

Abstract
The molecular mechanisms underlying the transition from recreational drug use to chronic addiction remain poorly understood. One molecule implicated in this process is ΔFosB, a transcription factor that accumulates in striatum after repeated drug exposure and mediates sensitized behavioral responses to psychostimulants and other drugs of abuse. The downstream transcriptional mechanisms by which ΔFosB regulates drug-induced behaviors are incompletely understood. We reported previously the chromatin remodeling mechanisms by which ΔFosB activates the expression of certain genes; however, the mechanisms underlying ΔFosB-mediated gene repression remain unknown. Here, we identify c-fos, an immediate early gene rapidly induced in striatum after acute psychostimulant exposure, as a novel downstream target that is repressed chronically by ΔFosB. We show that accumulation of ΔFosB in striatum after chronic amphetamine treatment desensitizes c-fos mRNA induction to a subsequent drug dose. ΔFosB desensitizes c-fos expression by recruiting histone deacetylase 1 (HDAC1) to the c-fos gene promoter, which, in turn, deacetylates surrounding histones and attenuates gene activity. Accordingly, local knock-out of HDAC1 in striatum abolishes amphetamine-induced desensitization of the c-fos gene. In concert, chronic amphetamine increases histone H3 methylation on the c-fos promoter, a chromatin modification also known to repress gene activity, as well as expression levels of the H3 histone methyltransferase, KMT1A (lysine methyltransferase 1A, formerly SUV39H1). This study reveals a novel epigenetic pathway through which ΔFosB mediates distinct transcriptional programs that may ultimately alter behavioral plasticity to chronic amphetamine exposure.
Keywords: addiction, amphetamine, striatum, chromatin, histone modification, gene regulation
Introduction
Repeated use of psychostimulants such as amphetamine and cocaine often results in a transition from recreational drug use to a chronically addicted state (Hyman et al., 2006). One mechanism implicated in this process involves the transcription factor ΔFosB, a highly stable splice product of the immediate early gene fosB, which dimerizes with Jun family proteins to form functional AP-1 transcriptional complexes (McClung et al., 2004). ΔFosB accumulates several-fold in striatum after repeated exposure to drugs of abuse, and this accumulation has been linked to increased cocaine reward, locomotor sensitization, and self-administration (Kelz et al., 1999; Colby et al., 2003; McClung et al., 2004), which together suggest a role in the neural mechanisms involved in transitioning between recreational and addicted drug use. According to this hypothesis, ΔFosB functions in a positive feedback loop by increasing drug-seeking behaviors, which, in turn, induce more ΔFosB. One key outstanding question is how does ΔFosB mediate its effects on drug-related behaviors? Genome-wide microarray studies in mice that overexpress ΔFosB in striatum provided the first insight into potential downstream targets (McClung and Nestler, 2003). This study suggested that ΔFosB can serve as a transcriptional activator or repressor, depending on the target gene. However, the study examined transcripts regulated in an overexpression setting, so it is not clear which of these genes are direct, physiological ΔFosB targets.
We recently identified the cyclin-dependent kinase 5 (cdk5) gene as a direct target for endogenous ΔFosB, which promotes Cdk5 transcription in striatum (Kumar et al., 2005). However, the mechanisms involved in the repression of target genes by ΔFosB have remained elusive. One attractive candidate is c-fos, a gene that is induced dramatically by acute psychostimulants but only weakly after repeated exposure (Hope et al., 1992; Persico et al., 1993; Steiner and Gerfen, 1993), when levels of ΔFosB and ΔFosB-containing AP-1 complexes are high (Hope et al., 1992, 1994). Because the c-fos gene contains an AP-1-like site in its proximal promoter (Morgan and Curran, 1989), it is a plausible candidate for ΔFosB-mediated repression. Induction of c-fos is traditionally viewed as an early marker of neural activation because it is rapidly and transiently induced in response to a variety of stimuli (Morgan and Curran, 1989). The c-fos gene is also important for behavioral responses to cocaine, because mice lacking c-fos in dopamine D1 receptor-containing neurons, the neuronal cell type where ΔFosB is induced by psychostimulants (McClung et al., 2004), have reduced behavioral sensitization to cocaine (Zhang et al., 2006). These findings led us to investigate whether ΔFosB controls c-fos gene activity after chronic amphetamine exposure. We describe here a novel epigenetic mechanism by which ΔFosB accumulation in response to chronic amphetamine feeds back to desensitize c-fos induction to subsequent drug doses. This novel interplay between ΔFosB and chromatin remodeling events on the c-fos promoter may be an important homeostatic mechanism to regulate an animal's sensitivity to repeated drug exposure.
