

Summary
The Trypanosoma brucei genome is colonized by the site-specific non-LTR retrotransposon SLACS, or spliced leader-associated conserved sequence, which integrates exclusively into the spliced leader (SL) RNA genes. Although there is evidence that the RNA interference (RNAi) machinery regulates SLACS transcript levels, we do not know whether RNAi deficiency affects the genomic stability of SLACS, nor do we understand the mechanism of SLACS transcription. Here, we report that prolonged culturing of RNAi-deficient T. brucei cells, but not wild-type cells, results in genomic rearrangements of SLACS. Furthermore, two populations of SLACS transcripts persist in RNAi-deficient cells: a full-length transcript of approximately 7 kb and a heterogeneous population of small SLACS transcripts ranging in size from 450 to 550 nt. We provide evidence that SLACS transcription initiates at the +1 of the interrupted SL RNA gene and proceeds into the 5′ UTR and open reading frame 1 (ORF1). This transcription is carried out by an RNA polymerase with α-amanitin sensitivity reminiscent of SL RNA synthesis and is dependent on the SL RNA promoter. Additionally, we show that both sense and antisense small SLACS transcripts originate from ORF1 and that they are associated with proteins in vivo. We speculate that the small SLACS transcripts serve as substrates for the production of siRNAs to regulate SLACS expression.
Introduction
Mobile genetic elements, such as transposons and retrotransposons, have shaped the evolution of eukaryotes from yeast to human. While previously considered selfish disruptors of the genome, more recent research has revealed a role for these elements in regulating host gene expression, facilitating recombination and promoting genetic diversity (Hasler and Strub, 2006; Volff, 2006). Additional evidence that the RNA interference (RNAi) pathway controls the silencing of mobile genetic elements (Vastenhouw and Plasterk, 2004) has thrust these genomic ‘parasites’ into the spotlight.
The genome of the early divergent eukaryote Trypanosoma brucei is colonized by two families of non-LTR retrotransposons, namely ingi (Kimmel et al., 1987) and SLACS, or spliced leader-associated conserved sequence (Aksoy et al., 1987; 1990; Carrington et al., 1987). While ingi integration does not appear to be site-specific, SLACS is known to integrate exclusively at nucleotide 11 of the 140 nt spliced leader (SL) RNA gene. A previous study detected nine copies of SLACS among the approximately 200 copies of the SL RNA gene in the T. brucei gambiense strain (Aksoy et al., 1990). SLACS is a 6.8 kb element and encodes two open reading frames (ORFs). ORF1 contains a nucleic acid binding domain with homology to retroviral gag domains, and ORF2 encodes a bifunctional polypeptide with domains that display homology to an endonuclease and a reverse transcriptase (Aksoy et al., 1990). As is characteristic of an element that integrates via an RNA intermediate, SLACS has a poly(dA) track at its 3′ end, and is flanked by a 49-nucleotide target duplication site, which is predicted to be a consequence of a staggered break in the DNA duplex made during non-LTR retrotransposon integration (Luan et al., 1993).
Homologues of the SLACS retrotransposon have been identified in all subspecies of T. brucei and more recently in Leishmania braziliensis (Peacock et al., 2007). Related SL RNA-specific retroelements have been characterized in protozoan parasites, such as Trypanosoma cruzi (CZAR) and Crithidia fasciulata (CRE1) (Gabriel et al., 1990; Villanueva et al., 1991), as well as in the metazoan Caenorhabditis elegans (Ne-SL-1) (Malik and Eickbush, 2000).
Although the sequence and integration site of SLACS elements in T. brucei have been described some time ago, little is known about SLACS expression. We have previously reported that SLACS transcripts are more stable and more abundant in RNAi-deficient T. brucei cells, providing evidence that the RNAi pathway regulates retroposon gene expression at transcriptional and post-transcriptional levels (Shi et al., 2004). RNAi-dependent transcriptional silencing has been reported in fungi (Nolan et al., 2005), plants (Lippman et al., 2003), worms (Vastenhouw et al., 2003), flies (Aravin et al., 2001) and human cells (Yang and Kazazian, 2006). Our observations that similar transcriptional silencing pathways may be active in T. brucei led us to investigate the genomic stability and mechanism of transcription of SLACS elements in RNAi-deficient cells. Two mechanisms for transcription of non-LTR retroposons have been proposed. The first involves transcription from an internal promoter encoded within the element’s 5′ UTR (Mizrokhi et al., 1988). The second predicts that non-LTR retroposons are expressed via co-transcription with an interrupted gene, though to the best of our knowledge, this has yet to be proven experimentally (Eickbush et al., 2000; Malik and Eickbush, 2000; Ye and Eickbush, 2006). Here, we report the detection of SLACS transcripts that initiate at the +1 nt of the interrupted SL RNA gene and show that this transcription relies on the activity of the upstream SL RNA promoter. We also describe a novel population of smaller SLACS transcripts originating from ORF1, leading us to speculate that they serve as substrates for the production of SLACS siRNAs, and thus are involved in the RNAi-dependent silencing of these elements.
Results
Genomic instability of SLACS in ago1-/- cells
As disruption of the RNAi pathway in nematodes led to mobilization of endogenous transposons (Tabara et al., 1999), we wanted to investigate whether RNAi deficiency in T. brucei had any effect on the genomic stability of SLACS elements. We previously established RNAi-deficient cells by replacing both argonaute 1 (AGO1) alleles in the wild-type YTat 1.1 strain with drug-resistance markers (Shi et al., 2004). Thus, populations of wild-type YTat 1.1 and ago1-/- cells were maintained in culture continuously for approximately 820 and 640 generations respectively. The resulting cells were then cloned by limiting dilutions. Genomic DNA was prepared from each clone and digested with EcoRV, a restriction enzyme that cuts within SLACS elements but not within the SL RNA repeat unit. Following gel electrophoresis and Southern transfer, the membrane was hybridized with a SLACS-specific probe close to the 3′ end of the element to highlight fragments spanning from one SLACS element to the next, and thus displaying the number of SLACS elements present in the genome (schematically illustrated in Fig. 1C). This revealed about 15 distinct fragments (Fig. 1B, lane 1), which was in the range of our estimation of 16-18 copies of the SLACS element per haploid genome in our wild-type YTat 1.1 strain (data not shown).
Fig. 1.
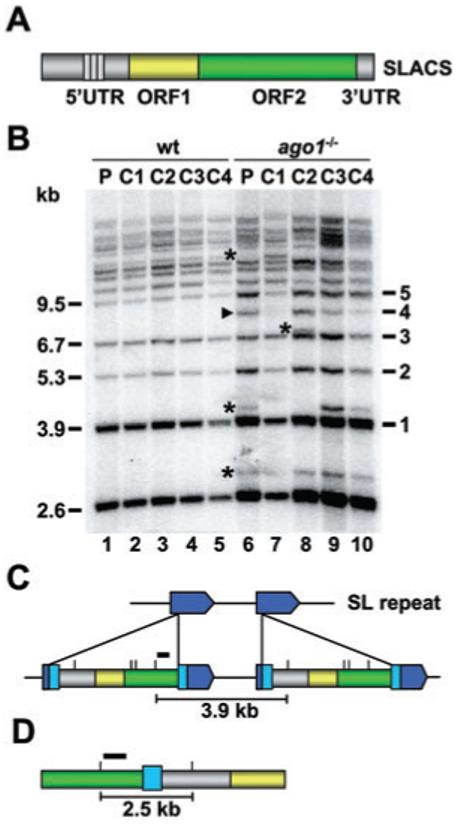
Genomic organization of SLACS in the SL RNA gene cluster of T. brucei rhodesiense.
A. Schematic representation of the SLACS element with the 5′ UTR, ORF1, ORF2 and 3′ UTR indicated in different colours. The 185 bp repeat region present in the 5′ UTR is shown by small rectangles. The poly(dA) tract immediately following the 3′ UTR has been omitted for clarity.
