

Abstract
Objective
Apolipoprotein A-I (apoAI) acts as an ABCA1-dependent acceptor of cellular phospholipids and cholesterol during the biogenesis of HDL, but this activity is susceptible to oxidative inactivation by myeloperoxidase. We tried to determine which residues mediated this inactivation and create an oxidant resistant apoAI variant.
Methods and Results
Mass spectrometry detected the presence of tryptophan, methionine, tyrosine, and lysine oxidation in apoAI recovered from human atheroma. We investigated the role of these residues in the myeloperoxidase mediated loss of apoAI activity. Site directed mutagenesis and chemical modification were used to create variants of apoAI which were tested for ABCA1-dependent cholesterol acceptor activity and oxidative inactivation. We previously reported that tyrosine modification is not required for myeloperoxidase induced loss of apoAI function. Lysine methylation did not alter apoAI's sensitivity to myeloperoxidase, while site-specific substitution of apoAI methionine to valine increased apoAI's sensitivity to myeloperoxidase. ApoAI tryptophan residues were identified as essential in apoAI function and oxidant sensitivity as substitution of all four apoAI tryptophan residues to leucine led to loss of function, but the conservative substitution to phenylalanine retained full function and was resistant to oxidative inactivation.
Conclusions
Tryptophan modification of apoAI is primarily responsible for the myeloperoxidase-mediated loss of apoAI's cholesterol acceptor activity.
High levels of high density lipoprotein (HDL) and apolipoprotein A-I (apoAI), the major HDL protein, are associated with decreased risk for cardiovascular disease 1,2. The protection against cardiovascular disease afforded by apoAI and HDL may in part be attributed to their role in reverse cholesterol transport, although they have additional anti inflammatory, antioxidant, and endothelial cell regenerative properties that may also play protective roles. However, all HDL may not be functionally equivalent. Fogelman and colleagues have reported that HDL samples from patients with cardiovascular disease have a diminished capacity to inhibit LDL induced monocyte migration in a co-culture system 3. Previously, work from our group and subsequently from the group led by Heinecke have found that apoAI serves as a preferred target for myeloperoxidase (MPO)1 catalyzed oxidative modification, leading to chlorination and nitration of specific tyrosine residues and concomitant loss of ABCA1-mediated cholesterol acceptor activity 4,5,6,7. In addition, elevated content of chlorotyrosine and nitrotyrosine residues are observed in plasma apoAI 4,8, and more highly in apoAI recovered from human atheroma 4,8.
We also reported that the degree of tyrosine modification of plasma apoAI, isolated from cardiology patients, correlates with its cholesterol acceptor activity 4. Thus, tyrosine modification of apoAI by MPO is associated with loss of apoAI function as an ABCA1 dependent acceptor of cellular lipids and can serve as a fingerprint to monitor the extent of apoAI modification. However, our prior studies using a tyrosine-free apoAI derivative (7YF, all seven tyrosines substituted by phenylalanine) show that it is equally susceptible to MPO mediated loss of function compared to wild type apoAI 9. We also demonstrated in vitro that MPO could modify apoAI lysine residues into lysine chloramines and aminoadipic acid, and apoAI tryptophan residues were converted into the mono- and di-oxygenated derivatives 9. It has also been previously shown that MPO modification of HDL led to a time and dose dependent decrease in bulk tryptophan fluorescence 10. However, whether such modifications occurred in vivo and might be responsible for oxidative inactivation of apoAI cholesterol efflux activity remains unknown. In the current study we report the presence of multiple site-specific tryptophan, methionine, and lysine modifications in apoAI isolated from human atheroma. We thus sought to determine the effects of altering the MPO sensitive lysine, methionine, and tryptophan residues in apoAI and whether such modifications might account for the observed oxidative inactivation of apoAI in vivo. We report here that replacement of the four apoAI tryptophan residues with leucines led to loss of its cholesterol acceptor function, while the replacement of tryptophan with phenylalanines not only preserved apoAI function, but rendered it resistant to MPO-mediated loss of cholesterol acceptor and lipid binding activities. The apoAI with tryptophan to phenylalanine substitutions, though it retained its activity, was still sensitive to MPO-mediated cross-linking and loss of α-helical content. The present studies thus suggest that apoAI tryptophan residues are responsible for MPO dependent oxidative loss of apoAI function in vivo.
Methods
Mass Spectrometry
Human atheroma derived apoAI was isolated by immunoaffinity chromatography as previously described 5. ApoAI was eluted in glycine buffer (pH 2.5) and subjected directly to trypsin digestion or first separated by SDS PAGE and subjected to in gel trypsin digestion. Mass spectrometry was performed and collision induced dissociation (CID) spectra were obtained, as previously described 5. Chlorotyrosine and 2-amino adipic acid analyses were performed after acid hydrolysis with heavy isotope internal standards as previously described 4,9 using duplicate assays of apoAI from human atheroma or from plasma of healthy volunteers isolated by immunoaffinity chromatography.
Site Directed Mutagenesis and Recombinant ApoAI Production
The pET-20b bacterial expression vector containing the cDNA of 6-His tagged recombinant human apoAI (rh-apoAI) was previously described 11. Point Mutations to tryptophan (8, 50, 72, 108) and methionine (86, 112, 148) residues were made using QuickChange Mutagenesis Kit from Stratagene (Carlsbad, CA), and confirmed by DNA sequencing. Plasmids were transformed into Escherichia coli strain BL21 (DE-3) pLysS and apoAI expression and purification was performed as described previously 9. rh-ApoAI was extensively dialyzed against PBS or MPO reaction buffer (60 mM sodium phosphate, 100 mM sodium chloride, 100 μM diethylenetriamine pentaacetic acid, pH 7.0) in order to remove any trace of imidazole, analyzed by SDS-PAGE, and found to be > 95% pure. Since Trp and Met substitution alters the protein OD280 and reactivity to the BCA or Lowry protein assays, protein concentrations were determined based upon free amines using the o-phthaldialdehyde (OPA) assay, with a human plasma-derived apoAI (Biodesign) standard, as previously described 12. Cleavage of the initial Met and His tag of rh-apoAI was performed by formic acid treatment 11, followed by FPLC purification.