Materials and Methods
RNA isolation and quantification.
Frozen brain tissue was thawed in TRIzol (Invitrogen) and processed according to the manufacturer's protocol. RNA was purified with RNeasy Micro columns (Qiagen). Total RNA was reverse-transcribed using Superscript III (Invitrogen). Real-time PCR was then run using SYBR Green (ABI) and quantified using the ΔΔCt method. For a complete list of primers, see supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Chromatin immunoprecipitation.
Chromatin was sonicated and then immunoprecipitated (supplemental Methods, available at www.jneurosci.org as supplemental material) using acetylated histone antibodies (Millipore), anti-histone deacetylase 1 (HDAC1) or anti-histone H3 dimethylated at lysine 9 (H3K9me2) (Abcam), anti-FosB (C terminus) (Kumar et al., 2005), anti-FosB (N terminus) (Santa Cruz Biotechnology), or a rabbit IgG control (Millipore). The immunoprecipitation was collected using Protein A beads from Millipore. After washing, chromatin was eluted from the beads and reverse cross-linked in the presence of proteinase K. DNA was then purified and quantified using real-time PCR.
Immunoprecipitation.
PC12 cells were transfected with V5-tagged HDAC1 (Montgomery et al., 2007), FosB, or ΔFosB as described previously (Carle et al., 2007). Cell lysates were split and incubated with either nonimmune IgG (Sigma) or anti-FosB antibodies (sc-48; Santa Cruz Biotechnology) overnight at 4°C. Immunoprecipitation was performed with Protein G beads (Sigma). The immunoprecipitated proteins were run with SDS-PAGE and analyzed by Western blotting using a custom polyclonal anti-FosB (N terminus) antibody (Carle et al., 2007) and anti-V5 antibody (Abcam). To determine whether HDAC1 and ΔFosB are binding partners in vivo, we used repeated electroconvulsive seizures to induce high levels of ΔFosB protein (Hope et al., 1994). Cortical tissue was dissected from chronic (seven, daily) seizure or sham-treated rats, lysed, and immunoprecipitated as described above with anti-HDAC1 antibodies (Abcam).
Laser capture microdissection.
Using stereotactic surgery, the ventral striata of mice were infected with an adeno-associated virus (AAV) expressing the indicated gene or green fluorescent protein (GFP) on opposite sides of the brain. After amphetamine treatment, frozen brains were processed into 8-μm-thick coronal sections and mounted onto membrane slides (Lieca). AAV-infected regions were laser-dissected (Leica) to exclude noninfected cells and processed with the PicoPure RNA extraction kit (MDS). RNA was amplified with the RiboAmp HS kit (MDS) and reverse-transcribed as described above. For complete details, see the supplemental Methods (available at www.jneurosci.org as supplemental material).
Results
ΔFosB desensitizes c-fos mRNA induction in striatum after chronic amphetamine exposure
To explore whether the desensitization of c-fos mRNA expression is a cellular adaptation controlled by ΔFosB, we treated rats with saline or acute or chronic amphetamine and let them withdraw in their home cage for 1–10 d. The rats were then analyzed 1 h after a saline or amphetamine challenge dose. As demonstrated previously (see Introduction), c-fos mRNA was induced fourfold in the striatum by acute amphetamine administration. In rats exposed previously to chronic amphetamine, however, the expression of c-fos in response to drug challenge was significantly attenuated for up to 5 d of drug withdrawal (Fig. 1A), a point at which ΔFosB remains elevated in this brain region (Hope et al., 1994). Additionally, in rats that were withdrawn from chronic amphetamine for 5 d, we found that basal c-fos mRNA expression was reduced below levels found in saline-treated controls (Fig. 1A). Importantly, the magnitude of c-fos induction to an amphetamine challenge was significantly attenuated at day 1 of withdrawal compared with saline-treated animals. Together, these findings demonstrate an effect of chronic amphetamine on both basal and induced c-fos mRNA levels, although with the two effects occurring with a complex time course.
