

Abstract
A diastereoselective [4 + 2]-annulation of vinyl carbodiimides with chiral N-alkyl imines has been developed to access the stereochemically rich polycyclic guanidine cores of the batzelladine alkaloids. Application of this strategy, together with additional key steps such as long-range directed hydrogenation and diastereoselective intramolecular iodo-amination, led to highly convergent total syntheses of (−)-batzelladine D and (+)-batzelladine A with excellent stereocontrol.
Introduction
Polycyclic guanidine natural products comprise an intriguing class of structurally diverse marine alkaloids possessing varied and potent biological activities. In the mid to late 1990s, nine novel tricyclic guanidine marine alkaloids were isolated as metabolites of the Crambe genus.1 This family of batzelladine alkaloids (1–9, Chart 1) is characterized by at least one stereochemically rich fused tricyclic guanidine core to which are appended additional guanidine fragments (R1–R6) of varying complexity. Members of the batzelladine alkaloids have exhibited biological activities that include potential antiviral activity in the inhibition of the binding of HIV gp120 to human CD4 (batzelladines A and B), as well as potential immunosuppressive activity in the induction of dissociation of protein tyrosine kinase p56lck from CD4 (batzelladines F–I).
Chart 1.
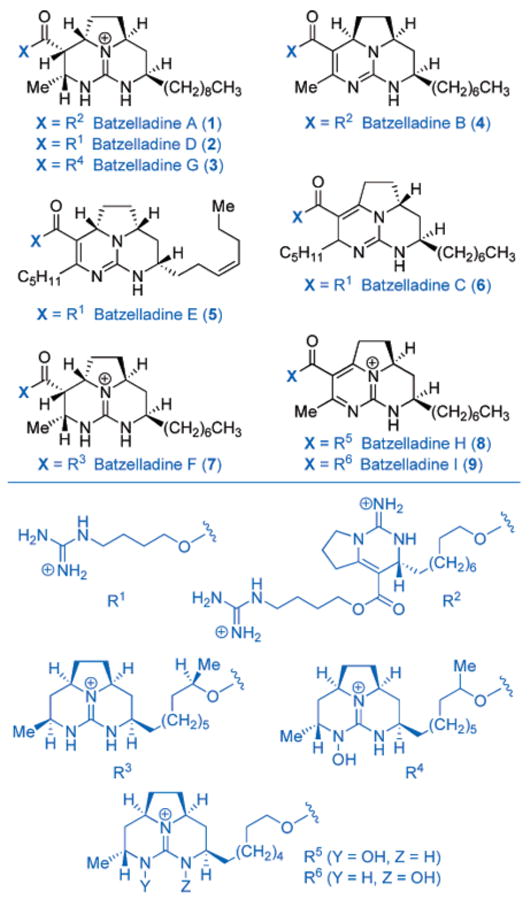
Over the past decade, considerable effort has been devoted to the synthesis of this class of alkaloids. The first total synthesis of any member of the batzelladine alkaloids was reported by Snider and Chen,2 involving a biomimetic strategy consisting of the condensation of isourea derivatives with multifunctional bis(enones) in the preparation of (±)-batzelladine E (5). A nonracemic synthesis of (−)-batzelladine D (2) by Overman and co-workers soon followed employing a versatile tethered Biginelli condensation approach that later served in their enantioselective synthesis and structure revision of (+)-batzelladine F (7).3 A completely distinct strategy involving a nitrone 1,3-dipolar cycloaddition was showcased by Nagasawa and co-workers first in their synthesis of (±)-batzelladine D (2) and later in the only report to date of the synthesis of (+)-batzelladine A (1) employing the same strategy.4 Other creative approaches to complex fragments of the batzelladines have included N-acylation,5 N-acyliminium cyclizations,6 biomimetic guanidine-bis(enone) condensations,7 formal aza-Diels–Alder reactions,8 and stereoselective radical cyclizations.9
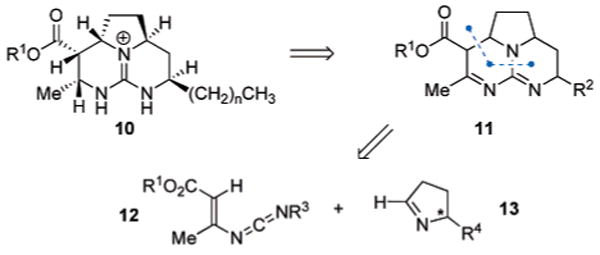