ApoAI Lysine Modifications
Human plasma derived apoAI was dialyzed against PBS and diluted to 0.5 mg/ml. Lysine reductive methylation was performed as previously described 12. Extent of lysine modification was determined by the OPA assay. ApoAI was then dialyzed against MPO reaction buffer and the protein concentration of lysine modified apoAI was determined using the BCA reagent.
ApoAI MPO and Hypochlorous Acid Modifications
MPO at a final concentration of 57 nM, prepared as previously described 4, was added to ApoAI at 100 mg/ml (3.5 μM) that had been extensively dialyzed against MPO reaction buffer. The reaction was initiated by adding hydrogen peroxide at varying mole ratios to apoAI in 4 aliquots at 15 min intervals at 37°C, and continuing the incubation for 90 min, at which time 2 mM L-methionine was added to quench the reaction. For chemical modification of apoAI, sodium hypochlorite (NaOCl) was added to 100 μg/ml ApoAI in MPO buffer at varying concentrations in 4 aliquots at 15 min intervals at 37°C. After a total incubation time of 60 min, 2 mM L-methionine was added to quench the reaction.
ABCA1-Dependent Cholesterol Efflux Assay
RAW 264.7 murine macrophage cells were labeled with [3H]cholesterol and treated with 0.3 mM 8Br-cAMP to induce ABCA1 activity, as previously described 13,14. The cells were washed and chased for 4 hr in serum-free medium in the presence of 0.3 mM 8Br-cAMP and the presence or absence of various apoAI preparations. The radioactivity in the chase media was determined after brief centrifugation to pellet debris. Radioactivity in the cells was determined by extraction in hexane:isopropanol (3:2) with the solvent evaporated in a scintillation vial prior to counting. The percent cholesterol efflux was calculated as 100 × (medium dpm) / (medium dpm + cell dpm).
Lipid Binding Activity Assay
Lipid binding activity of apoAI was assessed via the inhibition of phospholipase C (PLC) mediated aggregation of human low density lipoprotein, performed as previously described 12. We have previously shown that this assay give results similar to those observed by the DMPC dispersion clearance assay, but it is more sensitive and requires less apoAI 12. The final concentration of apoAI used in this assay was 12.5 μg/ml, which was sufficient to decrease the initial rate of LDL aggregation by ∼ 75%.
Detection of ApoAI Cross Links
250 ng of apoAI per lane was denatured in an SDS sample buffer, run on a 10% Tris glycine gel in the presence of SDS, and the protein was transferred to a PVDF membrane. The membrane was probed sequentially with goat anti-human apoAI primary antibody (1:1,000 dilution, DiaSorin, Stillwater, MN) and rabbit anti-goat-HRP conjugated antibody (1:10,000 dilution), and apoAI was visualized with an enhanced chemiluminescent substrate.
Results
ApoAI Modifications in Human Atheroma
We previously demonstrated the presence of nitro- and chlorotyrosine in apoAI isolated from human atheroma tissue 4. We extended these studies to determine whether we could also identify modified tryptophan, methionine, and lysine residues in apoAI isolated from human atheroma, all modifications which we or others have previously identified after in vitro MPO treatment of apoAI 9,15. Using tandem mass spectrometry, we were able to detect monohydroxytryptophan residues at all four tryptophan positions, 8, 50, 72, and 108, and dihydroxytryptophan at position 108 within apoAI (Figure 1 A-E). We previously identified mono- and di-hydroxytrytophan at residue W72 in in vitro MPO modified apoAI 9. In addition we detected methionine sulfoxide at residues 48 and 112 (Figure 1 D, E). To look for lysine modification by MPO-generated HOCl we used stable isotope dilution HPLC with online tandem mass spectrometry to quantify 2-aminoadipic acid, an end product of lysine oxidation by the MPO generated oxidant. 2-Aminoadipic acid levels were low but detectable in apoAI isolated from the plasma of six healthy volunteers, while the mean levels were strikingly elevated ∼16-fold in apoAI isolated from six human atheroma samples (Fig. 2, p=0.005 by a two-tailed t-test). Thus, tryptophan, methionine, and lysine oxidation of apoAI occur physiologically within human atheroma. We therefore sought to determine which of these modifications was responsible for yielding dysfunctional apoAI with a diminished capacity to accept cellular cholesterol.
Figure 1. Identification of modified apoAI in human atheroma by mass spectrometry.