Figure 1.
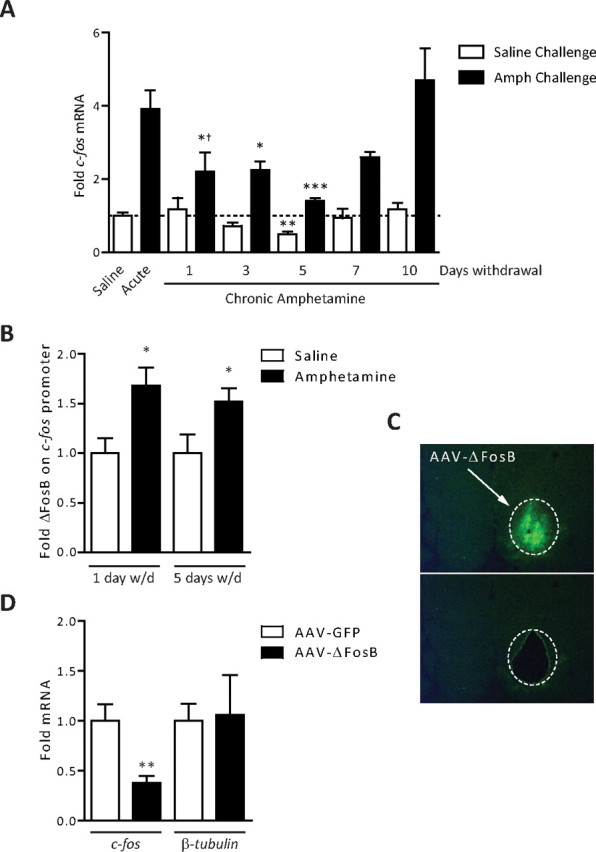
ΔFosB desensitizes c-fos mRNA induction in striatum after chronic amphetamine exposure. A, Rats were treated with saline, acute amphetamine (4 mg/kg), or chronic amphetamine (7 d), and allowed to withdraw for 1–10 d. Rats that received chronic amphetamine received a challenge dose of either saline or amphetamine (4 mg/kg) and were analyzed 1 h later. The levels of c-fos mRNA observed after an acute dose of amphetamine were significantly reduced in rats that were exposed previously to chronic amphetamine and withdrawn from the drug for 1–5 d (ANOVA, significant effect of drug challenge, F(1,35) = 132.57, p < 0.0001; significant effect of withdrawal, F(5,35) = 9.24, p < 0.0001; Bonferroni post hoc, 1 d withdrawal vs acute, *p < 0.05, 3 d withdrawal vs acute, **p < 0.01; 5 d withdrawal vs acute, ***p < 0.001; n = 3–5). Moreover, at 1 d of withdrawal, the fold induction of c-fos versus acute was significantly blunted (†p < 0.05), with a strong trend at 3 and 5 d of withdrawal. In rats treated with chronic amphetamine, after 5 d of withdrawal, there was a significant reduction in c-fos mRNA after a saline challenge compared with drug-naive controls (**p < 0.01). B, ChIP of striatal lysates found significantly more ΔFosB bound to the c-fos promoter after 1 and 5 d of withdrawal from chronic amphetamine (*p < 0.05, Student's t test; n = 4–5). C, Representative image of mouse striatum from the laser capture microscope before (top) or after (bottom) dissection of the infected region. D, Mice were given an acute dose of amphetamine and processed for laser capture microdissection. In striatum infected with AAV-ΔFosB compared with that infected with AAV-GFP, there was a significant decrease in c-fos mRNA (**p = 0.01, Student's t test; n = 3). No change was observed in β-tubulin mRNA (p > 0.05). Error bars indicate SEM.
