sialic acids by an aldol condensation reaction catalyzed by an E. coli K-12 sialic acid aldolase, and then activated to CMP-sialic acids by an N. meningitidis CMP-sialic acid synthetase (NmCSS). The sialic acid residue in the CMP-sialic acids was then transferred to para-nitrophenyl β-D-galactopyranoside (Galβ-pNP) by a multifunctional Pasteurella multocida sialyltransferase (PmST1) to form α2,3-linked sialosides or to Galβ-pNP and para-nitrophenyl N-acetamido-β-D-galactopyranoside (GalNAcβ-pNP) by a Photobacterium damsela α2,6-sialyltransferase (Pd2,6ST) to form α2,6-linked sialosides. These para-nitrophenol-tagged sialosides containing different naturally occurring sialic acid forms, different sialyl linkages, and different penultimate monosaccharides are the representative terminal disaccharide units of naturally occurring sialoglycoconjugates.
Scheme 2.

One-pot three-enzyme chemoenzymatic approach for the synthesis of sialoside libraries. R = NHAc, NHGc, or OH; R’ = H, Ac, or lactyl.
As shown in Table 1, using ManNAc, ManNAc derivatives, and mannose 1–5 as naturally occurring sialic acid precursors, α2,3-linked sialosides Siaα2,3Galβ-pNP 8–12 were synthesized in 75–91% yields when Galβ-pNP 6 was used as an acceptor and PmST1 was used as the sialyltransferase. From the same set of sialic acid precursors, α2,6-linked sialosides Siaα2,6Galβ-pNP 13–17 and Siaα2,6GalNAcβ-pNP 18–22 were synthesized in 78–92% and 33–92% yields, respectively, from Galβ-pNP 6 and GalNAcβ-pNP 7 acceptors when Pd2,6ST was used as the sialyltransferase. Except for the synthesis of Neu5,9Ac2α2,6GalNAcβ-pNP 19 and Neu5Ac9Ltα2,6GalNAcβ-pNP 20, all other sialosides were obtained efficiently with a yield equal or higher than 75%. The relatively low yields for the production of sialosides 19 and 20 were caused by the formation of deacetylated and delactylated byproducts respectively in longer incubation time required for the synthesis of these two compounds.
Table 1.
Sialoside libraries synthesized for sialidase substrate specificity assays using the one-pot three-enzyme chemoenzymatic approach shown in Scheme 2.
|
|
|
|||||
|---|---|---|---|---|---|---|
| Donor Precursor | ||||||
| Product using PmST1 | Yield(%) | Product using Pd2,6ST | Yield(%) | Product using Pd2,6ST | Yield(%) | |
|
| ||||||
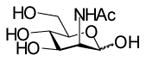
1 |
8 |
91 |

13 |
88 |

18 |
92 |

2 |
9 |
87 |

14 |
78 |

19 |
33 |

3 |
10 |
75 |

15 |
92 |

20 |
42 |

4 |
11 |
76 |

16 |
85 |

21 |
85 |
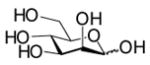
5 |
12 |
75 |

17 |
79 |
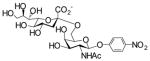
22 |
80 |
Due to the important roles of sialidases in many physiological and pathological processes, the investigation of sialidase substrate specificity has drawn a considerable interest. Previous studies, however, have been focused on using a limited number of synthetic compounds or glycoproteins, sialyloligosaccharides and glycolipids isolated from natural sources before or after glycan remodeling,[10b,12] which are heterogeneous compounds with varied carbohydrate structures and variable sites of glycosylation. Although these earlier studies have provided us some useful information, it is necessary to investigate the substrate specificities of sialidases with structurally defined substrates containing diverse naturally occurring sialic acid forms, different sialyl linkages, and different penultimate monosaccharides. These structurally defined sialosides containing naturally occurring sialic acid modifications are extremely difficult to isolate in homogeneous forms from natural sources.[13] Obtaining these compounds by pure organic glycosylation also poses a big challenge.[14] The synthesis of pNP-tagged sialoside libraries reported here using the highly efficient one-pot three-enzyme chemoenzymatic method established in our laboratory represents the first step towards the systematic investigation of the substrate specificity of a variety of sialidases.
A coupled enzyme colorimetric assay for high-throughput substrate specificity studies of sialidases
Using the sialoside libraries obtained, a 96-well plate based coupled enzymatic assay for high-throughput colorimetric screening of structures that can be recognized by different sialidases was developed (Figure 1). In this method, individual sialosides are incubated in triplicate with an appropriate amount of a sialidase of interest and an excess amount of exogalactosidase or hexosaminidase. If the sialoside is a substrate for the sialidase, the terminal sialic acid residue is cleaved by the enzyme to give Galβ-pNP or GalNAcβ-pNP, gwhich is quickly hydrolyzed by the excess amount of galactosidase or hexosaminidase in the reaction mixture to give para-nitrophenol (pNP) and Gal or GalNAc. By adjusting the pH of the reaction mixture to 9.6, the assay is stopped and the amount of the para-nitrophenolate formed is determined spectrophotometrically at A405 nm. The amount of the para-nitrophenolate formed is equivalent to that of the sialic acid released from the sialoside by the sialidase. A comparison of the color developed for different sialosides by different sialidases reveals the effect of the structural diversity of the sialoside substrate on the hydrolytic activity of different sialidases.
Figure 1.