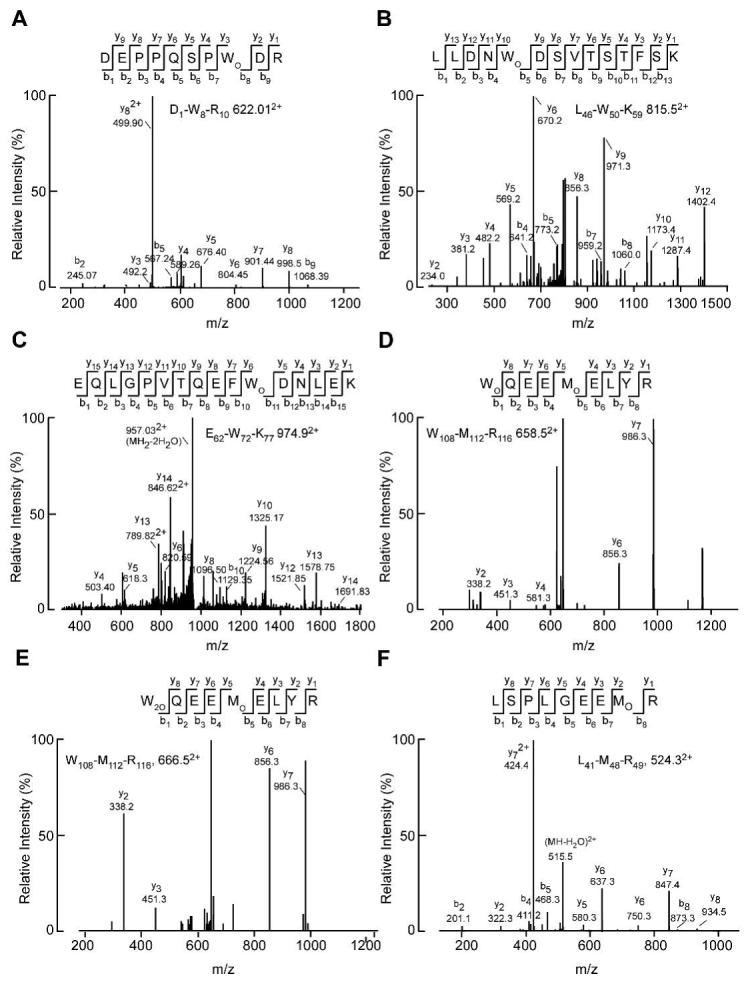
CID spectra were acquired after direct or in gel tryptic digest of imunnoaffinity purified apoAI derived from human atheroma. Doubly charged ions were detected and fragmented in an LC-tandem mass spectrometry experiment. A. Peptide D1-R10 containing monohydroxytryptophan at residue 8. B. Peptide L46-K59 containing monohydroxytryptophan at residue 50. C. Peptide E62-K77 containing monohydroxytryptophan at residue 72. D. Peptide W108-R116 containing monohydroxytryptophan at residue 108 and methionine sulfoxide at residue 112. E. The same peptide as in D, but the tryptophan at residue 108 is converted to dihydroxytryptophan. F. Peptide L41-R49 containing methionine sulfoxide as at residue 48.
Figure 2. Aminoadipic acid levels in plasma and lesion apoAI.
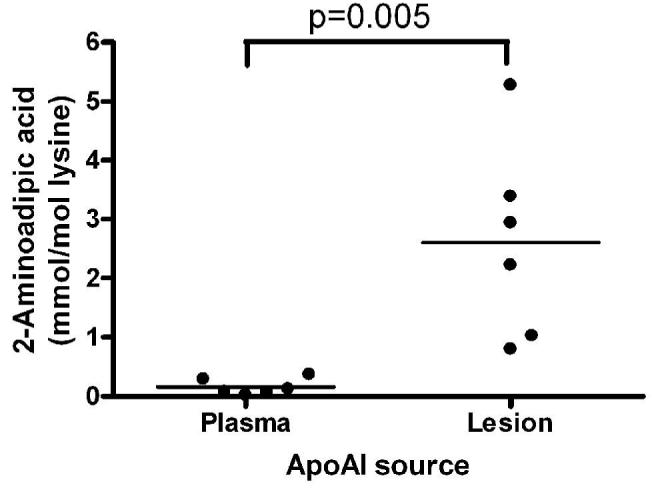
ApoAI was isolated by immunoaffinity chromatography from the plasma of six healthy subjects and from six atheroma samples. 2-aminoadipic acid levels were quantified by mass spectrometry after acid hydrolysis, and normalized to apoAI lysine content. Data show mean of duplicate determinations for each sample (p=0.005 by two tailed t-test).
ApoAI Lysine Modification
In the amphipathic structure of apoAI, the 21 lysine residues overwhelming reside on both sides of and adjacent to the hydrophobic face 16,17. Lysine modification by MPO is an attractive candidate to be responsible for MPO induced loss of apoAI function as we previously demonstrated that apoAI lysine residues can undergo modification by MPO 9, and that extensive chemical modification of apoAI lysine residues that alter its positive charge led to loss of apoAI cholesterol acceptor activity 12. However, we also found that lysine modification by reductive methylation, which retains the lysine positive charge, led to only modest reductions of apoAI function 12. ApoAI was subjected to reductive methylation, leading to 92% lysine modification, or control incubation and dialyzed extensively against MPO reaction buffer. Modification reactions were performed using catalytic amounts of MPO and increasing molar ratios of H2O2:apoAI. The reaction products were assayed for cholesterol acceptor activity using cholesterol labeled RAW264 macrophages that had been pretreated with a cAMP analogue to induce ABCA1. In the absence of H2O2 in the modification reaction, the methylated and nonmethylated control apoAI had robust and equivalent ABCA1-dependent cholesterol acceptor activity. With increasing doses of H2O2, the cholesterol acceptor activity of both the methylated and control apoAI samples declined in a similar fashion (Supplemental Figure I, please see http://atvb.ahajournals.org). In addition, the alpha helix content of these preparations was estimated by CD, and both methylated and control apoAI preparations were similarly susceptible to the MPO/H2O2 dose dependent reduction in alpha helix content (Supplemental Table I). Since reductive methylation of lysine's primary amine into a tertiary amine decreases its chemical reactivity but did not lead to protection of apoAI's function, apoAI lysine modification by MPO is unlikely to be responsible for apoAI's loss of function.