Within a considerable number of the batzelladine alkaloids, namely batzelladines A, D, F, and G, there exists a common tricyclic tetrahydropyrimidine core (10, Scheme 1) incorporating multiple stereogenic centers along its periphery. Consideration of this tetrahydropyrimidine core suggests a potentially expedient route toward its construction, in which simple retrosynthetic oxidation state adjustment of 10 would provide the tricyclic dihydropyrimidine intermediate 11. A direct [4 + 2]-disconnection along the highlighted C–C and C–N bonds in 11 would yield simple precursors in the form of a vinyl carbodiimide 12 and a chiral imine 13. Ideally, the lone chiral center in 13 would dictate the entire stereochemical course of the synthesis. Reports on annulations of vinyl carbodiimides with imines in N-heterocycle syntheses are rare,10 in which only achiral PhCH=NPh was used as a coupling partner with generally modest yields. We report herein our establishment of a diastereoselective [4 + 2]-annulation of vinyl carbodiimides with chiral N-alkylimines and its application to the nonracemic total syntheses of batzelladines D11 and A.
Scheme 1.
Results and Discussion
To assess the feasibility and stereochemical outcome of the diastereoselective [4 + 2]-annulation of vinyl carbodiimides and imines, model investigations commenced with the preparation of electron-deficient vinyl carbodiimides 16 and 17 (Scheme 2). 2-Butynoic acid methyl ester (14) was subjected to 1,4-conjugate addition with tetramethylguanidinium azide (TMGA) to afford the β-azido acrylate 15 as a mixture of geometrical isomers. The E-isomer 15 could be isolated in high purity (44%) and was thus employed in the initial model experiments for carbodiimide formation. Treatment of E-1512 with PPh3 led to conversion to its iminophosphorane derivative (81%), which allowed for its condensation by aza-Wittig reaction with either p-methoxyphenyl isocyanate or p-methoxybenzyl isocyanate to form the carbodiimides 16 (72%) and 17 (74%), respectively. Notably, both of these vinyl carbodiimides could be purified by silica gel chromatography. The model chiral imine coupling partner was derived from (S)-5-(hydroxymethyl)-2-pyrrolidinone (18), which, after TBDPS protection of the hydroxyl group (83%), could be directly reduced to the imine 19 with Schwartz's reagent (68%).13,14
Scheme 2 a.
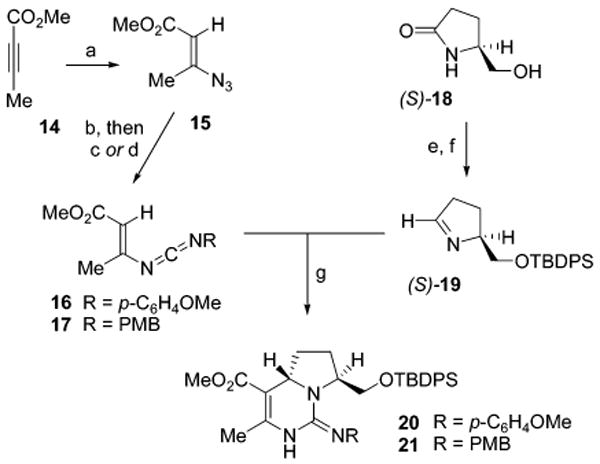
a Reagents and conditions: (a) (MeHN)2CNMe2N3, CHCl3, 44% (E); (b) PPh3, CH2Cl2, 81%; (c) p-MeOC6H4NCO, PhMe, 72%; (d) p-MeOC6H4-CH2NCO, PhMe, 85 °C, 74%; (e) TBDPSCl, imidazole, DMF, 83%; (f) Cp2ZrHCl, THF, −20 °C, 68%; (g) (ClCH2)2, 62% (R = PMP), 98% (R = PMB).