To determine whether ΔFosB accumulation after chronic amphetamine directly contributes to the desensitization of c-fos expression, we first performed chromatin immunoprecipitation (ChIP) for ΔFosB on the c-fos gene promoter in striatum. As shown in Figure 1B, the c-fos promoter has significantly more ΔFosB bound after chronic amphetamine exposure, an effect seen for at least 5 d of drug withdrawal. These data correlate ΔFosB occupancy on the c-fos promoter with the kinetics of reduced c-fos gene activity. Next, to directly test whether ΔFosB causes reduced c-fos induction in response to amphetamine challenge, we used an AAV vector to overexpress either ΔFosB, or GFP as a control, in the striatum. We then isolated the infected striatum by laser microdissection (Fig. 1C) and performed quantitative reverse transcription (qRT)-PCR for c-fos mRNA. We observed significantly less c-fos mRNA induced after an acute dose of amphetamine in the striatal tissue infected with AAV-ΔFosB compared with the contralateral side infected with AAV-GFP, whereas levels of β-tubulin mRNA remained unchanged (Fig. 1D). These data suggest that c-fos desensitization is mediated by accumulation of ΔFosB on its promoter after chronic amphetamine exposure.
ΔFosB recruits HDAC1 to the c-fos promoter to mediate c-fos gene repression
To explore the mechanisms by which ΔFosB mediates c-fos desensitization, we focused on the time point at which c-fos was most significantly repressed: 5 d of withdrawal from chronic amphetamine. A key mechanism involved in c-fos activation in response to a variety of stimuli, including cocaine (Kumar et al., 2005), is histone acetylation. We were therefore interested to determine whether histone acetylation on the c-fos gene promoter was also induced by acute amphetamine and whether repeated drug exposure attenuated this response. Indeed, acute amphetamine increased histone H4 acetylation on the c-fos promoter and, after chronic amphetamine treatment, this induction was no longer observed (Fig. 2A). Acetylation of H4 was specific, because no effect was observed for H3 (data not shown). These data suggest that reduced histone acetylation, associated with a more compact and inactive chromatin structure (Kouzarides, 2007), contributes to the desensitization of the c-fos gene after chronic amphetamine exposure. To directly test this hypothesis, we treated rats with chronic amphetamine and, after 5 d of withdrawal, administered the HDAC inhibitor, sodium butyrate, or its vehicle. We found that sodium butyrate reversed the amphetamine-induced repression of c-fos expression (Fig. 2B), directly supporting the idea that hypoacetylation of histones on the c-fos promoter is a key mechanism underlying desensitization of the gene.
Figure 2.
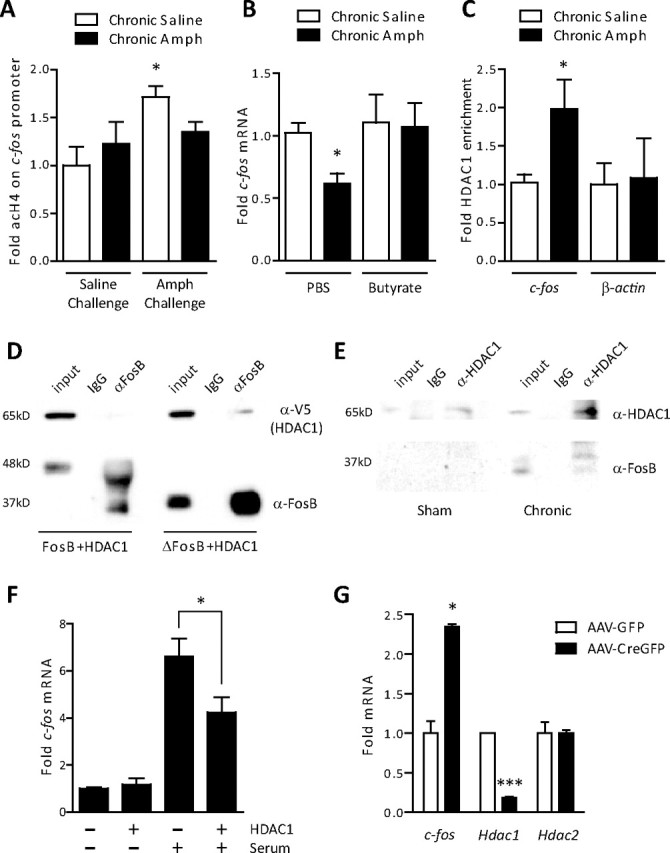