ApoAI Methionine Substitution
We then turned our attention to the three apoAI methionine residues, which were previously implicated by Heinecke and colleagues in the MPO induced loss of apoAI function 15. In order to substitute valine for all three methionines, we used recombinant human apoAI (rh-apoAI), which adds an additional methionine initiation codon and a 6-His tag to the N-terminus. We and others have previously demonstrated that rh-apoAI behaves similarly to plasma derived apoAI in its cholesterol acceptor activity, lipid binding activity, and its susceptibility to MPO mediated loss of function 11,15,18,9. Using site directed mutagenesis, we created an apoAI expression construct encoding a protein with the three internal methionines converted to valine (rh-apoAI 3MV). One cannot substitute for the initiating methionine; however, this methionine and the His tag can be chemically cleaved by formic acid 11. We determined that the rh-apoAI 3MV, regardless of whether the initiating methionine and His tag were intact or removed, had similar ABCA1-dependent cholesterol acceptor activity compared to wild type rh-apoAI. In addition, the rh-apoAI 3MV, with or without the N-terminal methionine, and wild type rh-apoAI were equally susceptible to a high dose (H2O2: apoAI = 15:1) MPO mediated loss of cholesterol acceptor activity (Figure 3A). MPO modifications of rh-apoAI and the 3MV variant were performed at varying and modest molar ratios of H2O2:apoAI, and the 3MV variant was paradoxically more sensitive to loss of cholesterol acceptor activity at low molar ratios (Figure 3B). For example, at an H2O2:apoAI ratio of 1.4, wild type apoAI had a negligible loss of cholesterol acceptor activity, while the 3MV variant lost ∼ half of its cholesterol acceptor activity. Thus, we conclude that the methionine residues in apoAI, instead of playing a role in oxidative impairment of apoAI function, actually play a protective role by absorbing oxidants, a function previously suggested by Stocker and colleagues 19.
Figure 3. Methionine to valine substituted apoAI has increased susceptibility to MPO mediated loss of function.
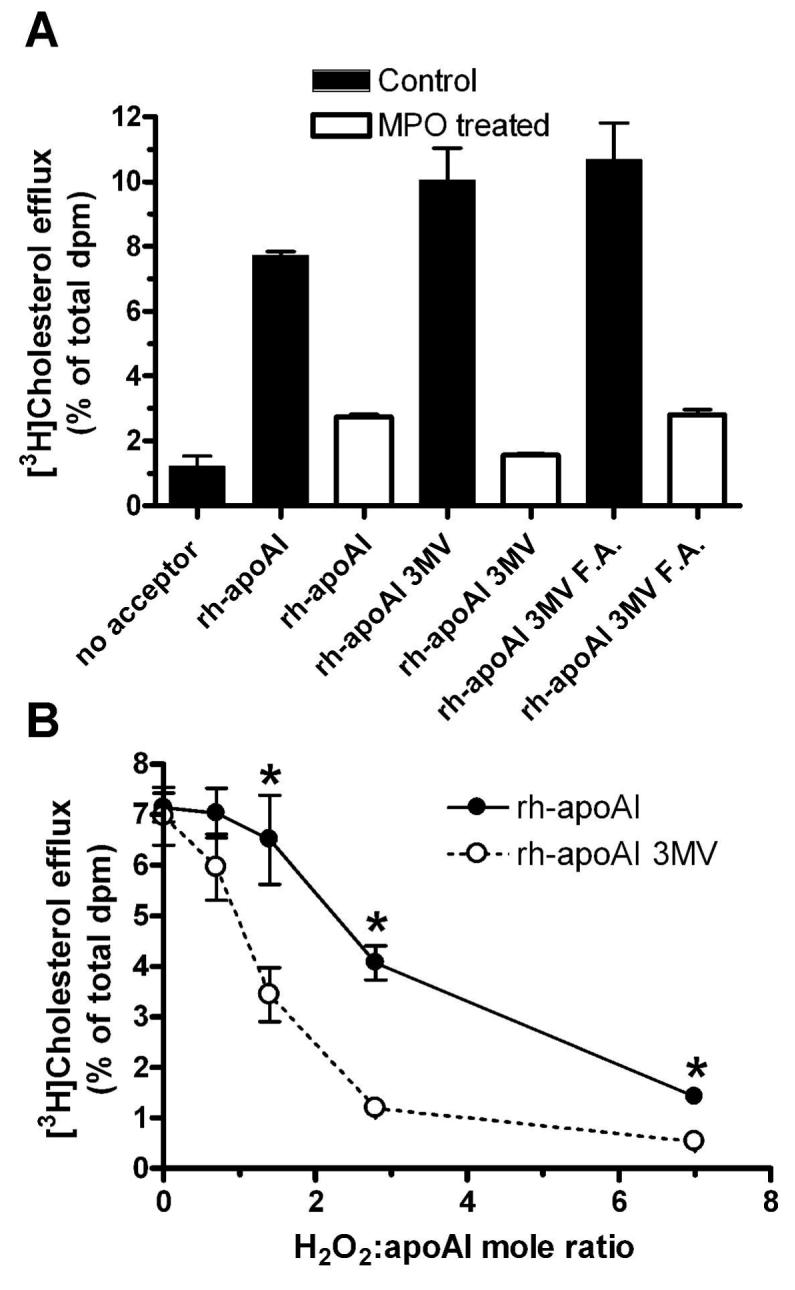
A. High dose MPO modification, at an H2O2:apoAI mole ratio of 15:1, was performed on rh-apoAI, rh-apoAI 3MV (3 internal Met substituted with Val), and rh-apoAI 3MV treated with formic acid (rh-apoAI 3MV F.A.), which deletes the initiation Met and 6-His tag. Cellular cholesterol efflux activity was determined as the Methods section. Data are mean ± S.D. of duplicate determinations. B. rh-ApoAI (filled circles, solid line) and rh-apoAI 3MV (open circles, dashed line) were subjected to modification by MPO at varying H2O2: apoAI mole ratios. These proteins were then assayed for ABCA1 dependent cellular cholesterol acceptor activity as described in Fig. 3. Data are means ± S.D. of triplicate determinations. *, p<0.01 vs. rh-apoAI 3MV at the same H2O2: apoAI mole ratio, by two tailed t-test.