B. Southern blot analysis of wild-type (wt) and ago1-/- genomic DNA digested with EcoRV and hybridized with a probe indicated by a black bar in panels C and D. The wild-type (P, lane 1) and ago1-/- populations (P, lane 6) were cloned, and four randomly selected clones were analysed for both cell types (C1-C4, lanes 2-5 and lanes 7-10). Black arrow heads and asterisks indicate new bands appearing in ago1-/- cells after being maintained in culture for 8 months. Approximate sizes (kb) and the number of SL RNA repeats separating SLACS elements are indicated on the left and right respectively.
C. Schematic representation of two SLACS elements integrated into two adjacent SL RNA genes, shown in dark blue. The 49 bp duplication of the target sequence, generated as a consequence of SLACS insertion, is shown in light blue. Small vertical lines above the SLACS elements specify EcoRV restriction sites and the black bar identifies the probe used for the Southern blot analysis shown in panel B. And 3.9 kb is the predicted distance between EcoRV sites in two adjacent SLACS.
D. Schematic representation of the cloned 2.5 kb genomic fragment, with two tandem SLACS separated by the 49 bp target site duplication. The black bar identifies the probe used for Southern blot analysis shown in panel D. Drawings are not to scale.
Interestingly, whereas there were no apparent differences in the banding pattern of the wild-type population and the resulting clonal cell lines, the ago1-/- population showed variations from the wild-type pattern, and the four randomly selected ago1-/- clones displayed three distinct arrangements of SLACS (Fig. 1B, lanes 6-10). These alterations involved the appearance of individual bands, as well as changes in the relative intensity of existing bands. Whereas we do not know what most new bands represent, the 8.1 kb band (indicated by an arrow head) is consistent with two SLACS elements separated by four SL repeat units. Thus it appeared that in the absence of an RNAi silencing pathway, SLACS elements are subjected to rearrangements, which could be based on transpositional and/or recombinational mechanisms.
The Southern blot in Fig. 1B also yielded information about the genomic organization of SLACS elements in our wild-type T. brucei rhodesiense strain. Assuming that two adjacent SL RNA genes, separated by 1.4 kb, are interrupted by SLACS, EcoRV digestion and subsequent hybridization as outlined above will generate a predicted fragment of 3.9 kb (Fig. 1C). Curiously, our Southern blots revealed the presence of a strongly hybridizing band of 2.6 kb (Fig. 1B). To clarify this issue, we cloned the 2.6 kb EcoRV fragment and determined its sequence. As expected, one end of the DNA sequence started at the last EcoRV site in ORF2, proceeded through the 3′ UTR into a stretch of 23 A residues, and then continued with the 49 bp of the duplicated target SL sequence. However, instead of continuing through the SL RNA gene repeat, the duplicated SL sequence was followed by the beginning of the SLACS element up to the first EcoRV restriction site (illustrated in Fig. 1D). This configuration of two adjacent SLACS separated by 49 bp of SL sequences is not unique to our T. brucei rhodesiense strain, as bioinformatics analysis revealed a similar organization in the sequenced genome of T. brucei strain TREU927/4 GUTat10.1 (see for example NCBI accession number AQ639458).
A distinct population of small SLACS transcripts originates from the 3′ end of ORF1
Spliced leader-associated conserved sequence elements are 6.8 kb in length and encode a long 5′ UTR, as well as two ORFs (Fig. 1A). We previously reported the detection of transcripts approximately the size of a complete SLACS element (Shi et al., 2004). Furthermore, we observed that these SLACS transcripts are more stable and more abundant in ago1-/- cells, which lack the protein AGO1 and are unable to perform RNA interference (Shi et al., 2004). However, these previous studies used RNA isolation methods that selected against smaller-sized transcripts. Therefore, to investigate whether other SLACS transcripts persist in T. brucei, we performed Northern blot analysis using total RNA isolated from wild-type and ago1-/- cells with the TRIzol method. This method of RNA isolation does not discriminate against small RNAs and therefore, transcripts < 500 nt will be represented in these preparations. Confirming our previous results, we detected RNA of approximately 7 kb corresponding to full-length SLACS transcripts using a radiolabelled probe complementary to the entire SLACS ORF1 (Fig. 2A). Northern blots using probes complementary to a portion of the SLACS 5′ UTR (nt 1-1356), and a portion of SLACS ORF2 (nt 3378-6281) also displayed the high-molecular-weight RNA, providing additional evidence that these RNA molecules represent full-length SLACS transcripts (data not shown). Some variation in size of these transcripts was observed, which is most likely due to differences in the number of a 185 bp repeat sequence contained within the 5′ UTR of SLACS (Fig. 1A). Indeed, in performing Southern blot analysis to enumerate the number of these repeats in different SLACS elements in the YTat 1.1 genome, we found that the number of repeats varies from 3 to 14 (Fig. S1).
Fig. 2.
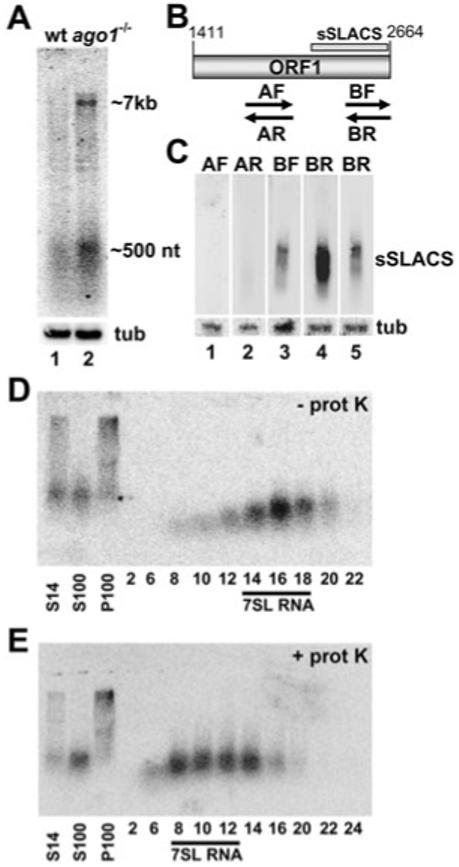
Small SLACS transcripts in ago1-/- cells.
A. Northern blot analysis of SLACS transcripts in wild-type (wt, lane 1) and ago1-/- cells (lane 2) using a probe designed against SLACS ORF1. Approximate sizes of full-length SLACS transcripts (∼7 kb) and the sSLACS transcripts (∼500 nt) are indicated. α-Tubulin (tub) served as a control for RNA recovery and loading (lower panel).
B. Schematic representation of SLACS ORF1. The origin of the sSLACS is indicated by a bar and the PCR-synthesized single-stranded probes used in panel C are shown below ORF1. The drawing is to scale.
C. Northern blot analysis of sSLACS transcripts. RNA from ago1-/- cells was hybridized with probe AF (lane 1) and AR (lane 2), which detect antisense and sense transcripts respectively, between SLACS nt 1755-2054 (see panel B), and with probes BF (lane 3) and BR (lane 4), which detect antisense and sense transcripts respectively, between SLACS nt 2359-2661 (see panel B). A shorter exposure of the BR blot is provided at the far right. α-Tubulin (tub) served as a control for RNA recovery and loading (lower panel).
D. A post-nuclear supernatant from ago1-/- cells (S14) was separated by high-speed centrifugation in a soluble (S100) and pellet fraction (P100), and the S100 was applied to a 10-30% glycerol density gradient. Selected fractions were Northern-blotted with a probe complementary to SLACS ORF1. The fractions containing 7SL RNA are indicated.
E. An equivalent amount of the S100 fraction that was analysed in D was digested with proteinase-K prior to glycerol density gradient centrifugation. The fractions containing 7SL RNA are indicated.