Recruitment of HDAC1 mediates ΔFosB action on c-fos. A, ChIP of striatal lysates revealed a significant increase in acetylated histone H4 on the promoter of c-fos 1 h after a challenge dose of amphetamine in drug-naive rats (ANOVA, significant effect of drug, F(1,12) = 6.26, p < 0.05; Bonferroni post hoc, *p < 0.05; n = 5). This increase was not observed in rats exposed previously to chronic amphetamine (Amph) (p > 0.05). B, The HDAC inhibitor, sodium butyrate (400 mg/kg), reversed the amphetamine-induced reduction in c-fos mRNA observed after 5 d of withdrawal (ANOVA, significant effect of butyrate, F(1,28) = 5.29, p < 0.05; Bonferroni post hoc, chronic amphetamine plus PBS vs chronic saline plus PBS, *p < 0.05, chronic amphetamine plus butyrate vs chronic saline plus PBS or butyrate, p > 0.05; n = 4–9). C, ChIP of striatal lysates revealed significantly more HDAC1 bound to the c-fos promoter (*p < 0.05, Student's t test; n = 5–6), but not the promoter of β-actin (p > 0.05), after 5 d of withdrawal from chronic amphetamine. D, HDAC1 was transfected into PC12 cells with either full-length FosB or ΔFosB. HDAC1 selectively immunoprecipitated with ΔFosB, not full-length FosB. E, In rats that received chronic electroconvulsive seizures (7 daily seizures), a condition known to increase ΔFosB several-fold, immunoprecipitation of HDAC1 pulled down significant levels of ΔFosB. This interaction was not observed in sham-treated animals. Blots are representative of two to three experiments. F, Serum stimulation increased c-fos mRNA significantly less in cells transfected with HDAC1 than with GFP (*p < 0.05; n = 3 independent experiments). G, Floxed HDAC1 mice whose striata were infected with either AAV-GFP or AAV-CreGFP on opposite sides of the brain were treated with chronic amphetamine (7 d, 4 mg/kg) and 5 d of withdrawal. We found significantly higher c-fos expression in cells infected with AAV-CreGFP, where HDAC1 had been floxed out, than in cells expressing AAV-GFP after a 2 mg/kg amphetamine challenge (*p < 0.05; n = 2–3). Significantly less Hdac1 mRNA was observed in AAV-Cre-infected neurons (***p < 0.001), whereas Hdac2 expression was unaffected (p > 0.05). Error bars indicate SEM.
To understand how ΔFosB inhibits histone acetylation on the c-fos promoter, we investigated whether ΔFosB interacts with enzymes that reduce histone acetylation, namely, HDACs. We first explored HDAC1 and HDAC2 because these enzymes form complexes with a variety of transcription factors to repress gene expression (Grozinger and Schreiber, 2002). Because preliminary ChIP studies identified significant HDAC1 binding on the c-fos promoter (see below), but no detectable HDAC2 (data not shown), we performed coimmunoprecipitation experiments to determine whether ΔFosB physically interacts with HDAC1. Indeed, we found that immunoprecipitation of ΔFosB also pulled down HDAC1 in PC12 cells (Fig. 2D). Importantly, this interaction is specific for ΔFosB because full-length FosB, which does not accumulate after chronic psychostimulant administration (Hope et al., 1994), did not interact with HDAC1. We performed the reverse experiment in vivo by inducing large amounts of ΔFosB with electroconvulsive seizures. Consistent with our cell culture data, immunoprecipitation with an antibody against HDAC1 pulled down ΔFosB from brain tissue (Fig. 2E).