ApoAI Tryptophan Substitutions
We altered all four tryptophans to either leucine (rh-apoAI 4WL) or phenylalanine (rh-apoAI 4WF) for functional characterization and oxidant sensitivity testing. Our data revealed that the aromatic or bulky nature of the tryptophan residues seemed to be crucial for apoAI's cholesterol acceptor activity, as the 4WL variant lost the majority of this activity over a wide range of apoAI doses, while the 4WF variant retained this activity (Figure 4A). Thus, the 4WF variant was competent for physiological lipidation by cellular ABCA1. In addition, the 4WF variant was equally competent compared to wild type rh-apoAI in the clearance of a DMPC:cholesterol (90:10 mole %) emulsion (Supplemental Figure II), demonstrating that the 4WF variant was able to interact with lipids in a cell-free context. We also prepared rHDL by cholate dialysis using POCP and the wild type or 4WF apoAI. Both yielded a similar pattern of rHDL discs estimated by non-denaturing gels at ∼ 9.8, 12, and 17 nm, without any lipid free apoAI remaining (Supplemental Figure III A). We tested the wild type and 4WF rHDL, and both were equally competent to mediate ABCA1-independent cholesterol efflux from RAW264.7 cells (Supplemental Figure III B), without ABCA1 dependent acceptor activity, as expected for fully lipidated apoAI. We examined the predicted alpha helix content of these proteins by CD and found that the rh-apoAI had 56% alpha helix, while the 4WL and 4WF variants both had increased estimated alpha helix contents of 68% and 71%, respectively. Thus, the loss of efflux and lipid binding activity of the 4WL variant cannot be attributed to loss of helical content.
Figure 4. Effect of tryptophan substitutions to phenylalanine or leucine on rh-apoAI function.
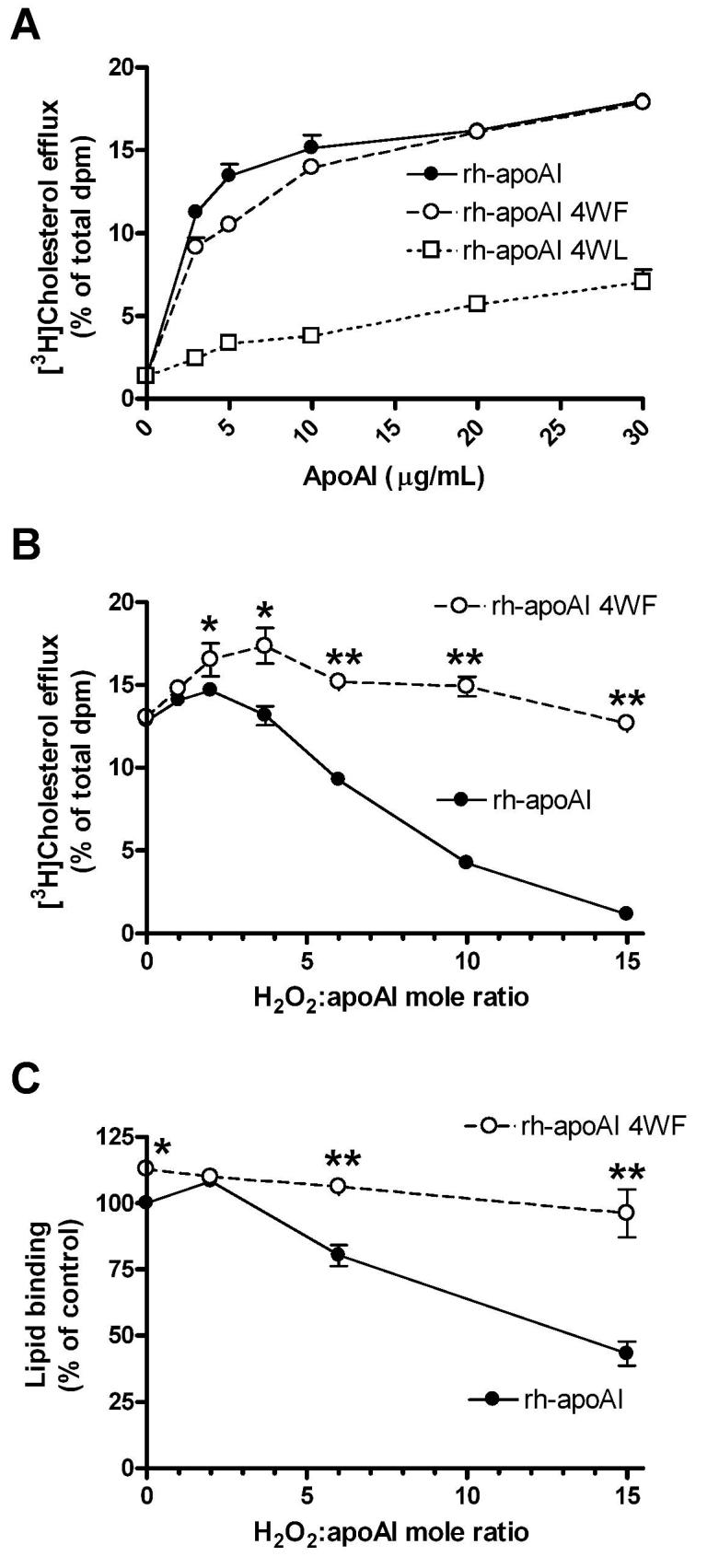
A. Varying concentrations of rh-ApoAI (closed circles, solid line), rh-apoAI 4WF (open circles, dashed line), and rh-apoAI 4WL (open squares, dotted line) were assayed for ABCA1 dependent cellular cholesterol acceptor activity. The 4WF variant largely retained this activity, while the 4WL variant lost this activity. Data are means ± S.D. of triplicate determinations. B. rh-ApoAI (filled circles, solid line) and rh-apoAI 4WF (open circles, dashed line) were subjected to modification by MPO at varying H2O2: apoAI mole ratios. These proteins were then assayed for ABCA1 dependent cellular cholesterol acceptor activity. Data are means ± S.D. of triplicate determinations. *, p<0.05; and **, p<0.0001 vs. rh-apoAI at the same H2O2: apoAI mole ratio, respectively, by two tailed t-test. C. rh-ApoAI (filled circles, solid line) and rh-apoAI 4WF (open circles, dashed line) were subjected to modification by MPO at varying H2O2: apoAI mole ratios. These proteins were then assayed for lipid binding activity as described in the Methods section. Data are normalized to the lipid binding activity of rh-apoAI treated in the absence of H2O2. Data are means ± S.D. of triplicate determinations. *, p<0.05; and **, p<0.001 vs. rh-apoAI 4WF at the same H2O2: apoAI mole ratio, respectively, by two tailed t-test.