The ORF1 probe also detected a population of low-molecular-weight RNAs, which we have named small SLACS (sSLACS). They are reproducibly visualized as a smear by Northern blot analysis and range in size from about 450 to 550 nt. Preliminary Northerns showed that probes directed against the 5′ UTR or ORF2 did not detect the sSLACS (data not shown). Based on this observation, we designed probes to determine the polarity of the sSLACS and to further delineate the origin of these RNAs (Fig. 2B). To address the first point, we synthesized single-stranded probes by PCR. Probes BF and BR, corresponding to nt 2359-2661 from SLACS ORF1, both hybridized to sSLACS, albeit the pattern was distinct (Fig. 2C, lanes 3 and 4). Sense transcripts detected by probe BR appeared to be more abundant than antisense transcripts detected by probe BF. However, we cannot exclude the possibility that this disparity between sense and antisense sSLACS was due to differences in hybridization efficiencies of the two probes. On the other hand, probes AF and AR, designed against an upstream region (nt 1755-2054), did not detect the sSLACS (Fig. 2C, lanes 1 and 2). Next, we designed a series of 22- to 24-nt-long oligonucleotide probes covering the 3′ end region of ORF1 to determine the boundaries of sSLACS hybridization. The results of this mapping revealed that the sSLACS originated exclusively from the 3′ end of ORF1 and specifically between nt 2110-2617 of SLACS (indicated in Fig. 2B and data not shown).
Retroposon transcripts associate in vivo with their self-encoded reverse transcriptase/endonuclease (Hohjoh and Singer, 1996). We therefore wished to determine whether SLACS transcripts are present in the form of a ribonucleoprotein particle (RNP). A post-nuclear supernatant from ago1-/- cells was centrifuged at 100 000 g to yield a soluble (S100) and pellet fraction (P100). While the full-length SLACS transcripts sedimented in the P100 fraction, probably as a large ribonucleoprotein complex, the sSLACS were present predominantly in the S100 fraction (Fig. 2D and E). The sSLACS-containing S100 fraction was further separated on a 10-30% glycerol density gradient. As shown in Fig. 2D, sSLACS transcripts were mainly detected in fractions 14-18. In order to approximate the molecular mass of the sSLACS, the gradient fractions were probed by Northern blot for the T. brucei 7SL RNA, which in vivo is found in an 11S RNP (Michaeli et al., 1992). As 7SL RNA also sedimented in fractions 14-18, we concluded that the sSLACS had an apparent molecular weight of 11S. Digestion of the S100 extract with proteinase-K prior to glycerol density fractionation resulted in a clear shift of sSLACS, as well as 7SL RNA, to a lower molecular weight (Fig. 2E), demonstrating that sSLACS were in a ribonucleoprotein complex. The nature of this RNP was further investigated by challenging the stability of the particle with increasing salt concentrations (Fig. S2). For these experiments, we used the 7SL RNP as a reference, as this particle has been shown to be resistant to salt concentrations as high as 500 mM (Liu et al., 2003). Whereas the majority of sSLACS cofractionated with the 7SL RNP in a 10-30% glycerol density gradient containing 120 mM KCl (Fig. S2, upper panel), we also detected a proportion of sSLACS in lighter fractions. A similar fractionation profile was observed, when the salt concentration was raised to 250 mM, with a noticeable increase of sSLACS fractionating lighter than the 7SL RNP (Fig. S2, middle panel). Finally, performing the fractionation in 500 mM KCl further increased the proportion of sSLACS being lighter than the 7SL RNP (Fig. S2, lower panel), indicating that proteins dissociate from the sSLACS RNP under these conditions.
Characteristics of SLACS transcription
So far, nothing is known how SLACS RNAs are generated, i.e. whether transcription initiates from an internal promoter located within the 5′ UTR, or whether SLACS is expressed via co-transcription with the interrupted SL RNA gene. To begin to address this issue, we first examined newly synthesized RNA originating from SLACS elements. Nascent 32P-radiolabelled RNA was harvested from lysolecithin-permeabilized ago1-/- cells and hybridized to nitrocellulose filters containing strand-specific single-stranded DNA probes of equal size (300 bp) covering the beginning of the 5′ UTR (probe 1, nt 6-316), the beginning of ORF1 (probe 2, nt 1453-1751), the beginning of ORF2 (probe 3, nt 2783-3083), the middle of ORF2 (probe 4, nt 4618-4919) and the end ORF2 (probe 5, nt 6295-6591). As shown in Fig. 3A, the strongest hybridization occurred at the beginning of the 5′ UTR, and hybridization intensity dropped about 15-fold between probe 1 and probe 2. DNAs representing ORF2 hybridized weakly and with approximately similar intensities, and the signals were approximately 20- to 60-fold less intense than what we observed at the 5′ end of the SLACS element. Hybridizing newly synthesized RNA to an SL RNA probe revealed that SL RNA transcription, when normalized on the basis of copy numbers (∼200 SL RNA genes versus ∼17 SLACS elements), was about equal to that detected by the 5′ UTR probe. We also detected low levels of antisense transcripts that hybridized to probes 1 and 2, curiously, the same region where we observed the most sense transcription.
Fig. 3.
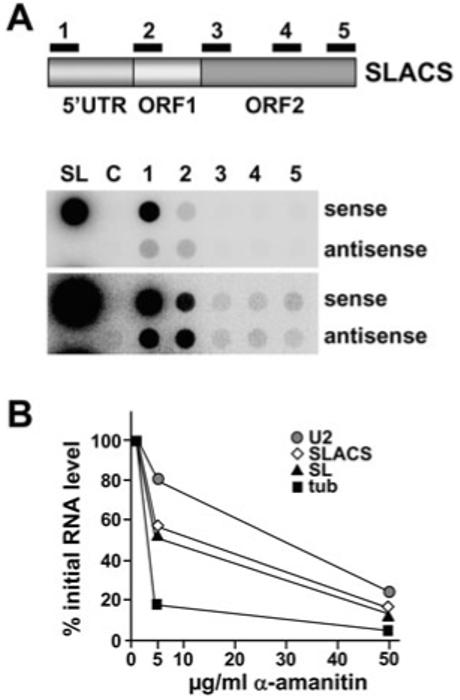
SLACS transcription in ago1-/- cells.
A. Dot blot hybridization of newly synthesized RNA DNA fragments of about 300 bp, cloned into M13 vectors and spotted onto a nitrocellulose membrane. The numbers refer to the regions indicated in the accompanying graphical representation of SLACS. Drawing is not to scale. SL, spliced leader RNA gene probe; C, vector control. Two different exposure times are shown.
B. α-Amanitin sensitivity of SLACS transcription. [α-32P]-RNA was synthesized in permeable cells in the presence of increasing concentrations of α-amanitin as indicated, hybridized to cloned DNAs immobilized onto nitrocellulose filters, quantified and expressed as percentages of the level of transcription measured in the absence of the inhibitor. U2, U2 snRNA; tub, α-tubulin.
Next, we set out to understand the nature of the RNA polymerase (Pol) responsible for SLACS transcription. Lysolecithin-permeabilized ago1-/- cells were treated with increasing concentrations of the inhibitor α-amanitin, and synthesis of various RNAs was analysed by hybridizing newly synthesized 32P-labelled RNA to different gene probes immobilized on nitrocellulose filters. As expected, Pol II-mediated transcription of the β-tubulin genes decreased by over 80% at the low α-amanitin concentration of 5 μg ml-1, and was almost completely abrogated at 50 μg ml-1 α-amanitin (Fig. 3B). Under the same conditions, synthesis of the U2 snRNA by Pol III was reduced by 20% and 70% in the presence of 5 and 50 μg ml-1 α-amanitin respectively. As previously reported, transcription of the SL RNA genes by Pol II (Gilinger and Bellofatto, 2001) had an intermediate sensitivity to the inhibitor with an IC50 of approximately 20 μg ml-1 (Laird et al., 1985; Ullu and Tschudi, 1990). In the presence of 5 and 50 μg ml-1 of the inhibitor, transcription of SLACS decreased by 50% and 80% respectively (Fig. 3B), thus matching the α-amanitin sensitivity of SL RNA gene transcription. A comparable result was obtained when we monitored SLACS transcription with a probe originating from the beginning of the 5′ UTR or the beginning of ORF1, whereas the hybridization signals further downstream (probes 3-5 in Fig. 3A) were too weak for a reliable analysis.