A 96-well plate based colorimetric high-throughput screening method for sialidase substrate specificity studies. Sialoside libraries containing sialosides with different sialic acid forms, different sialyl linkages, and different penultimate monosaccharides were incubated in triplicate with different sialidases and an excess amount of a β-galactosidase or a hexosaminidase at 37 °C for 20–60 min. The reaction was stopped by adding CAPS buffer to adjust pH to 9.6. The formation of the p-nitro phenolate was monitored by measuring the A405 nm using a microplate reader. The absorbance of the solution correlates to the amount of the sialic acid cleaved by the sialidase, and to the hydrolytic activity of the sialidase.
Although a similar coupled enzyme colorimetric assay was reported by Paulson and coworkers,[10g] the method was limited to two compounds Neu5Acα2,3Galβ-pNP and Neu5Acα2,6Galβ-pNP. It was, therefore, only limited to the determination of linkage specificity of sialidases. Furthermore, the reported method was based on assays using cuvettes instead of a 96-well plate format and thus was not suitable for high-throughput screening development. The addition of β-galactosidase after the sialidase reaction further complicated the assay procedures.
Previous high-throughput screening methods[15] using fluorophore, chromophore, or chemilluminescent compound labelled sialic acids have several limitations. Firstly, the chemical synthesis of a library of α-sialosides containing naturally occurring sialic acid residue directly linked to a fluorophore, a choromophore, or a chemilluminescent compound poses difficulties. Therefore, most of these studies are limited to a few naturally occurring sialic acid forms.[12c] Secondly, these compounds preclude the study of the effect of sialyl linkage and the penultimate monosaccharide on sialidase specificity. In contrast, the coupled enzyme colorimetric assay reported here allows for the systematic study of the effect of the sialic acid structures, sialyl linkages, and the structures of the penultimate monosaccharide residues on the sialidase substrate specificity using a convenient and commonly accessible 96-well plate based high-throughput screening format.
Substrate specificity of seven bacterial sialidases
Seven different bacterial sialidases, including commercially available sialidases from Arthrobacter ureafaciens, Clostridium perfingens, Streptococcus sp. IID, Vibrio cholera, Salmonella typhimurium, and Streptococcus pneumoniae, as well as a multifunctional Pasteurella multocida sialyltransferase (PmST1) which also possesses sialidase activity,[11] were used as model systems to test the application of the sialoside libraries and the 96-well plate based high-throughput colorimetric screening method. As shown in Figure 2, four bacterial sialidases including those from Arthrobacter ureafaciens (Figure 2A), Clostridium perfingens (Figure 2B), Streptococcus sp. IID (Figure 2C), and Vibrio cholera (Figure 2D) can cleave both α2,3- and α2,6- linked sialosides with high efficiency. Sialidases from Salmonella typhimurium (Figure 2E), Streptococcus pneumoniae (Figure 2F) and PmST1 (Figure 2G) can only cleave α2,3-linked sialosides efficiently.
Figure 2.