Both rh-apoAI and the 4WF variant were then subjected to the MPO/Cl−/H2O2 oxidation system at increasing doses of H2O2. Figure 4B shows the result of a study representative of four different experiments using two independent preparations of each protein. As previously observed, the ABCA1-dependent cholesterol acceptor activity of wild type apoAI was inhibited by increasing MPO induced oxidation; however, the 4WF variant maintained this activity even as an H2O2:apoAI mole ratio of 15 (Figure 4B). The cell free lipid binding activity of rh-apoAI 4WF was also resistant to MPO mediated inhibition, compared to rh-apoAI (Figure 4C).
MPO modification of apoAI leads to extensive cross linking resulting in dimers, multimers, and presumably intramolecular cross links as well, and we previously have shown that the MPO mediated apoAI cross linking pattern was not altered in the variant with all 7 tyrosine residues converted to phenylalanine 9. Upon subjecting rh-apoAI and the 4WF variant to MPO/Cl−/H2O2 oxidation at increasing doses of H2O2 (using the identical protein products that were used for efflux in Figure 4B), we observed altered migration of these proteins in denaturing gels consistent with intermolecular cross linking (Figure 5). However, the migration patterns were different, with the 4WF variant giving a sharp predominant band at ∼70 kD, while the wild type protein yielded a less distinct predominant zone between 55 and 65 kD (Figure 5). The migration of the monomer was altered for both proteins, which could be indicative of intramolecular cross links or other amino acid modifications. Although the 4WF variant is resistant to MPO mediated loss of cholesterol acceptor activity, this variant was more susceptible to MPO induced cross linking, particularly at low doses of H2O2 (Figure 5). We also subjected these MPO modified proteins to structural analysis by CD (Supplemental Table II), and found that both were susceptible to loss of alpha helical content, although the 4WF variant started with a higher value.
Figure 5. Tryptophan to phenylalanine substituted apoAI is still susceptible to cross linking by MPO.
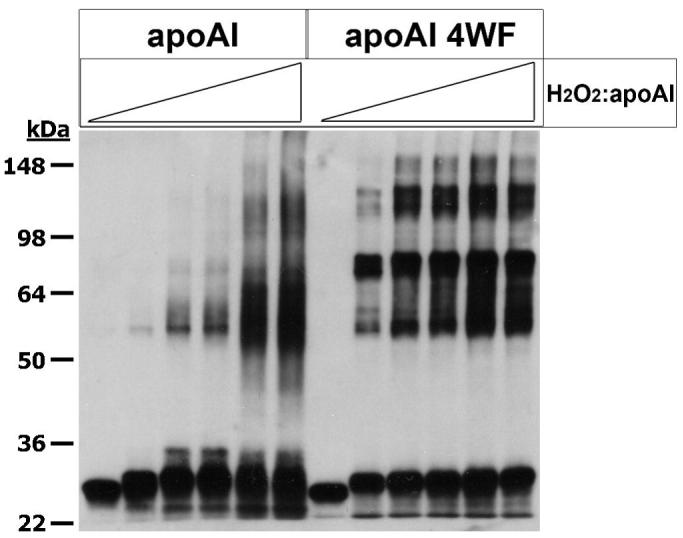
rh-ApoAI, and rh-apoAI 4WF were subjected to MPO modification at the following mole ratios of H2O2:apoAI; 0, 1, 2, 3, 5, and 12.5 (from left to right). ApoAI cross linking was qualitatively assessed by Western blotting as described in the Methods section. The migration of molecular weight standards is shown on the left side.
The MPO/Cl−/H2O2 oxidation system generates HOCl 20, the active reagent of bleach, and we and others have previously demonstrated that HOCl treatment of apoAI results in loss of cholesterol acceptor and lipid binding activity 5,6. Thus, we subjected wild type apoAI and the 4WF variant to increasing doses of HOCl. Similar to the findings with the MPO modification system, the cholesterol acceptor activity of the 4WF variant was resistant to this treatment while the efflux activity of wild type rh-apoAI was impaired by increasing doses of HOCl (Figure 6).
Figure 6. Tryptophan to phenylalanine substituted apoAI is resistant to HOCl mediated loss of function.

rh-ApoAI (filled circles, solid line) and rh-apoAI 4WF (open circles, dashed line) were subjected to modification at varying HOCl: apoAI mole ratios. These proteins were then assayed for ABCA1 dependent cellular cholesterol acceptor activity. Data are means ± S.D. of triplicate determinations. **, p<0.0001 vs. rh-apoAI at the same HOCl:apoAI mole ratio, respectively, by two tailed t-test.