SLACS transcripts initiate at the +1 nt of the interrupted SL RNA gene in vivo
The results of the above experiments indicated that transcription of the SLACS element up to position 1751 is carried out by a polymerase with an α-amanitin sensitivity typical of the Pol II responsible for SL RNA synthesis. As this could mean that SLACS transcription is directed by the promoter of the interrupted SL RNA gene, we next set out to determine the 5′ end of these SLACS transcripts. Poly(A)+ RNA isolated from ago1-/- cells was reverse-transcribed with an oligonucleotide complementary to the very end of the SLACS 5′ UTR (nt 1386-1409), and then PCR-amplified in combination with an oligonucleotide complementary to the first 29 nt of the SL sequence present at the very 5′ end of all mRNAs in trypanosomes. This generated three major fragments ranging in size from 1.4 to 1.6 kb, and subsequent sequencing revealed that the three fragments were basically identical, except that they differed in the number of the 185 bp repeat present in the 5′ UTR (data not shown). Most importantly, the very 5′ end contained the first 60 nt of the SL coding region fused to the beginning of the SLACS 5′ UTR, i.e. the predicted sequences, if transcription initiated at the +1 of the interrupted SL RNA gene. Nevertheless, as this approach was biased towards transcripts with the SL sequence at the very 5′ end, we performed primer extension analysis with total RNA isolated from ago1-/- cells and a α-32P end-labelled oligonucleotide complementary to nt 1-28 of the SLACS 5′ UTR. As can be seen in Fig. 4, this produced an 88-nt-long cDNA (lane 4), which places the 5′ end at the +1 of the SL RNA gene. Curiously, primer extension of the SLACS RNA at the low deoxyribonucleotide triphosphate concentrations used did not result in characteristic primer extension stops at the 5′ end of the SL, which are indicative of a fully modified cap 4 structure (Mandelboim et al., 2002). The presence of this cap structure was evident when SL RNA from wild-type or ago1-/- cells was primer-extended with an oligonucleotide complementary to nt 65-88 of the SL RNA (Fig. 4, lanes 2 and 3). As the SLACS primer extension was reminiscent of hypomethylated SL RNA, which was obtained from cells grown in the presence of the methylation inhibitor sinefungin (lane 1), our result would suggest that the 5′ end of SLACS transcripts does not possess a fully modified cap structure.
Fig. 4.
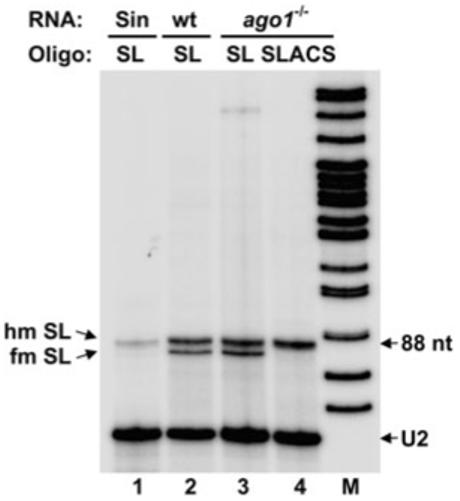
SLACS transcripts initiate at the +1 nt of the interrupted SL RNA gene. Total RNA isolated from sinefungin-treated (sin, lane 1), wild-type (wt, lane 2) and ago1-/- cells (lanes 3 and 4) was primer-extended with an oligonucleotide complementary to nt 65-88 of the SL RNA (SL, lanes 1-3) and an oligonucleotide complementary to SLACS nt 1-28 (SLACS, lane 4). Primer extension stops corresponding to fully modified (fm) and hypomodified (hm) SL RNA are indicated. M, α-32P-labelled MspI digest of pBR322. A U2 snRNA-specific primer was included in all the reactions to control for RNA amounts and quality (U2).
SLACS transcription is dependent on the SL RNA promoter
So far our results are consistent with SLACS being co-transcribed with the interrupted SL RNA gene. To determine whether the SL RNA promoter was essential for this expression, transcription of different plasmid constructs was analysed in vitro in a homologous cell-free extract (Günzl et al., 1995). We have previously used this system to accurately transcribe the T. brucei SL RNA gene, which was tagged at position +51 relative to the transcription start site by the addition of 19 nt (pGS-Lins19) to allow specific detection of transcripts by primer extension with an oligonucleotide complementary to the tag. Similarly, a SLACS expression construct, containing 240 bp of SL upstream sequences, 60 bp of the SL coding region and 1.3 kb of the SLACS 5′ UTR, was marked by the insertion of 19 nt at position 51 of the SL coding region. This construct (SLACS) was linearized at a unique BglI restriction site at position 234 in the SLACS 5′ UTR and transcribed in vitro in the presence of [α-32P]-GTP. As shown in Fig. 5A (lane 3), this reaction produced a major transcript of approximately 250 bp, which places the transcription initiation site in the vicinity of the +1 nt of the interrupted SL RNA. The absence of other specific bands suggested that there are no major internally initiated SLACS transcripts produced in this in vitro system [two minor and smaller RNAs visible in lane 3 were also present in the control reactions (lanes 1 and 2)]. To accurately map the transcription start site, we performed primer extension analysis with an oligonucleotide complementary to the tag (Fig. 5B, lane 1). This gave rise to one major band (72 nt) corresponding to accurate initiation at position +1 of the interrupted SL RNA gene.
Fig. 5.
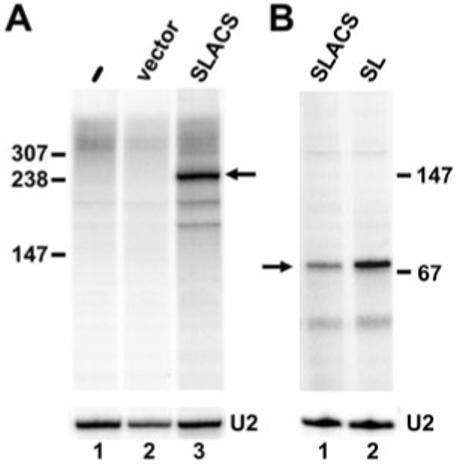
SLACS in vitro transcription.
A. In vitro transcription reactions containing no added DNA (lane 1), the pXS2 backbone vector (lane 2) or pSLACS (lane 3) were performed in the presence of [α-32P]-GTP, and RNA products were analysed on an 8% polyacrylamide-7 M urea gel.
B. In vitro transcription reactions were carried out in the absence of radiolabelled nucleotides, and RNA was analysed by primer extension with an end-labelled oligonucleotide complementary to a 19 nt tag engineered in both the SLACS and SL templates (SLtag). U2 primer extension served as a loading control in both panels.
To determine if the SL promoter was essential for expression of SLACS sequences, we engineered several point mutations in the SL promoter upstream element 1 (USE1) located between nt -71 and -62. Previous studies have demonstrated that these mutations effectively abrogated the ability of the SL RNA promoter to recruit the Pol II machinery, and thus severely affected SL RNA transcription (Günzl et al., 1997). As predicted, mutations in USE1 reduced transcription from the tagged SL construct to almost undetectable levels (compare lanes 3 and 4 in Fig. 6). Similarly, transcription of the SLACS construct containing a mutant USE1 (SLACSmut) was severely inhibited (compare lanes 5 and 6) and comparable to the effect on SL RNA transcription: 20- and 25-fold reduction for SL RNA and SLACS transcription respectively. In addition, we observed an inhibition of SLACS expression conferred by the USE1 mutation, when in vitro run-off transcription reactions were performed in the presence of [α-32P]-GTP (Fig. S3). Taken together, these results provided evidence that SLACS transcription is dependent on the SL RNA promoter.
Fig. 6.
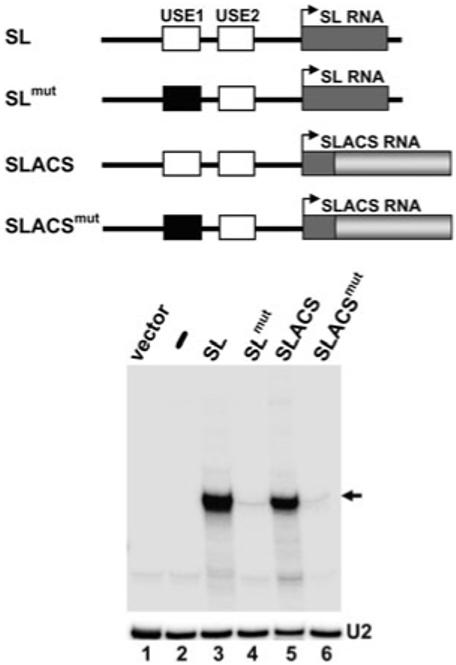
SLACS transcription is dependent on the upstream SL promoter. In vitro transcription reactions were carried out with the constructs schematically shown above the autoradiograph, and RNA was primer-extended with the SLtag oligonucleotide as described in Fig. 5. Drawings are not to scale. U2 primer extension served as a loading control (U2).