Based on these findings that ΔFosB and HDAC1 physically interact in vitro and in vivo, we hypothesized that, after chronic amphetamine, ΔFosB recruits HDAC1 to the c-fos gene promoter. Indeed, ChIP of striatal lysates found significantly higher levels of HDAC1 on the c-fos promoter after chronic amphetamine exposure (Fig. 2C), whereas amphetamine did not alter HDAC1 binding to the β-actin gene promoter. To directly determine whether HDAC1 was sufficient to attenuate c-fos induction, we transfected human embryonic kidney 293T cells with HDAC1 or GFP and stimulated them with 5% serum (supplemental Methods, available at www.jneurosci.org as supplemental material). We found that serum-induced c-fos expression was significantly blunted in cells overexpressing HDAC1 (Fig. 2F). These studies were extended in vivo by using floxed HDAC1 mice infected with AAV-GFP on one side of their striatum and AAV-CreGFP to induce local knock-out of the hdac1 gene in the contralateral striatum. AAV-CreGFP reduced Hdac1 mRNA expression in the infected tissue (isolated by laser microdissection) by >75% compared with AAV-GFP injected controls, whereas Hdac2 expression remained unchanged (Fig. 2G). Mice were then treated with chronic amphetamine followed by drug withdrawal for 5 d. The mice were analyzed 30 min after amphetamine challenge and the infected striatal regions were microdissected. We found that amphetamine induced significantly more c-fos mRNA in striatal tissue infected with AAV-CreGFP compared with AAV-GFP (Fig. 2G), demonstrating that HDAC1 is necessary for chronic amphetamine-induced repression of c-fos expression. These data suggest that ΔFosB accumulation in rats after chronic amphetamine treatment results in more ΔFosB binding to the c-fos promoter, recruitment of HDAC1, less histone acetylation, and ultimately less activity of the gene.
Histone methylation is elevated on the c-fos promoter after chronic amphetamine exposure
Repression of gene activity often involves several epigenetic modifications that occur in parallel (Kouzarides, 2007; Tsankova et al., 2007). One of the best-characterized histone modifications associated with reduced gene activity is methylation of H3K9. This histone modification, when found on promoter regions, is associated with transcriptional repression by recruiting corepressors such as HP1 (heterochromatin protein 1) (Kouzarides, 2007). We therefore analyzed whether hypoacetylation of the c-fos gene, seen after chronic amphetamine administration, is also associated with alterations in H3K9 methylation. Consistent with this hypothesis, ChIP performed on striatal tissue from rats treated with chronic amphetamine revealed that H3K9me2 was significantly increased on the c-fos promoter (Fig. 3A), an effect not observed on the β-actin gene promoter. One of the key enzymes that mediates H3K9 methylation is KMT1A (lysine methyltransferase 1A, formerly SUV39H1), which raised the question of whether the expression of this enzyme was regulated by chronic amphetamine exposure. We performed qRT-PCR on the striatum of rats treated with chronic amphetamine and observed a significant upregulation of Kmt1a/Suv39h1 mRNA, whereas the distinct chromatin modifying enzyme, Hdac5, remained unaffected (Fig. 3B). Unlike HDAC1, however, coimmunoprecipitation experiments did not reveal any detectable interaction between ΔFosB and KMT1A/SUV39H1, nor were we able to identify significant enrichment of the methyltransferase on the c-fos promoter by ChIP (data not shown). Regardless, these findings suggest that upregulation of KMT1A/SUV39H1 may hypermethylate H3 at c-fos and contribute to the mechanisms reducing c-fos gene activity after chronic amphetamine exposure.
Figure 3.

Histone methylation after chronic amphetamine exposure. A, ChIP of striatal lysates found significantly more H3K9me2 bound to the c-fos promoter after 5 d of withdrawal from chronic amphetamine (*p < 0.05, Student's t test; n = 3), whereas no change occurred on the β-actin promoter. B, Rats treated with chronic amphetamine and analyzed after 5 d of withdrawal have significantly higher Kmt1a/Suv39h1 mRNA levels compared with saline-treated rats (*p < 0.05; n = 3), whereas Hdac5 levels remain unchanged (p > 0.05). Error bars indicate SEM.