We used a quantitative mass spectrometry method, with heavy isotope internal standards, to detect total chlorotyrosine from MPO modified rh-apoAI 4WF (H2O2:apoAI = 15:1); and, we detected 1.2 mole% conversion of tyrosine into chlorotyrosine, comparable to the highest levels of chlorotyrosine detected in apoAI recovered from human atheroma 4. Thus, the apoAI 4WF variant had fully functional efflux capacity (see Fig. 4B) at physiological levels of tyrosine chlorination found within the highly oxidative environment of human atheroma tissues.
Discussion
ApoAI is a selective target for MPO mediated modification as demonstrated by its high chlorotyrosine content in human plasma and atheroma 4,5. Moreover, it is clear that in vitro modification of apoAI by MPO leads to reduced activity of apoAI as both a lipid binding protein and an acceptor of cellular lipids through the ABCA1 pathway 4,5,7,6. This has led to the concept that apoAI in some subjects may be dysfunctional, and in fact apoAI's cholesterol acceptor activity varies in different subjects and is correlated with tyrosine chlorination, which is a unique indicator of MPO mediated modification 4,21. These basic findings have been independently observed by our group and Heinecke's group. However, the precise mechanism by which MPO modification renders apoAI dysfunctional is controversial. We previously reported that a tyrosine-free apoAI variant was equally susceptible as wild type apoAI to MPO mediated loss of apoAI function; and, we identified MPO induced in vitro modifications of lysine and tryptophan residues of unknown physiological and functional consequences 9. In the current work, we identified site-specific in vivo modifications of tryptophan, methionine, and lysine in atheroma derived apoAI and then used chemical modification and site directed mutagenesis to determine which of these residues is associated with the MPO mediated loss of apoAI function. We found that replacing apoAI tryptophan residues with phenylalanine led to formation of a fully functional apoAI variant with marked resistance against oxidative inactivation by pathophysiological exposure levels of MPO-generated oxidants.
Using model peptides, Heinecke and colleagues reported that a lysine downstream of a tyrosine in a YXXK peptide motif can increase tyrosine chlorination by MPO or HOCl 22. Shao et al. then used site directed mutagenesis of apoAI lysine 195 to arginine, and found that this led to decreases in susceptibility to MPO or HOCl mediated tyrosine 192 chlorination 15. They also suggested that altering glutamate 198 to methionine, creating a YXXMXXK motif, inhibited MPO mediated chlorination of tyrosine 192, consistent with protein bound methionine acting as a scavenger for MPO generated reactive halogenating species 15. This was supported also by substitution of methionine 112 to lysine (going from a MXXY to a KXXY motif), which increased tyrosine 115 chlorination by MPO 15. They also demonstrated that MPO treatment of apoAI led to the modification of its three methionine residues to methionine sulfoxide, and that this could be reversed by adding the enzyme methionine sulfoxide reductase 15. However, Shao et al. did not directly test whether methionine substitution altered apoAI's sensitivity to the MPO mediated loss of function, but they did find that treatment of MPO modified apoAI with the enzyme methionine sulfide reductase could partially restore apoAI's cholesterol acceptor activity 15. In our studies, we directly observed that apoAI methionine residues play a protective scavenging role, as we found a markedly increased susceptibility of the methionine substituted 3MV apoAI variant to low doses of H2O2 in the complete MPO chlorination system. Thus, our site directed substitution and cholesterol efflux data clearly show that neither methionine nor tyrosine serve as the oxidant sensitive residue involved in MPO-dependent apoAI inactivation. We also performed chemical modification of apoAI lysine residues, which failed to alter apoAI's sensitivity to MPO mediated loss of function.
We substituted all four apoAI tryptophan residues with either leucine or phenylalanine. The apoAI 4WL variant lost its lipid binding and cholesterol accepting activities, while the 4WF variant retained these activities. Since tryptophan and phenylalanine are the most hydrophobic residues on the Wimley and White scale 23, our results imply that highly hydrophobic and bulky residues are required at the tryptophan positions on the nonpolar face of apoAI for its lipid binding and accepting functions. The global replacement of tryptophan with phenylalanine, which is far less susceptible to oxidative modification by MPO, created an apoAI that was clearly resistant to the MPO mediated loss of function, but still susceptible to other modifications that lead to cross linking. In regard to the MPO mediated cross linking of apoAI, in the current work we observed an altered cross linking pattern comparing the 4WF variant with wild type apoAI, while in our prior study we did not observe an alteration of the cross linking pattern comparing the tyrosine-free 7YF variant 9. These combined data suggests that: 1) tryptophan residues either contribute directly to the cross links observed in wild type apoAI, or that tryptophan substitution alters the tertiary structure and this alters the preferred sites of cross linking; and 2) tyrosine residues do not directly participate in the cross links observed in wild type apoAI.
We conclude that tryptophan oxidation, which we observed in apoAI isolated from human atheroma, is likely to be the causative alteration that results in the production of dysfunctional apoAI. Although, we cannot exclude the possibility that the 4WF variant's oxidation resistance may be due to an altered tertiary structure of this variant leading to protection of some other sensitive residue. All of our in vitro MPO treatments were performed with lipid-free apoAI; similar to pre β particles formed in vivo during lipoprotein remodeling. It is these lipid free and lipid poor apoAI particles that are capable to participate in ABCA1 mediated lipid efflux and thus play an important and physiological role in reverse cholesterol transport 24,25,26,27. We speculate that the 4WF apoAI variant would be a better therapeutic reagent, compared to wild type apoAI or apoAI Milano 28, to promote the regression of plaques, a location where the levels of MPO generated oxidants as well as modified apoAI are high.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health Grants HL66082 (J.D.S.), P50 HL077107 (S.L.H. and J.D.S), and PO1 HL076491 (S.L.H.). D.-Q. Peng was the recipient of an American Heart Association Fellowship Award (0525386B).
Footnotes
Publisher's Disclaimer: Disclaimer: The manuscript and its contents are confidential, intended for journal review purposes only, and not to be further disclosed.