Probing the transcriptional machinery associated with SLACS
Our finding that SLACS transcription is directed by the SL promoter raises the question of how a transcriptional machinery dedicated to the synthesis of a short transcript, i.e. the 139-nt-long SL RNA, manages to elongate transcription into the SLACS element. As SL RNA gene and SLACS transcription rely on identical promoter elements (Fig. 6), we considered it unlikely that different transcription factors would assemble on these two promoters. Thus, we hypothesized that the SLACS transcription unit itself might recruit specific factors involved in transcription elongation. To begin to address this possibility, we searched the trypanosomatid databases for possible candidates, and identified two putative subunits of FACT (facilitates chromatin transcription). Human FACT promotes the dislocation of one H2A-H2B dimer from a nucleosome without requirement for ATP, and consequently the resulting nucleosome structure is more favourable for Pol II elongation (Belotserkovskaya et al., 2003; 2004). FACT in humans is a two-subunit complex comprised of p140 and SSRP1 (Orphanides et al., 1999) with the yeast homologues Spt16 and Pob3. Putative T. brucei homologues of the small (Pob3) and large subunit (Spt16) of FACT were assigned to a 61 kDa protein (GeneDB Accession No. Tb10.61.2370) and 112 kDa (GeneDB Accession No. Tb03.5L5.860) protein respectively (J.-P. Ruan and C. Tschudi, unpublished and Martinez-Calvillo et al., 2007). Downregulation of TbSpt16 and TbPob3 mRNAs by RNAi revealed that both proteins are encoded by essential genes (J.-P. Ruan and C. Tschudi, unpublished).
To monitor the association of the T. brucei Pob3 and Spt16 polypeptides with DNA in living cells, we used our previously established chromatin immunoprecipitation (ChIP) method (Ruan et al., 2004). We generated two procyclic T. brucei cell lines, each expressing solely an epitope-tagged version of either Pob3 or Spt16. Following immunoprecipitation with an epitope-specific antibody and reversal of the cross-link, samples were analysed by PCR using primers amplifying the region from nucleotide -56 to +82 of the SL RNA transcription unit, as well as a region upstream of the gene (-530 to -608), which, as we have previously shown (Ruan et al., 2004), does not recruit factors required for transcription. As can be seen in Fig. 7A, neither Pob3 nor Spt16 were recruited to the SL RNA gene at a detectable level. However, most interestingly, both Spt16 and Pob3 were found associated with SLACS (Fig. 7B and data not shown; the double bands observed at SLACS regions 1 and 3 are a consequence of sequence heterogeneity, i.e. nucleotide deletions/insertions, in the 5′ UTR and ORF2, respectively, of SLACS elements). In particular, it appeared that these two proteins are recruited in the vicinity of the SL promoter upstream of SLACS (region 1 in Fig. 7B), although our ChIP data do not allow us to distinguish between SL promoter and SLACS 5′ UTR sequences. Furthermore, Spt16 was also found over 3000 bp downstream of the promoter (region 3 in Fig. 7B), suggesting that it might travel with the elongating polymerase, similar to what has been shown in other organisms (Belotserkovskaya et al., 2004).
Fig. 7.
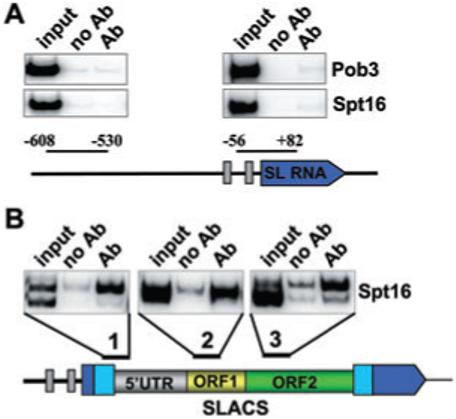
Spt16 association with SLACS, but not the SL RNA gene. ChIP was performed with antibodies directed against the BB2 epitope present on Spt16 or Pob3, and precipitates were analysed by PCR with primers specific for the regions indicated (Ab). A control with no antibody (no Ab) and an aliquot of the total input (input) is also shown. The double bands observed at SLACS regions 1 and 3 are a consequence of sequence heterogeneity, i.e. nucleotide deletions/insertions, in the 5′ UTR and ORF2, respectively, of SLACS elements.
Discussion
The site-specific non-LTR retroposon SLACS was first described in 1987, and was shown to insert exclusively into the tandemly repeated SL coding sequence, thus generating a 49 bp duplication (Aksoy et al., 1987). Whereas it was originally recognized that SLACS has many attributes of a retroposon, including an ORF2 encoding a putative bifunctional polypeptide with domains that display homology to an endonuclease and a reverse transcriptase and a short 3′ UTR immediately followed by a poly(dA) tail, our knowledge about its expression has remained rather limited. In particular, it has been difficult to detect SLACS RNA (Aksoy et al., 1990). A first clue was provided when the analysis of siRNAs in T. brucei revealed constitutively expressed SLACS-derived siRNAs, hinting that the RNAi machinery controls the levels of retroposon transcripts (Djikeng et al., 2001). This implication was further substantiated when RNAi-deficient cells, generated by a genetic knock-out of AGO1, failed to accumulate SLACS siRNAs and concomitantly revealed an increase in SLACS transcript abundance in steady-state RNA (Shi et al., 2004). In the study presented here, we took advantage of the RNAi-deficient cells to ask how SLACS is expressed.
The vast majority of protein-coding genes in T. brucei are transcribed by a typical Pol II with an exquisite sensitivity to the inhibitor α-amanitin. Quite surprisingly, our studies in permeabilized cells revealed that the response of SLACS transcription to this inhibitor closely resembles the sensitivity of Pol II-mediated expression of the SL RNA (Fig. 3C). Our inhibitor assays were restricted to the first 1.5 kb of the SLACS element, as a result of very low abundance of nascent SLACS transcripts further downstream. Nevertheless, our data provided evidence that an SL RNA-typical Pol II transcribed the 5′ UTR and elongated into ORF1 of SLACS, possibly connecting SLACS transcription to the SL RNA gene promoter. Indeed, we detected SLACS transcripts initiating at the +1 of the SL RNA coding sequence and extending into the 5′ UTR by primer extension and RT-PCR. These findings were corroborated by in vitro transcription analysis, which established that SLACS expression is dependent on the SL RNA gene promoter. We should note that this is not the first report that the SL RNA promoter can drive the transcription of a protein-coding gene: the SL RNA promoter from the related protozoan parasite Leptomonas seymouri was found sufficient to express a chloramphenicol acetyl transferase reporter construct (Bellofatto and Cross, 1989).
Factors identified so far to be required for SL RNA transcription include an unusual small nuclear RNA-activating protein complex, composed of TRF4 and two divergent TFIIA subunits, as well as trypanosome homologue of TFIIB and TFIIH (Ruan et al., 2004; Das et al., 2005; Schimanski et al., 2005; 2006; Palenchar et al., 2006; Lee et al., 2007). Whereas the transcriptional machinery assembling on the SL RNA promoter normally makes a short 139-nt-long transcript, we were curious to probe whether the same or a different machinery elongates through the 5′ UTR into ORF1 of SLACS and possibly to the end of the SLACS element. One scenario is that specific elongation factors are recruited to the SLACS unit. Indeed, we found that the T. brucei putative homologues of chromatin factors Pob3 and Spt16 are present at the SLACS locus, but were not found associated with the SL RNA genes. At present, we do not know the actual site of the initial recruitment, as our ChIP analysis cannot distinguish between the SL RNA promoter and the beginning of the SLACS 5′ UTR. It is unlikely that the promoter elements per se are involved, as both SL RNA and SLACS transcription initiation rely on identical elements. More probable is that distinct chromatin structures at the SL and SLACS transcription units are key determinants to allow recruitment of these factors to one unit but not the other. Finally, although our data, including RT-PCR, primer extension and in vitro transcription, are consistent with SLACS transcription initiating at the +1 of the SL RNA gene and proceeding into the SLACS element, it is important to point out that we cannot entirely rule out the presence of internal initiation sites. This scenario cannot be overlooked, as Pol II transcription initiation of protein coding regions in trypanosomatids remains an enigma.