Exposure of the vinyl carbodiimide 16 or 17 to the imine 19 resulted in successful [4 + 2]-annulation in both cases to provide exclusively the (S,S)-diastereomer of the bicyclic dihydropyrimidines 20 and 21 (62% and 98%, respectively). Detailed NMR analysis of the dihydropyrimidine products confirmed the anti-stereochemical configuration of the pyrrolidine substructure within both 20 and 21. The high efficiency and degree of diastereoselectivity in this annulation process highlight the potential for its application to the synthesis of batzelladines D and A, both of which share a common anti-fused tricyclic guanidine core.
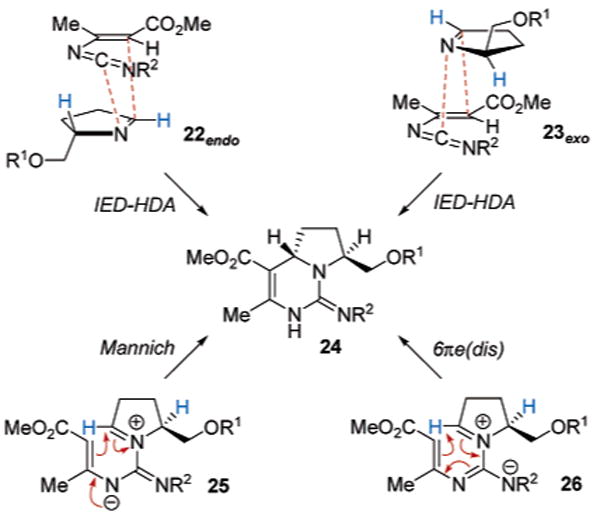
The mechanism by which the formation of the bicyclic dihydropyrimidines 20 and 21 occurs is unclear. Be it a concerted or stepwise process, the anti-stereochemical outcome could be rationalized primarily on the basis of steric factors. For example, in the case of an asynchronous concerted [4 + 2]-cycloaddition (Scheme 3), the inverse electron-demand hetero-Diels–Alder (IED-HDA) may proceed through an endo (22) or exo (23) transition state. In either case, approach of the diene should preferentially occur on the imine dienophile face opposite to that of the alkyl ether side chain, thereby committing to the anti-cycloadduct 24 following tautomerization. Alternatively, a stepwise process could entail initial nucleophilic addition of the imine to form the guanidine zwitterion 25, followed by an intramolecular Mannich closure in which the π-nucleophile approaches from the less sterically encumbered face of the iminium group. An additional stepwise process invokes the zwitterionic resonance structure of the initial adduct 25 in the form of 26, which incorporates a conjugated 1,3,5-triene substructure, thereby setting the stage for a 6π-electrocyclic rearrangement. If such a process were thermally induced, disrotatory closure would likely be torquo-selective wherein the sterically demanding alkyl ether substituent adopts an “out-orientation” to again secure formation of the anti-cycloadduct 24 after tautomerization.
Scheme 3.
Despite the different mechanistic pathways by which the annulation process can proceed, the outcome nonetheless appeared ideally suited for a stereoselective synthesis of the batzelladine core 10, especially with the presence of the β-oriented alkyl ether side chain within 21 (Scheme 4). Previous approaches at employing hydrogenation protocols to establish the C7 and C15 stereochemistry15 within the tetrahydropyrimidine core of the batzelladine alkaloids were met with significant difficulties.3 However, the presence of the Lewis basic oxygen functionality in the C11 side chain of 21 offered unique opportunities to explore a long-range directed hydrogenation process to secure these two C-stereocenters. Thus, the TBDPS ether within 21 was removed (TBAF, 97%), and the vinylogous carbamate 27 was subjected to hydrogenation with Crabtree's catalyst16 under 400 psi H2. The β-guanidino dihydropyrimidine product 28 was formed as a single stereoisomer (81%), whose structure was verified through X-ray analysis. The free hydroxyl functionality in 27 was found to be critical in this stereoselective reduction, as attempts at Crabtree hydrogenation of the silyl ether 21 under otherwise identical conditions led to no reaction, and Rh/Al2O3-catalyzed hydrogenation of 27 led to 2:1 dr, favoring the undesired C7/C15 configurations in addition to reduction of the PMB group.