Discussion
This study identified c-fos as a novel downstream target gene of ΔFosB in the striatum after chronic amphetamine administration. We provide direct evidence that endogenous ΔFosB binds to the c-fos promoter in vivo, where ΔFosB recruits HDAC1 to deacetylate surrounding histones and reduce the transcriptional activity of the c-fos gene. Both pharmacological inhibition of HDACs and the inducible knock-out of HDAC1 were sufficient to alleviate c-fos desensitization and elevate c-fos expression in the striatum of chronic amphetamine-treated animals. We also found concurrent increases in repressive histone methylation at H3K9 on the c-fos promoter, an adaptation associated with amphetamine-induced upregulation of the histone methyltransferase, KMT1A/SUV39H1. Together, these findings provide fundamentally new insight into the mechanisms by which ΔFosB represses the activity of certain genes and illustrates a novel interplay between two key pathways that control behavioral responses to psychostimulants: ΔFosB induction (McClung et al., 2004) and chromatin remodeling (Tsankova et al., 2007). Our findings show how these two pathways converge on the c-fos promoter after chronic amphetamine exposure to alter activity of the gene.
We first observed desensitization of c-fos mRNA expression after chronic cocaine treatment >15 years ago (Hope et al., 1992), but little mechanistic insight has been available into how such profoundly different transcriptional responses could occur between acute versus chronic drug exposure. In our effort to understand downstream actions of ΔFosB, we revisited control of c-fos expression because of this differential regulation between acute and chronic psychostimulants exposure. Because ΔFosB is elevated several-fold after chronic drug exposure, this differential induction of c-fos mRNA, as well as an AP-1-like site in the c-fos proximal promoter, suggested a potential regulatory role for ΔFosB. This also made the c-fos gene an attractive candidate with which to study the repressive effects of ΔFosB on gene expression (McClung and Nestler, 2003).
Chronic amphetamine attenuated c-fos mRNA induction or its baseline levels in striatum for ∼5 d of drug withdrawal, a time course that is consistent with the stability of ΔFosB (Hope et al., 1994) and its occupancy on the c-fos promoter. Although ΔFosB can be detected after even longer periods of withdrawal, it gradually declines over time (Hope et al., 1994; Nye et al., 1995) and may be insufficient to maintain repression of the c-fos gene much beyond the 5 d time point. Nevertheless, the time course of c-fos desensitization is complex, with suppression of its fold-induction by an amphetamine challenge maximal at 1 d of withdrawal, but suppression of its basal levels maximal at 5 d of withdrawal. Our ChIP data show that ΔFosB is bound to the c-fos promoter at both time points, suggesting that the differential activity of the c-fos gene observed between 1 and 5 d of withdrawal may be caused by additional transcriptional regulators recruited to the gene with a very complicated time course. Additional studies are needed to understand the detailed mechanisms involved.
The behavioral significance of ΔFosB-mediated c-fos desensitization may be homeostatic, because mice that lack the c-fos gene in dopamine D1 receptor-containing neurons show reduced behavioral responses to cocaine (Zhang et al., 2006). Moreover, HDAC inhibitors, which block ΔFosB-mediated desensitization of c-fos, increase an animal's sensitivity to the behavioral effects of cocaine (Kumar et al., 2005; Renthal et al., 2007). These findings suggest that although the net effect of ΔFosB is to promote sensitized behavioral responses to psychostimulants (Kelz et al., 1999; Colby et al., 2003), it also initiates a novel transcriptional program through c-fos desensitization to limit the magnitude of these same behaviors. ΔFosB would, in effect, titrate behavioral responses to psychostimulants through a complex series of downstream transcriptional events, involving the induction or repression of numerous target genes (McClung and Nestler, 2003), which, in addition to the gene encoding c-Fos as shown here, also include the AMPA glutamate receptor subunit GluR2 (Kelz et al., 1999), the serine-threonine kinase Cdk5 (Bibb et al., 2001), and the opioid peptide dynorphin (Zachariou et al., 2006), among others (McClung and Nestler, 2003). Some of these genes are activated by ΔFosB (where ΔFosB recruits transcriptional coactivators) (Kumar et al., 2005), whereas others are repressed by ΔFosB (where ΔFosB, as shown here, recruits transcriptional corepressors). A major effort of future research is to identify the factors that determine whether ΔFosB activates or represses a target gene when it binds to the gene promoter.