The effect of structural variation of sialosides on the hydrolytic activity of seven bacterial sialidases: A) Arthrobacter ureafacien sialidase; B) Clostridium perfingens sialidase; C) Streptococcus sp IID sialidase; D) Vibrio cholera sialidase; E) Salmonella typhimurium sialidase; F) Streptococcus pneumoniae sialidase; G) PmST1. Except for the Salmonella typhimurium (Figure 2E) and Streptococcus pneumoniae (Figure 2F) sialidases, for which only Siaα2,3Galβ-pNP 8–12, Neu5Acα2,6Galβ-pNP 13, and Neu5Acα2,6GalNAcβ-pNP 18 were used for screening, all other sialidases were screened using all the sialosides (8–22) synthesized in this report. White columns, Siaα2,3Galβ-pNP; grey columns, Siaα2,6Galβ-pNP; black columns, Siaα2,6GalNAcβ-pNP. Note: The Salmonella typhimurium (Figure 2E), Streptococcus pneumoniae (Figure 2F), and PmST1 (Figure 2G) sialidases do not have detectable activity (<1% activity) on cleaving α2,6-linked sialosides. Except for Streptococcus pneumoniae (Figure 2F) and PmST1 (Figure 2G) sialidases, all other sialidases that have been tested do not have detectable activity (<1% activity) on cleaving terminal KDN residue from sialosides.
Among all of the seven bacterial sialidases tested, only PmST1 (Figure 2G) can cleave KDN from KDNα2,3-linked galactoside quite efficiently, at 8.5% of the cleavage rate for Neu5Acα2,3Gal terminated structure. The Streptococcus pneumoniae sialidase (Figure 2F) also cleaves KDN from KDNα2,3Galβ-pNP 12, although at a much slower rate (0.7%) compared to that for Neu5Acα2,3Galβ-pNP 8. All other sialidases, however, have no hydrolytic activity on KDN-terminated sialosides. PmST1, therefore, can be used as a powerful and specific α2,3-sialidase in cleaving α2,3-linked sialosides on cell surface or in glycoconjugates for glycan structural analysis or modifications.
Among four bacterial sialidases (Figure 2A - 2D) that can cleave both α2,3- and α2,6- linked sialosides, the sialyl linkage affects the hydrolytic activity of the sialidases only moderately with less than three fold difference (compare the white and grey columns in Figure 2A-2D). Except for the Arthrobacter ureafaciens sialidase (Figure 2A) which prefers to cleave α2,6-linked sialyl linkages, all other three sialidases favor the α2,3-linked sialyl linkages. The penultimate monosaccharide also affects the hydrolytic activity of the sialidases moderately with less than three fold difference (compare the grey and black columns in Figure 2A-2D). The 9-O-modification at Neu5Ac also affects the hydrolysis activity of these sialidases to a similar extent. For example, 9-O-acetylation and 9-O-lactylation decreases the rate of sialoside hydrolysis by the sialidases. Such rate drop is related to the size of the substituent on the 9-O-position of the Neu5Ac with the bigger the substituent, the slower the hydrolysis rate. The exceptions are the effect of Neu5Ac 9-O-modification on hydrolysis of Neu5Acα2,3Galβ-pNP by Vibrio cholera sialidase and the effect of Neu5Ac 9-O-modification on the hydrolysis of Neu5Acα2,6GalNAcβ-pNP by Streptococcus sp. IID and Vibrio cholera sialidases. For the Vibrio cholera sialidase, both 9-O-acetylation and 9-O-lactylation on the Neu5Ac residue in Neu5Acα2,3Galβ-pNP and Neu5Acα2,6GalNAcβ-pNP decrease the hydrolysis of the sialidase to the same extent. While for Streptococcus sp. IID sialidase, these modifications actually increase the rate of Neu5Acα2,6GalNAcβ-pNP hydrolysis by the sialidase. Substituting the N-acetyl group at C-5 of the Neu5Ac in Neu5Acα2,3Galβ-pNP and Neu5Acα2,6Galβ-pNP to an N-glycolyl (has an extra hydroxyl group) has very little effect on the hydrolytic activity of the Vibrio cholera sialidase but has quite dramatic effect for other three sialidases for which less than 50% activity remains. Replacing the N-acetyl group at C-5 of the Neu5Ac in Neu5Acα2,6GalNAcβ-pNP by an N-glycolyl group, however, decreases the activity of all four sialidases extensively with less than 50% activity retained. KDN-terminated sialosides (have a hydroxyl group at C-5 of the sialic acid residue) are completely resistant to the hydrolysis by these four sialidases, indicating the importance of the amido group at C-5 of the sialic acid residue in sialoside substrates to sialidases.