Scheme 4 a.
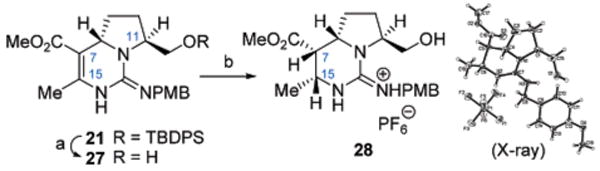
a Reagents and conditions: (a) TBAF, THF, 97%; (b) [Ir(cod)pyr(PCy3)]PF6, H2 (400 psi), CH2Cl2, 81%.
Synthesis of (−)-Batzelladine D
The promising results of these initial annulation investigations prompted efforts to apply this approach to the total synthesis of the simplest of the batzelladine alkaloids, namely batzelladine D (2, Chart 2). Although the stereoselective production of tetrahydropyrimidine 28 (Scheme 4) suggested the direct use of this intermediate in the total synthesis efforts, advancement of 28 to the tricyclic core of batzelladine D was not possible owing to functional group incompatibilities during its late-stage elaboration to close the third ring. As a result, a new imine coupling partner was prepared to overcome these difficulties (Scheme 5). In this sequence, the chiral imine was again derived from (S)-5-(hydroxymethyl)-2-pyrrolidinone (18), in which the primary hydroxyl was activated (TsCl, Et3N, 85%) and displaced with cyanide to form nitrile 29 (91%). Ethanolysis of the nitrile (89%) and reduction to the primary alcohol (LiBH4, 91%) allowed for its subsequent conversion to the TBDPS ether 30 (88%), which then underwent amide reduction with Cp2ZrHCl13 to afford the chiral imine 31 (66%), an intermediate corresponding to the single carbon homologation derivative of the original imine 19.
Chart 2.
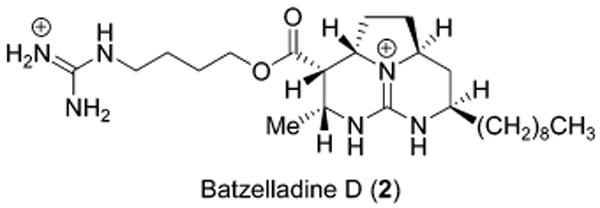
Scheme 5 a.
a Reagents and conditions: (a) TsCl, TEA, DMAP, CH2Cl2, 85%; (b) KCN, CH3CN, 85 °C, 91%; (c) HCl(g), EtOH, 65 °C, 89%; (d) LiBH4, MeOH, THF, 0 °C, 91%; (e) TBDPSCl, imidazole, DMF, 88%; (f) Cp2ZrHCl, THF, −20 °C, 66%.