Together, our findings identify a novel epigenetic mechanism through which ΔFosB mediates part of its transcriptional effects in the striatum after chronic amphetamine exposure. This study also provides important new insight into the basic transcriptional and epigenetic mechanisms in vivo involved in the desensitization (i.e., tolerance) of a crucial gene for psychostimulant-induced behavioral responses.
Footnotes
This work was supported by grants from the National Institute on Drug Abuse.
References
- Bibb JA, Chen J, Taylor JR, Svenningsson P, Nishi A, Snyder GL, Yan Z, Sagawa ZK, Ouimet CC, Nairn AC, Nestler EJ, Greengard P. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410:376–380. doi: 10.1038/35066591. [DOI] [PubMed] [Google Scholar]
- Carle TL, Ohnishi YN, Ohnishi YH, Alibhai IN, Wilkinson MB, Kumar A, Nestler EJ. Proteasome-dependent and -independent mechanisms for FosB destabilization: identification of FosB degron domains and implications for DeltaFosB stability. Eur J Neurosci. 2007;25:3009–3019. doi: 10.1111/j.1460-9568.2007.05575.x. [DOI] [PubMed] [Google Scholar]
- Colby CR, Whisler K, Steffen C, Nestler EJ, Self DW. Striatal cell type-specific overexpression of DeltaFosB enhances incentive for cocaine. J Neurosci. 2003;23:2488–2493. doi: 10.1523/JNEUROSCI.23-06-02488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- Hope B, Kosofsky B, Hyman SE, Nestler EJ. Regulation of immediate early gene expression and AP-1 binding in the rat nucleus accumbens by chronic cocaine. Proc Natl Acad Sci U S A. 1992;89:5764–5768. doi: 10.1073/pnas.89.13.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kelz MB, Chen J, Carlezon WA, Jr, Whisler K, Gilden L, Beckmann AM, Steffen C, Zhang YJ, Marotti L, Self DW, Tkatch T, Baranauskas G, Surmeier DJ, Neve RL, Duman RS, Picciotto MR, Nestler EJ. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–276. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ. DeltaFosB: a molecular switch for long-term adaptation in the brain. Brain Res Mol Brain Res. 2004;132:146–154. doi: 10.1016/j.molbrainres.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Stimulus-transcription coupling in neurons: role of cellular immediate-early genes. Trends Neurosci. 1989;12:459–462. doi: 10.1016/0166-2236(89)90096-9. [DOI] [PubMed] [Google Scholar]
- Nye HE, Hope BT, Kelz MB, Iadarola M, Nestler EJ. Pharmacological studies of the regulation of chronic FOS-related antigen induction by cocaine in the striatum and nucleus accumbens. J Pharmacol Exp Ther. 1995;275:1671–1680. [PubMed] [Google Scholar]
- Persico AM, Schindler CW, O'Hara BF, Brannock MT, Uhl GR. Brain transcription factor expression: effects of acute and chronic amphetamine and injection stress. Brain Res Mol Brain Res. 1993;20:91–100. doi: 10.1016/0169-328x(93)90113-4. [DOI] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Steiner H, Gerfen CR. Cocaine-induced c-fos messenger RNA is inversely related to dynorphin expression in striatum. J Neurosci. 1993;13:5066–5081. doi: 10.1523/JNEUROSCI.13-12-05066.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Bolanos CA, Selley DE, Theobald D, Cassidy MP, Kelz MB, Shaw-Lutchman T, Berton O, Sim-Selley LJ, Dileone RJ, Kumar A, Nestler EJ. An essential role for DeltaFosB in the nucleus accumbens in morphine action. Nat Neurosci. 2006;9:205–211. doi: 10.1038/nn1636. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang L, Jiao H, Zhang Q, Zhang D, Lou D, Katz JL, Xu M. c-Fos facilitates the acquisition and extinction of cocaine-induced persistent changes. J Neurosci. 2006;26:13287–13296. doi: 10.1523/JNEUROSCI.3795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]