One puzzling feature of SLACS expression was exposed, when we performed nuclear run-on assays in permeabilized T. brucei ago1-/- cells to evaluate RNA polymerase density along the retroposon. These experiments showed that polymerase density in the early region of the 5′ UTR was similar to the SL RNA gene, but functionally engaged RNA polymerase density dropped precipitously as transcription was assayed along the SLACS element (Fig. 3). Thus, sequences beyond the 5′ UTR appear to be poor substrates for transcriptional elongation, which could be due to slow elongation rate, stalling of the RNA polymerase complex or premature dissociation. One consideration is that there are termination sites in the SLACS element. SL RNA transcription terminates in an extensive T-tract, and mutational analysis has shown that more than six T residues are required for this process to be efficient in vivo (Sturm et al., 1999). In SLACS, the termination signals of the interrupted SL RNA gene are located several kb downstream from the transcription initiation site past the poly(dA) tract. Furthermore, there are no T tracts longer than five residues in ORF1 or ORF2. However, there is one copy of six T residues in the 185 bp repeat in the 5′ UTR, which could result in a partial stalling of the polymerase. Alternatively, experiments investigating LINE-1 (L1) elements in the human genome (Han et al., 2004) have shown that poor expression of L1 RNA was correlated with a strong adenosine-rich bias in the sense strand. This was elegantly proven by synthesizing L1 with an altered base content without changing the amino acid sequence (Han and Boeke, 2004). Reducing the adenosine content to 26%, as compared with 40% in the wild-type element, overcame the transcriptional defect and also resulted in high-frequency retrotransposition (Han and Boeke, 2004; Han et al., 2004). One proposal from these studies was that this elongation defect hinders the spread of L1 elements in the human genome. It is possible that T. brucei has evolved a similar mechanism for controlling the proliferation of the SLACS retroposon among its essential SL RNA gene repeats. Indeed, the 5′ UTR of SLACS has an unusually high frequency of AT residues, as compared with ORF1 and ORF2 (Fig. S4). Specifically, the SLACS 5′ UTR contains 39% adenosine residues, resulting in an AT content of 61%, as compared with 44% in the remainder of the element. Taken together, it is therefore likely that transcription through the SLACS ORFs is inefficient because of an elongation defect in the 5′ UTR, and therefore inhibiting the robust expression of SLACS RNA and proteins.
To the best of our knowledge, there have been no reports in other eukaryotes of a population of retroposon-encoded, less-than-full-length transcripts like the sSLACS of about 500 nt we describe here. Thus, these transcripts may be a unique adaptation of these retroposons, or of the trypanosome in response to these elements. While it is common for truncated versions of retroelements to arise as a consequence of recombination events, Southern blots and bioinformatics analysis did not reveal the presence of truncated forms of SLACS in the T. brucei genome (C. Tschudi, unpubl. data). Furthermore, we observed that the steady-state sSLACS transcripts originate exclusively from the 3′ end of ORF1 (Fig. 2B), and that both sense and antisense transcripts can be detected by Northern blot analysis. Previously, we have cloned a population of endogenous siRNAs and found that the majority of SLACS-derived siRNAs contained sequences from SLACS ORF1 and the 5′ end of SLACS ORF2 (Djikeng et al., 2001). It is tantalizing that these potentially double-stranded sSLACS transcripts come from the same region of the SLACS coding sequence as the majority of siRNAs, and thus could serve as substrates for Dicer for the production of siRNAs. Currently, we do not know how the sSLACS transcripts are made. One possibility is that they originate from independent transcription initiation events. Alternatively, the sSLACS might be processing products of full-length SLACS transcripts. Such a processing pathway has been observed in plants, where it plays an important role in amplifying the RNAi response (Axtell et al., 2006; Baulcombe, 2007). We are currently investigating the presence of such a pathway in T. brucei.
Preliminary experiments isolating poly(A)+ RNA showed that the sSLACS transcripts are not polyadenylated, which brought into question why these transcripts are not degraded by exonucleases. One possibility is that proteins bind to these transcripts and occlude them from nucleolytic cleavage and indeed, we showed that the sSLACS are found in an RNP. There are reports of full-length retroelement transcripts associating with proteins in vivo, namely their self-encoded reverse transcriptase/endonuclease and in one case, the host-expressed poly(A) binding protein (West et al., 2002). However, the formation of an RNP containing a subset of shorter retroposon transcripts, such as the sSLACS, is novel. Naturally, we were keen to get a handle on the protein components in the sSLACS RNP. An estimation of the abundance of this RNP in trypanosome cells revealed that the sSLACS RNP was over 40 times less abundant than the U2 RNP, thus presenting a daunting challenge for biochemical purification. We are therefore left with educated guesses regarding the sSLACS proteins. It would be rather unlikely that the sSLACS are associating with the SLACS reverse transcriptase/endonuclease, as such an interaction would not result in a productive reintegration, and thus would not be a desirable adaptation for a retroelement. On the other hand, if the sSLACS serve as substrates for siRNA production, then it is possible that the sSLACS transcripts associate with components of the RNAi machinery, such as Dicer or an RdRP-like protein, and this continues to be under investigation.
Finally, based on variable restriction enzyme digestion patterns in ago1-/- cells, we provide evidence for genomic rearrangements of the SLACS element in the absence of the RNAi pathway. It is worth noting that organisms like T. brucei and L. braziliensis have both non-LTR retrotransposons and components of the RNAi machinery encoded in their genomes. However, related organisms like T. cruzi, which harbours several retrotransposon families, including L1Tc and the SL RNA-specific CZAR retrotransposon, do not have a functional RNAi pathway. This might suggest that T. cruzi lost the RNAi pathway at one point and consequently evolved some other mechanism to protect its genome from mobile genetic elements. At present, we can only speculate what genetic mechanism accounts for the genomic rearrangements of the SLACS element in RNAi-deficient T. brucei cells. The hypervariability observed in clonal cell lines could be explained by unequal crossing-over within the tandem arrays of the SL RNA genes, a mechanism commonly associated with the evolution of highly repetitive tandem sequences. Alternatively, we have to consider transposition, translocation or non-chromatid gene conversion events. Nevertheless, whatever the underlying mechanism is, the observed genomic instabilities were caused by RNAi deficiency, thus providing yet another opening for investigating the function of RNAi in T. brucei.
Experimental procedures
Trypanosome cell lines
Trypanosoma brucei rhodesiense Ytat 1.1 and ago1-/- procyclic cell lines were maintained in SM medium (Cunningham, 1977) containing 10% fetal bovine serum, and construction of the ago1-/- cell line has been described previously (Shi et al., 2004). Populations of wild-type YTat 1.1 and ago1-/- cells were maintained in culture for approximately 8 months, or about 820 generations for YTat 1.1 cells with a 7 h doubling time, and about 640 generations for ago1-/- cells with a 9 h doubling time.
Southern blot and copy number analysis
Ten to thirty micrograms of T. brucei YTat 1.1 and ago1-/- genomic DNA was digested with restriction enzymes over-night, followed by phenol chloroform extraction and ethanol precipitation. Digested DNA was separated by gel electrophoresis on a 0.7% agarose gel at 25 V overnight. DNA was transferred to a nitrocellulose membrane and hybridized with a radiolabelled probe directed against SLACS nt 4679-4980. Copy number analysis was performed by separating dilutions of known concentrations of a 1.2 kb SLACS ORF1 double-stranded PCR fragment on an agarose gel alongside dilutions of YTat 1.1 genomic DNA digested with PvuII. Digestion of genomic DNA by PvuII produces an ORF1-containing band of 1330 nt. Following transfer to a nitrocellulose membrane, the DNAs were hybridized to a radiolabelled 1.2 kb ORF1 probe. The copy number was estimated by comparing the ORF1 PCR product hybridization intensity with that of the PvuII-digested SLACS genomic DNA bands.