Preparation of an appropriately protected vinyl carbodiimide employed 1,4-but-2-ynoic acid benzyl ester (32, Scheme 6), which was subjected to 1,4-conjugate addition with TMGA to afford the β-azido acrylate 33 as a mixture of geometrical isomers (84%; 2:1 E:Z). In the earlier model investigations into the key [4 + 2]-annulation reaction (Scheme 2), only the E-isomers of the vinyl carbodiimides 16/17 were employed in the successful study. Since the formation of the alkenyl azide 33 was nearly nonstereoselective, it was prudent to determine the suitability of both isomers in the key annulation for the efficient synthesis of batzelladine D (2). Thus, both the E- and Z-isomers of 33 (Scheme 6) were separated by silica gel chromatography and subjected to Staudinger-aza-Wittig condensation with p-methoxybenzyl isocyanate to form the vinyl carbodiimides E-34 and Z-34 with comparable efficiency (73% and 67%, respectively). Treatment of either E-34 or Z-34 with 2-(2-O-TBDPS-ethyl)-3,4-dihydro-2H-pyrrole (31) led to the formation of a single diastereomer of the dihydropyrimidine 35 (86% from E-34 and 88% from Z-34), incorporating the expected anti-stereochemistry. It is worth noting that the formation of 35 may arise from either (1) both E- and Z-isomers of 34 undergoing the annulation or (2) in situ interconversion of the E/Z isomers of 34 wherein one preferentially undergoes dihydropyrimidine formation. Nevertheless, this sequence does illustrate that the low stereoselectivity in the conversion of 32 → 33 is not a liability given that both isomers comparably converge to the bicylic guanidine 35.
Scheme 6 a.
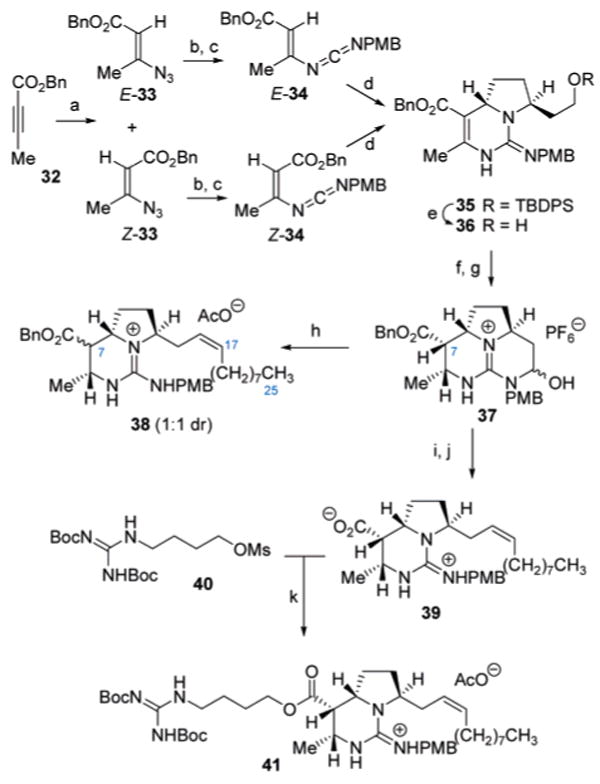
a Reagents and conditions: (a) (MeHN)2CNMe2N3, CHCl3, 84% (2:1 E:Z); (b) PPh3, CH2Cl2, 75% (E), 58% (Z); (c) p-MeOC6H4CH2NCO, PhMe, 85 °C, 73% (E), 67% (Z); (d) 31, (ClCH2)2, 23 °C, 86% from E-34, 88% from Z-34; (e) TBAF, THF, 99%; (f) [Ir(cod)pyr(PCy3)]PF6, H2 (400 psi), CH2Cl2, 80%; (g) IBX, DMSO, CH3CN, 98%; (h) Me(CH2)7CH=PPh3, THF; (i) 10% Pd(OH)2/C, AcOH, H2 (1 atm), MeOH, 99%; (j) Me(CH2)7-CH=PPh3, THF, 50 °C, 72%; (k) (BocHN)2C=N(CH2)4OMs, Cs2CO3, DMF, 40 °C, 93%.