Although the sialidases from Salmonella typhimurium (Figure 2E) and Streptococcus pneumoniae (Figure 2F), as well as PmST1 (Figure 2G) are specific for α2,3-linked sialosides, Salmonella typhimurium sialidase can also cleave α2,6-linked sialosides at a much slower rate (more than 200 fold slower) than the cleavage of α2,3-linked sialosides (data not shown). The latter observation corroborates well with that reported previously.[16] In addition, this sialidase cleaves Neu5Acα2,6GalNAcβ-pNP 18 at a rate about five folds slower than that of the cleavage of Neu5Acα2,6Galβ-pNP 13 (data not shown). For the three sialidases (Figure 2E-2G) that are specific for α2,3-linked sialosides, 9-O-substitution decreases the rate of sialidase hydrolysis, but it seems unrelated to the type of the substitution. For example, 9-O-acetylation and 9-O-lactylation decrease the rate of sialoside hydrolysis by individual sialidases to about the same extent. Similar to that described earlier for the sialidases from Arthrobacter ureafaciens, Clostridium perfingens, Streptococcus sp. IID, and Vibrio cholera (Figure 2A-2D), substituting the N-acetyl group at C-5 of Neu5Ac by an N-glycolyl group decreases the hydrolytic activity of both Salmonella typhimurium and Streptococcus pneumoniae sialidases (Figure 2E-2F). In contrast, PmST1 (Figure 2G) prefers to cleave the Neu5Gc at an 18 fold higher rate than that of the Neu5Ac cleavage.
Overall, 9-O-acetylation on Neu5Ac has little effect on the hydrolysis of sialosides, irrespective of the acceptor sugar or the sialyl linkage, by sialidases from Arthrobacter ureafaciens (Figure 2A), Streptococcus sp. IID (Figure 2C), and Streptococcus pneumoniae (Figure 2F) (the hydrolytic activity decreases to 60–80%), but decreases the hydrolytic activity of sialidases from Clostridium perfingens (Figure 2B), Vibrio cholera (Figure 2D), Streptococcus pneumoniae (Figure 2E), and PmST1 (Figure 2G) to 20–40%. However, 9-O-lactylation on Neu5Ac dramatically reduces the hydrolysis rate of both Arthrobacter ureafaciens (Figure 2A) and Clostridium perfingens (Figure 2B) sialidases. This modification is very well tolerated by Streptococcus sp. IID (Figure 2C) and Streptococcus pneumoniae (Figure 2F) sialidases.
The results obtained in this study for commercially available sialidases are comparable to those reported previously.[2,10g] It is interesting to point out that the Clostridium perfingens sialidase shows little difference in cleaving Neu5Ac from either α2,3- or α2,6- linked GalβpNP sialosides on the 96-well plate based colorimetric assays. High-performance liquid chromatography (HPLC) based hydrolysis studies carried out for the Clostridium perfingens sialidase using Neu5Acα2,3LacMU and Neu5Acα2,6LacMU as substrates indicate that the enzyme prefers to cleave the α2,3-sialyl linkage in Neu5Acα2,3LacMU (Neu5Ac α2,3-linked to 4-methylumbelliferyl-β-D-lactoside) over the α2,6-sialyl linkage in Neu5Acα2,6LacMU (data not shown). This is in consistent to a previous report which showed the preferential cleavage of Neu5Acα2,3Lac over Neu5Acα2,6Lac by this enzyme.[17] This result indicates the influence of the extended carbohydrate structures linked to the sialic acid in the sialoside substrate (Galβ1,4Glc in lactose versus Galβ) to the hydrolytic activity of the sialidase.