Permeabilized cells
ago1-/- procyclic trypanosomes were lysolecithin-permeabilized as described previously (Ullu and Tschudi, 1990). After a 15 min incubation at 28°C, RNA was extracted using the TRIzol method (Invitrogen), precipitated with ethanol, resuspended in H2O and hybridized to DNA that had been spotted onto nitrocellulose membranes. Membranes were hybridized overnight at 68°C in an aqueous buffer (5× SET, 10× Denhardts, 1% SDS, 10 μg ml-1 yeast RNA), and then washed three times for 30 min each at 68°C in 2× SSC and 0.1% SDS, followed by a 30 min incubation in RNase A (10 μg ml-1) in 2× SSC. Blots were exposed to a PhosphorImager screen, developed and analysed using OptiQuant software (Perkin Elmer).
Preparation of S100 cell extracts and in vitro transcription reactions
Five litres of Ytat 1.1 trypanosome cells was harvested by centrifugation at 3500 r.p.m. Cells were washed 3× in cell wash buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 3 mM MgCl2) and lysed by manual douncing using a 7 ml glass dounce homogenizer until about 90% of cells were broken, as determined by visual examination. After centrifugation at 4000 r.p.m., the packed cell volume was noted, and cells were resuspended in 0.8 vol. of 1× transcription buffer (150 mM sucrose, 20 mM potassium l-glutamate, 20 mM HEPES-KOH, pH 7.2, 3 mM MgCl2, 10 μg ml-1 leupeptin, 1 mM DTT). Cell homogenates were spun twice at 14 000 r.p.m., and the supernatants were spun twice at 200 000 g. S100 extracts were snap-frozen in liquid nitrogen and stored at -80°C. In vitro transcription reactions were done essentially as described (Nakaar et al., 1997). Briefly, 15 μl of extract was added to a transcription mix containing 20 mM potassium l-glutamate, 20 mM HEPES-KOH, pH 7.9, 3 mM MgCl2, 2.5% PEG 8000, 20 mM creatine phosphate, 0.48 mg ml-1 creatine kinase, 2 mM ATP, 0.8 mM UTP, 0.8 mM CTP, 0.8 mM GTP, 10 μg ml-1 leupeptin and 1 μg DNA template. For some experiments, 0.8 mM GTP was replaced with [α-32P]-GTP to produce radiolabelled transcripts. The transcription reactions were incubated at 28°C for 30 min. RNA was extracted by the TRIzol method (Invitrogen) and precipitated with ethanol. In vitro transcribed RNA was primer-extended with an end-labelled SL-tag oligonucleotide (TTCCATGGTATGGCGCCAG) (Günzl et al., 1997). The resultant cDNA was separated on a 10% polyacrylamide-7M urea gel and visualized by autoradiography. Radiolabelled RNA was resuspended directly in urea buffer, analysed on an 8% polyacrylamide-7 M urea gel and visualized by autoradiography.
Plasmid constructions
The pSLACS plasmid was constructed by PCR amplifying a 1.75 kb DNA fragment from the SL RNA locus from 240 bp upstream of the +1 nt of the SL RNA coding sequence to the 3′ end of the SLACS 5′ UTR, using a forward primer with the sequence GAGGGTACCTTATGGTTTTCTTGCATATC and a reverse primer with the sequence GAGCTCGAGTTTTTTGCGACCAATTTTTATTTG. The underlined KpnI and XhoI sites were used for cloning into the pXS2hygro vector. The pGSLins19 construct was described previously (Günzl et al., 1997). Single-stranded DNA probes were created by inserting portions of the SLACS sequence into the M13mp18 and M13mp19 cloning vectors.
Other procedures
Glycerol density gradients were carried out as described previously (Djikeng et al., 2003). Briefly, 109 cells were harvested, washed twice with cell wash, lysed with 1.2% NP-40 and centrifuged for 6 min at 14 000 r.p.m. The S14 fractions were then centrifuged for 45 min at 53 000 r.p.m. (100 000 g). The S100 supernatant was layered on a 10-30% glycerol gradient and centrifuged for 16 h at 40 000 r.p.m. ChIP was done as described (Ruan et al., 2004). SLACS1F (CTAACGCTATTATTAGAACAG) and SLACS1R (TGAGGATTTTCCTTCCTTTCC) primers were used to amplify immunoprecipitated DNA from region 1, which spans from nt 3 of the SL RNA to SLACS nt 93. SLACS2F (CCGACAAAGAGGTCATAAG AAAAT) and SLACS2R (CTACGGAAGTCTGT GTAGAAACCA) primers amplified region 2, which spans from SLACS nt 1089-1212. Primers SLACS3F (ATTGTGGGGTAAATGAGCTAATGG) and SLACS 3R (TAGACCTCCATATCTGTCCAGGAG) were used to amplify region 3, SLACS nt 2721-2874.
Supplementary Material
Acknowledgements
This work was supported by Public Health Service Grants AI28798 and AI56333 to E.U. and AI43594 to C.T.
References
- Aksoy S, Lalor TM, Martin J, Van der Ploeg LH, Richards FF. Multiple copies of a retroposon interrupt spliced leader RNA genes in the African trypanosome, Trypanosoma gambiense. EMBO J. 1987;6:3819–3826. doi: 10.1002/j.1460-2075.1987.tb02718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy S, Williams S, Chang S, Richards FF. SLACS retrotransposon from Trypanosoma brucei gambiense is similar to mammalian LINEs. Nucleic Acids Res. 1990;18:785–792. doi: 10.1093/nar/18.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr Biol. 2001;11:1017–1027. doi: 10.1016/s0960-9822(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Axtell MJ, Jan C, Rajagopalan R, Bartel DP. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127:565–577. doi: 10.1016/j.cell.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Baulcombe DC. Molecular biology. Amplified silencing. Science. 2007;315:199–200. doi: 10.1126/science.1138030. [DOI] [PubMed] [Google Scholar]
- Bellofatto V, Cross GA. Expression of a bacterial gene in a trypanosomatid protozoan. Science. 1989;244:1167–1169. doi: 10.1126/science.2499047. [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Saunders A, Lis JT, Reinberg D. Transcription through chromatin: understanding a complex FACT. Biochim Biophys Acta. 2004;1677:87–99. doi: 10.1016/j.bbaexp.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Carrington M, Roditi I, Williams RO. The structure and transcription of an element interspersed between tandem arrays of mini-exon donor RNA genes in Trypanosoma brucei. Nucleic Acids Res. 1987;15:10179–10198. doi: 10.1093/nar/15.24.10179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham I. New culture medium for maintenance of tsetse tissues and growth of trypanosomatids. J Protozool. 1977;24:325–329. doi: 10.1111/j.1550-7408.1977.tb00987.x. [DOI] [PubMed] [Google Scholar]
- Das A, Zhang Q, Palenchar JB, Chatterjee B, Cross GA, Bellofatto V. Trypanosomal TBP functions with the multisubunit transcription factor tSNAP to direct spliced-leader RNA gene expression. Mol Cell Biol. 2005;25:7314–7322. doi: 10.1128/MCB.25.16.7314-7322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A, Shi H, Tschudi C, Ullu E. RNA interference in Trypanosoma brucei: cloning of small interfering RNAs provides evidence for retroposon-derived 24-26-nucleotide RNAs. RNA. 2001;7:1522–1530. [PMC free article] [PubMed] [Google Scholar]
- Djikeng A, Shi H, Tschudi C, Shen S, Ullu E. An siRNA ribonucleoprotein is found associated with polyribosomes in Trypanosoma brucei. RNA. 2003;9:802–808. doi: 10.1261/rna.5270203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickbush DG, Luan DD, Eickbush TH. Integration of Bombyx mori R2 sequences into the 28S ribosomal RNA genes of Drosophila melanogaster. Mol Cell Biol. 2000;20:213–223. doi: 10.1128/mcb.20.1.213-223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel A, Yen TJ, Schwartz DC, Smith CL, Boeke JD, Sollner-Webb B, Cleveland DW. A rapidly rearranging retrotransposon within the miniexon gene locus of Crithidia fasciculata. Mol Cell Biol. 1990;10:615–624. doi: 10.1128/mcb.10.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilinger G, Bellofatto V. Trypanosome spliced leader RNA genes contain the first identified RNA polymerase II gene promoter in these organisms. Nucleic Acids Res. 2001;29:1556–1564. doi: 10.1093/nar/29.7.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günzl A, Tschudi C, Nakaar V, Ullu E. Accurate transcription of the Trypanosoma brucei U2 small nuclear RNA gene in a homologous extract. J Biol Chem. 1995;270:17287–17291. doi: 10.1074/jbc.270.29.17287. [DOI] [PubMed] [Google Scholar]