The synthesis of (−)-batzelladine D (2) continued with removal of the TBDPS ether in 35 to expose the primary hydroxyl group (TBAF, 99%) in 36 for directed hydrogenation of the vinylogous carbamate functionality. Even with the further extended hydroxyl group in 36, the Ir+-catalyzed reduction16 proceeded with complete stereocontrol to provide the corresponding tetrahydropyrimidine (80%), which efficiently underwent IBX oxidation to provide the tricyclic guanidine hemiaminal 37 (98%). At this juncture, advancement of 37 through the direct attachment of the C7–C2515 fragment via selective Wittig fragment coupling proved troublesome. For example, treatment of hemiaminal 37 with the ylide derived from nonyltriphenylphosphonium bromide led to stereoselective formation of the cis-alkene 38, although with unavoidable C7 epimerization of the pseudo-axial ester substituent. To remedy this situation, the benzyl ester in 37 was first subjected to hydrogenolysis (99%), allowing for cis-selective Wittig olefination17 with excess ylide (thereby generating the carboxylate anion in situ) to afford the alkene acid 39 after acidification (72%). Attachment of the guanidine-containing side chain of batzelladine D (2) was then accomplished by carboxylate O-alkylation with (BocHN)2C=N(CH2)4OSO2Me18 to form 41 in 93% yield.
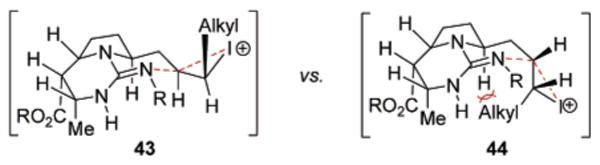
Closure of the remaining ring in (−)-batzelladine D was effected by intramolecular iodo-amination of the alkene in 41 (Scheme 7), employing I2 in the presence of K2CO3. Preferential 6-exo-cyclization (70%) proceeded with complete regio- and stereocontrol to establish the desired C13-(R)-configuration15 in 42 after reductive deiodination19 (89%). The high stereoselectivity of this final ring closure can be rationalized by the preferential formation and reaction of the iodonium diastereomer 43 (Chart 3), directly derived from oxidation of the cis-alkene. Cyclization in this manifold would place the alkyl group in a pseudo-equatorial orientation. Conversely, cyclization to form the undesired C13-(S)-diastereomer would arise from formation and reaction of the iodonium diastereomer 44, which places the alkyl group in a pseudo-axial orientation to experience pronounced A1,3-like strain. It is worth noting that the current data does not discount the possibility of iodine-induced alkene E/Z isomerization prior to cyclization. Unfortunately, further elucidation of the stereocontrol elements in the cyclization was hampered by difficulties both in the determination of the configuration of the newly formed iodine-bearing stereocenter in the immediate iodo-amination product and in the independent synthesis of the trans-alkene isomer of 41 to probe its oxidative cyclization. Nevertheless, the favorable outcome of this final cyclization event permitted the global deprotection of 42 (Scheme 7) with aqueous TFA to conclude the synthesis of (−)-batzelladine D (2, 82%).
Scheme 7 a.
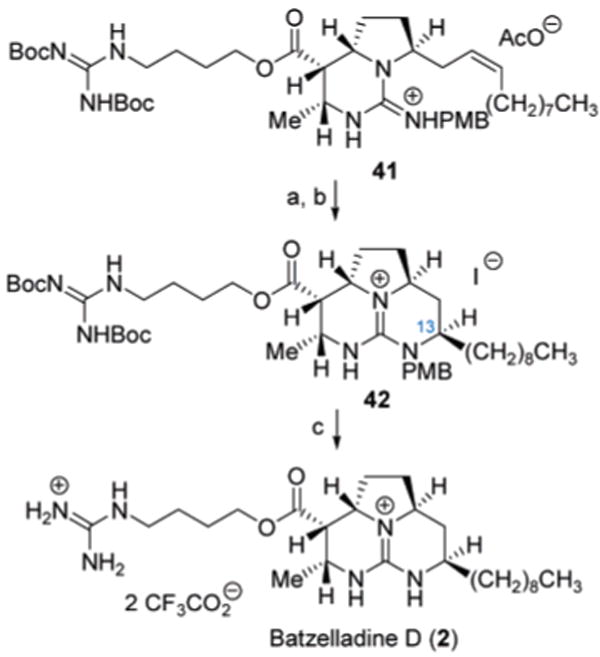
a Reagents and conditions: (a) I2, K2CO3, DME, 70%; (b) 10% Pd/C, Et3N, H2 (1 atm), EtOAc, 89%; (c) TFA, 82%.