It seems that the substrate specificity of a certain bacterial sialidase reflects its biological environment and biological function. For example, it is known that Pasteurella multocida is an important multi-species commensal and opportunistic pathogen that causes serious diseases in food animals and humans.[18] This may explain the flexible substrate specificity of PmST1 sialidase. The preference of the PmST1 towards Neu5Gc terminated structures, which are not presented in significant amount in humans but are widely distributed in animals, correlates well to the fact that Pasteurella multocida is mainly an animal pathogen. Another example is Vibrio cholera, a bacterium that causes cholera, an acute bacterial infectious disease of intestine. The flexible substrate specificity of Vibrio cholera sialidase correlates well to the fact that Neu5Ac and multiple acylated sialic acid (mainly Neu5,9Ac2) have been found in significant proportions (15–43%) in both α2,3- and α2,6- linkages to Gal and GalNAc residues in intestinal mucins of different parts of human intestine.[19] The broad specificity of the Vibrio cholera sialidase towards sialosides on gastro intestinal tract mucins would enable the bacterium to access the underlying epithelial cell surface (the site of bacterial invasion), hence increasing the ability of the bacterium in developing colonies in this area.
Sialidases have also been extensively used as reagents to demonstrate the presence and the roles of different types of sialic acids on sialoglycoconjugates in nature.[20] Sialidases with broad substrate specificity and those with substrate selectivity would both be valuable in glycan analysis studies. The sialoside libraries and the high-throughput screening method presented here have conveniently identified several bacterial sialidases that have flexibility towards the sialyl linkage and the penultimate monosaccharide on the substrates. These include sialidases from Arthrobacter ureafaciens, Clostridium perfingens, Streptococcus sp. IID, and Vibrio cholera. Sialidases that are specific to α2,3- sialyl linkages have also been identified from this study, including sialidases from Salmonella typhimurium and Streptococcus pneumoniae, as well as PmST1. PmST1 has also been shown to be the only sialidase among the seven bacterial sialidases that have been tested, that can efficiently cleave the terminal KDN in KDN-terminated sialosides. All these results provide us better knowledge in selecting a particular sialidase as a suitable reagent for practical glycan analysis of biological samples.
Conclusion
Studies of the detailed substrate specificity of sialidases have been impeded by the lack of diverse structurally defined sialosides. We report herein the synthesis of sialoside libraries using an efficient one-pot three-enzyme chemoenzymatic method and the application of these sialosides in a 96-well plate based rapid screening platform for examining the substrate specificity of various sialidases. Both synthetic and screening methods are highly efficient and applicable for better understanding sialidase mediated bio-processes and the significance of nature’s sialic acid diversity. The data obtained from this study indicate that the hydrolytic activity of different sialidases is fine tuned by the structure of the terminal sialic acid residue, the type of the sialyl linkage, and the structure of the penultimate monosaccharide in sialosides. The substrate specificity elucidated for individual sialidases also provides crucial information for sialidase selection in glycan analysis of biological samples.
Experimental Section
Materials
Arthrobacter ureafaciens and Streptococcus sp. IID sialidases were purchased from MP Biomedicals. Salmonella typhimurium sialidase and Streptococcus pneumoniae sialidase were bought from Prozyme. Clostridium perfingens sialidase (Type VI), Vibrio cholera sialidase (Type III), β-galactosidase from Aspergillus oryzae, and hexosaminidase from Canavalia ensiformis (jack bean) were purchased from Sigma. All of these enzymes were used without further purification. PmST1 was expressed in E. coli and purified as described previously.[11] Sodium pyruvate (Fisher Biotech), N-acetylmannosamine (ManNAc, Sigma), mannose (Acros Organics), para-nitrophenyl β-D-galactopyranoside (Galβ-pNP, Sigma), para-nitrophenyl N-acetamido-β-D-galactopyranoside (GalNAcβ-pNP, Senn Chemicals AG), and cytidine 5’-triphosphate disodium salt (CTP, Sigma) were from commercially available sources. Silica gel 60 Å (40–63 μm, Sorbent technologies) was used for flash column chromatography. Analytical thin-layer chromatography was performed on silica gel plates 60 GF254 (Sorbent technologies) using anisaldehyde stain for detection. Gel filtration chromatography was performed using a column (100 cm ×2.5 cm) packed with BioGel P-2 Fine resins (Bio-Rad, Hercules, CA). 96-Well plates for the sialidase assays were from Corning.