- Günzl A, Ullu E, Dorner M, Fragoso SP, Hoffmann KF, Milner JD, et al. Transcription of the Trypanosoma brucei spliced leader RNA gene is dependent only on the presence of upstream regulatory elements. Mol Biochem Parasitol. 1997;85:67–76. doi: 10.1016/s0166-6851(96)02816-2. [DOI] [PubMed] [Google Scholar]
- Han JS, Boeke JD. A highly active synthetic mammalian retrotransposon. Nature. 2004;429:314–318. doi: 10.1038/nature02535. [DOI] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429:268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- Hasler J, Strub K. Alu elements as regulators of gene expression. Nucleic Acids Res. 2006;34:5491–5497. doi: 10.1093/nar/gkl706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohjoh H, Singer MF. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 1996;15:630–639. [PMC free article] [PubMed] [Google Scholar]
- Kimmel BE, ole-MoiYoi OK, Young JR. Ingi, a 5.2-kb dispersed sequence element from Trypanosoma brucei that carries half of a smaller mobile element at either end and has homology with mammalian LINEs. Mol Cell Biol. 1987;7:1465–1475. doi: 10.1128/mcb.7.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW, Kooter JM, Loosbroek N, Borst P. Mature mRNAs of Trypanosoma brucei possess a 5′-cap acquired by discontinuous RNA synthesis. Nucleic Acids Res. 1985;13:4253–4266. doi: 10.1093/nar/13.12.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Nguyen TN, Schimanski B, Günzl A. SL RNA gene transcription in Trypanosoma brucei requires transcription factor TFIIH. Eukaryot Cell. 2007;6:641–649. doi: 10.1128/EC.00411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z, May B, Yordan C, Singer T, Martienssen R. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. Plos Biol. 2003;1:E67. doi: 10.1371/journal.pbio.0000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Ben-Shlomo H, Xu YX, Stern MZ, Goncharov I, Zhang Y, Michaeli S. The trypanosomatid signal recognition particle consists of two RNA molecules, a 7SL RNA homologue and a novel tRNA-like molecule. J Biol Chem. 2003;278:18271–18280. doi: 10.1074/jbc.M209215200. [DOI] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Malik HS, Eickbush TH. NeSL-1, an ancient lineage of site-specific non-LTR retrotransposons from Caenorhabditis elegans. Genetics. 2000;154:193–203. doi: 10.1093/genetics/154.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelboim M, Estrano CL, Tschudi C, Ullu E, Michaeli S. On the role of exon and intron sequences in trans-splicing utilization and cap 4 modification of the trypanosomatid Leptomonas collosoma SL RNA. J Biol Chem. 2002;277:35210–35218. doi: 10.1074/jbc.M201910200. [DOI] [PubMed] [Google Scholar]
- Martinez-Calvillo S, Saxena A, Green A, Leland A, Myler PJ. Characterization of the RNA polymerase II and III complexes in Leishmania major. Int J Parasitol. 2007;37:491–502. doi: 10.1016/j.ijpara.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaeli S, Podell D, Agabian N, Ullu E. The 7SL RNA homologue of Trypanosoma brucei is closely related to mammalian 7SL RNA. Mol Biochem Parasitol. 1992;51:55–64. doi: 10.1016/0166-6851(92)90200-4. [DOI] [PubMed] [Google Scholar]
- Mizrokhi LJ, Georgieva SG, Ilyin YV. jockey, a mobile Drosophila element similar to mammalian LINEs, is transcribed from the internal promoter by RNA polymerase II. Cell. 1988;54:685–691. doi: 10.1016/s0092-8674(88)80013-8. [DOI] [PubMed] [Google Scholar]
- Nakaar V, Günzl A, Ullu E, Tschudi C. Structure of the Trypanosoma brucei U6 snRNA gene promoter. Mol Biochem Parasitol. 1997;88:13–23. doi: 10.1016/s0166-6851(97)00078-9. [DOI] [PubMed] [Google Scholar]
- Nolan T, Braccini L, Azzalin G, De Toni A, Macino G, Cogoni C. The post-transcriptional gene silencing machinery functions independently of DNA methylation to repress a LINE1-like retrotransposon in Neurospora crassa. Nucleic Acids Res. 2005;33:1564–1573. doi: 10.1093/nar/gki300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400:284–288. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- Palenchar JB, Liu W, Palenchar PM, Bellofatto V. A divergent transcription factor TFIIB in trypanosomes is required for RNA polymerase II-dependent spliced leader RNA transcription and cell viability. Eukaryot Cell. 2006;5:293–300. doi: 10.1128/EC.5.2.293-300.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan JP, Arhin GK, Ullu E, Tschudi C. Functional characterization of a Trypanosoma brucei TATA-binding protein-related factor points to a universal regulator of transcription in trypanosomes. Mol Cell Biol. 2004;24:9610–9618. doi: 10.1128/MCB.24.21.9610-9618.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Nguyen TN, Günzl A. Characterization of a multisubunit transcription factor complex essential for spliced-leader RNA gene transcription in Trypanosoma brucei. Mol Cell Biol. 2005;25:7303–7313. doi: 10.1128/MCB.25.16.7303-7313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimanski B, Brandenburg J, Nguyen TN, Caimano MJ, Günzl A. A TFIIB-like protein is indispensable for spliced leader RNA gene transcription in Trypanosoma brucei. Nucleic Acids Res. 2006;34:1676–1684. doi: 10.1093/nar/gkl090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Djikeng A, Tschudi C, Ullu E. Argonaute protein in the early divergent eukaryote Trypanosoma brucei: control of small interfering RNA accumulation and retroposon transcript abundance. Mol Cell Biol. 2004;24:420–427. doi: 10.1128/MCB.24.1.420-427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm NR, Yu MC, Campbell DA. Transcription termination and 3′-end processing of the spliced leader RNA in kinetoplastids. Mol Cell Biol. 1999;19:1595–1604. doi: 10.1128/mcb.19.2.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, et al. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–132. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- Ullu E, Tschudi C. Permeable trypanosome cells as a model system for transcription and trans-splicing. Nucleic Acids Res. 1990;18:3319–3326. doi: 10.1093/nar/18.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Plasterk RH. RNAi protects the Caenorhabditis elegans germline against transposition. Trends Genet. 2004;20:314–319. doi: 10.1016/j.tig.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Vastenhouw NL, Fischer SE, Robert VJ, Thijssen KL, Fraser AG, Kamath RS, et al. A genome-wide screen identifies 27 genes involved in transposon silencing in C. elegans. Curr Biol. 2003;13:1311–1316. doi: 10.1016/s0960-9822(03)00539-6. [DOI] [PubMed] [Google Scholar]
- Villanueva MS, Williams SP, Beard CB, Richards FF, Aksoy S. A new member of a family of site-specific retrotransposons is present in the spliced leader RNA genes of Trypanosoma cruzi. Mol Cell Biol. 1991;11:6139–6148. doi: 10.1128/mcb.11.12.6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volff JN. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28:913–922. doi: 10.1002/bies.20452. [DOI] [PubMed] [Google Scholar]
- West N, Roy-Engel AM, Imataka H, Sonenberg N, Deininger P. Shared protein components of SINE RNPs. J Mol Biol. 2002;321:423–432. doi: 10.1016/s0022-2836(02)00542-9. [DOI] [PubMed] [Google Scholar]
- Yang N, Kazazian HH., Jr L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol. 2006;13:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- Ye J, Eickbush TH. Chromatin structure and transcription of the R1- and R2-inserted rRNA genes of Drosophila melanogaster. Mol Cell Biol. 2006;26:8781–8790. doi: 10.1128/MCB.01409-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.