Chart 3.
Synthesis of (+)-Batzelladine A
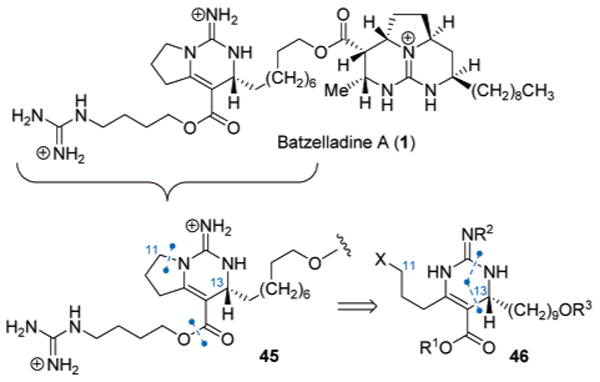
The successful application of the carbodiimide annulation strategy in the synthesis of the simplest member of batzelladine alkaloids (2) prompted investigation of this approach to more complex members of the family, namely the small-molecule binder to CD4,20 batzelladine A (1, Scheme 8). Unfortunately, the highlighted [4 + 2]-annulation approach does not immediately map onto the bicylic guanidine substructure 45 within 1. However, retrosynthetic rupture of the pyrrolidine at C11–N21 in 45 would generate a dihydropyrimidine intermediate 46, which should be amenable to the key [4 + 2]-disconnection wherein the absolute configuration of the lone C13-chiral center21 would be established.22
Scheme 8.
The preparation of a suitable vinyl carbodiimide substrate for the synthesis of the bicyclic guanidine substructure 45 within (+)-1 commenced (Scheme 9) with sequential O-benzylation (NaH, BnBr, 80%) and C-acylation (MeOC(O)Cl, 85%) of pent-4-yn-1-ol to afford the propargyllic ester 48. Treatment of alkyne 48 with TMGA led to conjugate 1,4-addition of azide to afford a 2:1 (E:Z) mixture of vinyl azides 49, which was then converted to vinyl carbodiimide 50 via Staudinger-aza-Wittig condensation with p-methoxybenzyl isocyanate (63%). The mixture of both geometrical isomers of 50 was exposed to chiral imine (R)-19, prepared similarly to that of its S-enantiomer (see Scheme 2). The annulation process proceeded at 23 °C to provide the dihydropyrimidine 51 (89%) as a single diastereomer. While it remains unclear whether the cyclization proceeded via a concerted or stepwise mechanism, the anti-stereochemistry of the product emerged as expected based on our previous experience with this reaction. Thus, annulation of chiral imine R-19 with vinyl carbodiimide 50 secured the required C13-R-configuration in 51 via relay of stereochemical information from the sacrificial stereogenic center originally obtained from chiral pyrrolidinone (R)-18.
Scheme 9 a.