Synthesis of sialic acid precursors
Chemical or enzymatic synthesis of ManNAc derivatives including ManN2,6Ac2 2, ManNAc6Lt 3, and ManNGc 4 was performed as described previously.[9,21]
Enzymatic Synthesis of Sialosides
General procedures for one-pot three-enzyme preparative synthesis of Neu5Ac, Neu5Gc, or KDN-terminated sialosides 8, 11, 12, 13, 16, and 17 using Galβ-pNP 6 as the acceptor: Galβ-pNP (1.0 equiv., 10 mM), a sialic acid precursor (e.g. ManNAc 1, ManNGc 4, or mannose 5, 1.2 equiv.), sodium pyruvate (6.0 equiv.), and CTP (1.3 equiv.) were dissolved in water in a 50 ml centrifuge tube containing Tris-HCl buffer (100 mM, pH 8.8) and MgCl2 (20 mM). After the addition of appropriate amounts of an E. coli sialic acid aldolase (0.2 – 0.75 mg), an N. meningitidis CMP-sialic acid synthetase (0.25 – 1.0 mg), and a sialyltransferase (0.05 – 0.2 mg of PmST1 or 0.1 –0.5 mg of Pd2,6ST), water was added to bring the final volume of the reaction mixture to 10 ml. The reaction was carried out by incubating the solution in an incubator for 4–8 h at 37°C with agitation at 125 rpm. The product formation was monitored by thin layer chromatography (TLC) using EtOAc:MeOH:H2O:HOAc = 5:2:1:0.1 (by volume) as the developing reagent and stained with p-anisaldehyde sugar stain. The reaction was stopped by adding the same volume of ice-cold ethanol and incubating at 4 °C for 30 min. The mixture was centrifuged to remove precipitates. The supernatant was concentrated and passed through a BioGel P-2 gel filtration column to obtain desired product. Silica gel purification (EtOAc:MeOH:H2O = 5:2:1) was applied when necessary to achieve further purification.
General procedures for one-pot three enzyme preparative synthesis of Neu5Ac, Neu5Gc, or KDN-terminated sialosides 18, 21, and 22 using GalNAcβ-pNP 7 as the acceptor: Most of the procedures were the same as described above for the synthesis of sialosides using Galβ-pNP as the acceptor. The differences are i) GalNAcβ-pNP (1.0 equiv., 7 mM) was used instead of Galβ-pNP; ii) 5% DMF was added to help the solublization of GalNAcβ-pNP; iii) the final volume of the reaction mixture was 15 ml; iv) the reaction was carried out at a longer time (6–12 h) at 37 °C with agitation at 125 rpm; and v) the enzymes were added in 2–3 portions over the reaction period. Reactions for synthesizing sialosides containing Neu5,9Ac2 and Neu5Ac9Lt 9, 10, 14, 15, 19, and 20 were carried out at pH 7.5 in Tris-HCl buffer (100 mM) containing the sialic acid precursor (ManN2,6Ac2 2 or ManNAc6Lt 3, 1.0 equiv.) and the acceptor (Galβ-pNP 6 or GalNAcβ-pNP 7, 1.5 equiv.). All other conditions and procedures were the same as described above. 1H NMR, 13C NMR, and high resolution mass spectroscopy (HRMS) data for the sialosides synthesized are included in Supplemental Data.
Sialidase substrate specificity assays
All sialidase assays were carried out at 37 °C in triplicate in a final volume of 100 μl containing substrate (35 nmoles, 35 μl of 1 mM stock solution), galactosidase (100 mU) (for sialylated Galβ-pNP) or hexosaminidase (50 mU) (for sialylated GalNAcβ-pNP) unless otherwise noted. The amount of excess galactosidase or hexosaminidase to be used was predetermined to ensure the complete and fast hydrolysis of the Galβ-pNP or GalNAcβ-pNP formed (data not shown). The conditions used for various sialidases were as follows: Arthrobacter ureafaciens sialidase (50 mM sodium acetate buffer, pH 4.8, 5 mU sialidase, and 100 mU hexosamindase), Clostridium perfingens