Author Disclosures Dao-Quan Peng: No disclosures Gregory Brubaker: No disclosures Zhiping Wu: No disclosures Lemin Zheng: No disclosures Belinda Willard: No disclosures Michael Kinter: No disclosures
References
- 1.Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986;256:2835–2838. [PubMed] [Google Scholar]
- 2.Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DJ, Bangdiwala S, Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- 3.Ansell BJ, Navab M, Hama S, Kamranpour N, Fonarow G, Hough G, Rahmani S, Mottahedeh R, Dave R, Reddy ST, Fogelman AM. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–2756. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 4.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen SL. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng L, Settle M, Brubaker G, Schmitt D, Hazen SL, Smith JD, Kinter M. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J Biol Chem. 2005;280:38–47. doi: 10.1074/jbc.M407019200. [DOI] [PubMed] [Google Scholar]
- 6.Bergt C, Pennathur S, Fu X, Byun J, O'brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF, Heinecke JW. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci U S A. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shao B, Bergt C, Fu X, Green P, Voss JC, Oda MN, Oram JF, Heinecke JW. Tyrosine 192 in Apolipoprotein A-I Is the Major Site of Nitration and Chlorination by Myeloperoxidase, but Only Chlorination Markedly Impairs ABCA1-dependent Cholesterol Transport. J Biol Chem. 2005;280:5983–5993. doi: 10.1074/jbc.M411484200. [DOI] [PubMed] [Google Scholar]
- 8.Pennathur S, Bergt C, Shao B, Byun J, Kassim SY, Singh P, Green PS, McDonald TO, Brunzell J, Chait A, Oram JF, O'brien K, Geary RL, Heinecke JW. Human atherosclerotic intima and blood of patients with established coronary artery disease contain high density lipoprotein damaged by reactive nitrogen species. J Biol Chem. 2004;279:42977–42983. doi: 10.1074/jbc.M406762200. [DOI] [PubMed] [Google Scholar]
- 9.Peng DQ, Wu Z, Brubaker G, Zheng L, Settle M, Gross E, Kinter M, Hazen SL, Smith JD. Tyrosine modification is not required for myeloperoxidase-induced loss of apolipoprotein A-I functional activities. J Biol Chem. 2005;280:33775–33784. doi: 10.1074/jbc.M504092200. [DOI] [PubMed] [Google Scholar]
- 10.Jerlich A, Hammel M, Nigon F, Chapman MJ, Schaur RJ. Kinetics of tryptophan oxidation in plasma lipoproteins by myeloperoxidase-generated HOCl. Eur J Biochem. 2000;267:4137–4143. doi: 10.1046/j.1432-1327.2000.01449.x. [DOI] [PubMed] [Google Scholar]
- 11.Ryan RO, Forte TM, Oda MN. Optimized bacterial expression of human apolipoprotein A-I. Protein Expr Purif. 2003;27:98–103. doi: 10.1016/s1046-5928(02)00568-5. [DOI] [PubMed] [Google Scholar]
- 12.Brubaker G, Peng DQ, Somerlot B, Abdollahian DJ, Smith JD. Apolipoprotein A-I lysine modification: effects on helical content, lipid binding and cholesterol acceptor activity. Biochim Biophys Acta. 2006;1761:64–72. doi: 10.1016/j.bbalip.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Smith JD, Miyata M, Ginsberg M, Grigaux C, Shmookler E, Plump AS. Cyclic AMP induces apolipoprotein E binding activity and promotes cholesterol efflux from a macrophage cell line to apolipoprotein acceptors. J Biol Chem. 1996;271:30647–30655. doi: 10.1074/jbc.271.48.30647. [DOI] [PubMed] [Google Scholar]
- 14.Oram JF, Lawn RM, Garvin MR, Wade DP. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275:34508–34511. doi: 10.1074/jbc.M006738200. [DOI] [PubMed] [Google Scholar]
- 15.Shao B, Oda MN, Bergt C, Fu X, Green PS, Brot N, Oram JF, Heinecke JW. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem. 2006;281:9001–9004. doi: 10.1074/jbc.C600011200. [DOI] [PubMed] [Google Scholar]
- 16.Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, Harvey SC. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 17.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 18.Smith JD, Le Goff W, Settle M, Brubaker G, Waelde C, Horwitz A, Oda MN. ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A-I. J Lipid Res. 2004;45:635–644. doi: 10.1194/jlr.M300336-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Garner B, Waldeck AR, Witting PK, Rye KA, Stocker R. Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem. 1998;273:6088–6095. doi: 10.1074/jbc.273.11.6088. [DOI] [PubMed] [Google Scholar]
- 20.Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem. 1976;251:1371–1374. [PubMed] [Google Scholar]
- 21.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99:2075–2081. doi: 10.1172/JCI119379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergt C, Fu X, Huq NP, Kao J, Heinecke JW. Lysine residues direct the chlorination of tyrosines in YXXK motifs of apolipoprotein A-I when hypochlorous acid oxidizes high density lipoprotein. J Biol Chem. 2004;279:7856–7866. doi: 10.1074/jbc.M309046200. [DOI] [PubMed] [Google Scholar]
- 23.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 24.Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J., Jr. Molecular and cellular physiology of apolipoprotein A-I lipidation by the ATP-binding cassette transporter A1 (ABCA1) J Biol Chem. 2004;279:7384–7394. doi: 10.1074/jbc.M306963200. [DOI] [PubMed] [Google Scholar]
- 25.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Mulya A, Lee JY, Gebre AK, Thomas MJ, Colvin PL, Parks JS. Minimal lipidation of pre-beta HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler Thromb Vasc Biol. 2007;27:1828–1836. doi: 10.1161/ATVBAHA.107.142455. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, Tall AR, Rader DJ. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–2224. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.