sialidase (50 mM sodium acetate buffer, pH 5.0, and 10 mU sialidase), Streptococcus sp. IID sialidase (10 mM CaCl2,100 mM sodium acetate buffer, pH 6.5, 25 mU sialidase), Vibrio cholera sialidase (150 mM NaCl, 9 mM CaCl2, 50 mM sodium acetate buffer, pH 5.5, and 3 mU sialidase), Salmonella typhimurium sialidase (100 mM NaCl, 50 mM sodium acetate buffer, pH 5.5, and 6 mU sialidase), Streptococcus pneumoniae sialidase (50 mM sodium phosphate buffer, pH 6.0, and 10 mU sialidase), and PmST1 (100 nmoles of substrate, 500 mU galactosidase, 50 mM sodium acetate buffer, pH 5.5, and 125 μg sialidase). The reaction mixtures were typically incubated for 20–60 min (the reaction time was varied for different sialidases in order to keep the A405 nm of the para-nitrophenolate formed after pH adjustment in the linear range). The assay was stopped by the addition of CAPS (N-cyclohexyl-3-aminopropanesulfonic acid, 130 μl, NaOH was used to adjust the pH) buffer (0.5 M, pH 9.6). The amount of the para-nitrophenolate formed is determined by measuring the A405 nm using a microplate reader (Biotek, model EL311).
Supplementary Material
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
Acknowledgments
We thank Prof. Claude F. Meares and Prof. Bruce D. Hammock for allowing us to use their microplate readers. This work was supported by Mizutani Foundation for Glycoscience and NIH R01GM076360.
References
- 1.Takada K, Hamada T, Hirota H, Nakao Y, Matsunaga S, van Soest RW, Fusetani N. Chem Biol. 2006;13:569–574. doi: 10.1016/j.chembiol.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Corfield T. Glycobiology. 1992;2:509–521. doi: 10.1093/glycob/2.6.509. [DOI] [PubMed] [Google Scholar]
- 3.Monti E, Bassi MT, Bresciani R, Civini S, Croci GL, Papini N, Riboni M, Zanchetti G, Ballabio A, Preti A, Tettamanti G, Venerando B, Borsani G. Genomics. 2004;83:445–453. doi: 10.1016/j.ygeno.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 4.a) Milner CM, Smith SV, Carrillo MB, Taylor GL, Hollinshead M, Campbell RD. J Biol Chem. 1997;272:4549–4558. doi: 10.1074/jbc.272.7.4549. [DOI] [PubMed] [Google Scholar]; b) Bonten E, van der Spoel A, Fornerod M, Grosveld G, d'Azzo A. Genes Dev. 1996;10:3156–3169. doi: 10.1101/gad.10.24.3156. [DOI] [PubMed] [Google Scholar]; c) Pshezhetsky AV, Richard C, Michaud L, Igdoura S, Wang S, Elsliger MA, Qu J, Leclerc D, Gravel R, Dallaire L, Potier M. Nat Genet. 1997;15:316–320. doi: 10.1038/ng0397-316. [DOI] [PubMed] [Google Scholar]; d) Achyuthan KE, Achyuthan AM. Comp Biochem Physiol B Biochem Mol Biol. 2001;129:29–64. doi: 10.1016/s1096-4959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 5.a) Kakugawa Y, Wada T, Yamaguchi K, Yamanami H, Ouchi K, Sato I, Miyagi T. Proc Natl Acad Sci USA. 2002;99:10718–10723. doi: 10.1073/pnas.152597199. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meuillet EJ, Kroes R, Yamamoto H, Warner TG, Ferrari J, Mania-Farnell B, George D, Rebbaa A, Moskal JR, Bremer EG. Cancer Res. 1999;59:234–240. [PubMed] [Google Scholar]; c) Miyagi T, Wada T, Yamaguchi K, Hata K. Glycoconj J. 2004;20:189–198. doi: 10.1023/B:GLYC.0000024250.48506.bf. [DOI] [PubMed] [Google Scholar]
- 6.Vimr ER. Trends Microbiol. 1994;2:271–277. doi: 10.1016/0966-842x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki Y. Biol Pharm Bull. 2005;28:399–408. doi: 10.1248/bpb.28.399. [DOI] [PubMed] [Google Scholar]
- 8.a) Angata T, Varki A. Chem Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]; b) Schauer R. Glycoconj J. 2000;17:485–499. doi: 10.1023/A:1011062223612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew Chem. 2006;118:4042–4048. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed Engl. 2006;45:3938–3944. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Paulson JC, Rogers GN. Methods Enzymol. 1987;138:162–168. doi: 10.1016/0076-6879(87)38013-9. [DOI] [PubMed] [Google Scholar]; b) Corfield AP, Higa H, Paulson JC, Schauer R. Biochim Biophys Acta. 1983;744:121–126. doi: 10.1016/0167-4838(83)90080-8. [DOI] [PubMed] [Google Scholar]; c) Baum LG, Paulson JC. Virology. 1991;180:10–15. doi: 10.1016/0042-6822(91)90003-t. [DOI] [PubMed] [Google Scholar]; d) Drzeniek R. Curr Top Microbiol Immunol. 1972;59:35–74. doi: 10.1007/978-3-642-65444-2_2. [DOI] [PubMed] [Google Scholar]; e) Drzeniek R. Histochem J. 1973;5:271–290. doi: 10.1007/BF01004994. [DOI] [PubMed] [Google Scholar]; f) Frisch A, Neufeld EF. Anal Biochem. 1979;95:222–227. doi: 10.1016/0003-2697(79)90209-4. [DOI] [PubMed] [Google Scholar]; f) Kodama H, Baum LG, Paulson JC. Carbohydr Res. 1991;218:111–119. doi: 10.1016/0008-6215(91)84090-2. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, Chokhawala H, Karpel R, Yu H, Wu B, Zhang J, Zhang Y, Jia Q, Chen X. J Am Chem Soc. 2005;127:17618–17619. doi: 10.1021/ja0561690. [DOI] [PubMed] [Google Scholar]
- 12.a) Paulson JC, Weinstein J, Dorland L, van Halbeek H, Vliegenthart JF. J Biol Chem. 1982;257:12734–12738. [PubMed] [Google Scholar]; b) Corfield AP, Sanderwewer M, Veh RW, Wember M, Schauer R. Biol Chem Hoppe-Seyler. 1986;367:433–439. doi: 10.1515/bchm3.1986.367.1.433. [DOI] [PubMed] [Google Scholar]; c) Kleineidam RG, Furuhata K, Ogura H, Schauer R. Biol Chem Hoppe-Seyler. 1990;371:715–719. doi: 10.1515/bchm3.1990.371.2.715. [DOI] [PubMed] [Google Scholar]; d) Corfield AP, Veh RW, Wember M, Michalski JC, Schauer R. Biochem J. 1981;197:293–299. doi: 10.1042/bj1970293. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xu GY, Suzuki T, Maejima Y, Mizoguchi T, Tsuchiya M, Kiso M, Hasegawa A, Suzuki Y. Glycoconj J. 1995;12:156–161. doi: 10.1007/BF00731360. [DOI] [PubMed] [Google Scholar]
- 13.Chappell MD, Halcomb RL. J Am Chem Soc. 1997;119:3393–3394. [Google Scholar]
- 14.Kiefel MJ, von Itzstein M. Chem Rev. 2002;102:471–490. doi: 10.1021/cr000414a. [DOI] [PubMed] [Google Scholar]
- 15.a) Potier M, Mameli L, Belisle M, Dallaire L, Melancon SB. Anal Biochem. 1979;94:287–296. doi: 10.1016/0003-2697(79)90362-2. [DOI] [PubMed] [Google Scholar]; b) Buxton RC, Edwards B, Juo RR, Voyta JC, Tisdale M, Bethell RC. Anal Biochem. 2000;280:291–300. doi: 10.1006/abio.2000.4517. [DOI] [PubMed] [Google Scholar]; c) Eschenfelder V, Brossmer R. Carbohydr Res. 1987;162:294–297. doi: 10.1016/0008-6215(87)80224-0. [DOI] [PubMed] [Google Scholar]; d) Liav A, Hansjergen JA, Achyuthan KE, Shimasaki CD. Carbohydr Res. 1999;317:198–203. doi: 10.1016/s0008-6215(99)00058-0. [DOI] [PubMed] [Google Scholar]; e) Fujii I, Iwabuchi Y, Teshima T, Shiba T, Kikuchi M. Bioorg Med Chem. 1993;1:147–149. doi: 10.1016/s0968-0896(00)82112-4. [DOI] [PubMed] [Google Scholar]
- 16.Hoyer LL, Roggentin P, Schauer R, Vimr ER. J Biochem (Tokyo) 1991;110:462–467. doi: 10.1093/oxfordjournals.jbchem.a123603. [DOI] [PubMed] [Google Scholar]
- 17.Cassidy JT, Jourdian GW, Roseman S. J Biol Chem. 1965;240:3501–3506. [PubMed] [Google Scholar]
- 18.a) Carter GR. Adv Vet Sci. 1967;11:321–379. [PubMed] [Google Scholar]; b) May BJ, Zhang Q, Li LL, Paustian ML, Whittam TS, Kapur V. Proc Natl Acad Sci USA. 2001;98:3460–3465. doi: 10.1073/pnas.051634598. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Steenbergen SM, Lichtensteiger CA, Caughlan R, Garfinkle J, Fuller TE, Vimr ER. Infect Immun. 2005;73:1284–1294. doi: 10.1128/IAI.73.3.1284-1294.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbe C, Capon C, Maes E, Rousset M, Zweibaum A, Zanetta JP, Michalski JC. J Biol Chem. 2003;278:46337–46348. doi: 10.1074/jbc.M302529200. [DOI] [PubMed] [Google Scholar]
- 20.a) Razi N, Varki A. Proc Natl Acad Sci USA. 1998;95:7469–7474. doi: 10.1073/pnas.95.13.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lewis AL, Nizet V, Varki A. Proc Natl Acad Sci USA. 2004;101:11123–11128. doi: 10.1073/pnas.0403010101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Butor C, Diaz S, Varki A. J Biol Chem. 1993;268:10197–10206. [PubMed] [Google Scholar]; d) Reuter G, Schauer R. In Guide to Techniques in Glycobiology. 1994;230:168–199. [Google Scholar]
- 21.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.