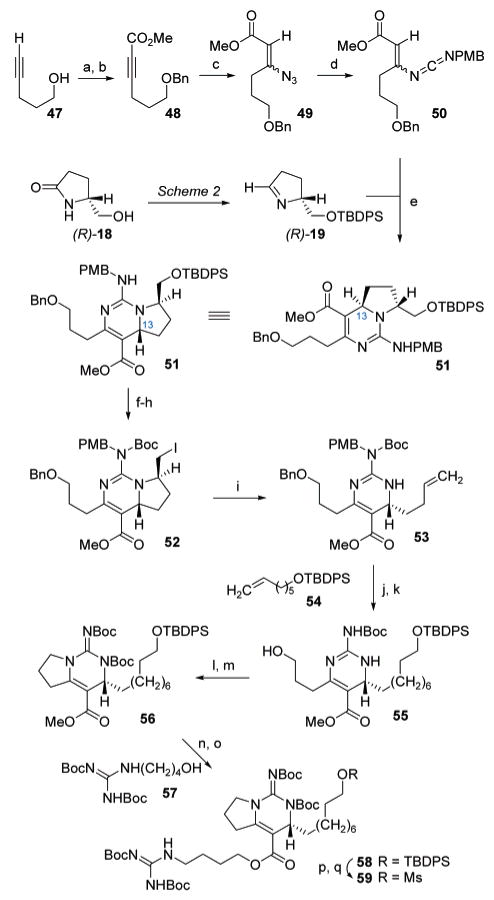
a Reagents and conditions: (a) BnCl, NaH, TBAI, THF, 70 °C, 80%; (b) nBuLi, methyl chloroformate, THF, −78 °C, 85%; (c) TMGA, CHCl3, 23 °C, 78% (2:1 E/Z); (d) PPh3, CH2Cl2, 23 °C; PMBNCO, PhMe, 80 °C, 63%; (e) (ClCH2)2, 23 °C, 89%; (f) NaH, Boc2O, DMAP, THF, 23 °C, 86%; (g) TBAF, AcOH, THF, 23 °C, 91%; (h) I2, PPh3, imidazole, PhMe, 23 °C, 97%; (i) t-BuLi, THF, −78 °C, 93%; (j) 54, Grubbs-G2, CH2Cl2, 45 °C, 73%; (k) 10% Pd(OH)2, AcOH, H2 (1 atm), MeOH, CH2Cl2, 23 °C, 90%; (l) DIAD, PPh3, PhMe, 23 °C, 93%; (m) NaH, Boc2O, DMAP, THF, 23 °C, 85%; (n) EtSLi, HMPA, 23 °C; (o) 57, BOPCl, TEA, CH2Cl2, 23 °C, 55% (two steps); (p) TBAF, THF, 23 °C, 92%; (q) MsCl, TEA CH2Cl2, 0 °C, 91%.
Construction of the bicyclic guanidine skeleton in batzelladine A continued as the guanidine group in 51 was fully protected by acylation with Boc2O (86%), allowing for conversion of the primary alkyl silyl ether to its iodo-counterpart 52 via sequential treatment with TBAF and I2/PPh3 (88%, two steps). Low-temperature lithium–halogen exchange of 52 occurred rapidly with concomitant β-elimination of the acylated guanidine functionality to cleave the pyrrolidine ring (93%). The resultant α-olefin in 53 served as a convenient handle with which to attach the C17–C2221 oligomethylene linker present in batzelladine A; thus, alkene 53 was exposed to the TBDPS ether of hept-6-en-1-ol (54) in the presence of Grubbs-G2 ruthenium catalyst23 to effect olefin cross-metathesis to provide a mixture of E- and Z-disubstituted alkene isomers (73%). Subsequent treatment with H2 and Pd(OH)2 led to benzyl ether hydrogenolysis, p-methoxybenzyl removal from the guanidine, and alkene hydrogenation in a single pot procedure to yield the primary alcohol 55 (90%). Closure of the pyrrolidine ring was accomplished by intramolecular aza-Mitsunobu (DIAD, PPh3, 93%),24 followed by acylation of the guanidine core with Boc2O to provide the bicyclic vinylogous carbamate 56 (85%). The remaining steps for elaboration of the bicyclic guanidine fragment of batzelladine A involved thiolate-induced removal of the vinylogous methyl carbamate followed by BOPCl-mediated esterification25 of (BocHN)2C=N(CH2)4OH (57) to provide the vinylogous carbamate 58 (55%, two steps). Subsequent conversion of the silyl ether in 58 to its methanesulfonate ester derivative (TBAF; MsCl, 84%, two steps) provided 59, an appropriately derivatized bicyclic guanidine core of batzelladine A.
The convergent final sequence in the assembly of both polycyclic guanidine fragments employed the advanced bicyclic guanidine construct 39, originally prepared (Scheme 6) for the synthesis of batzelladine D. The cesium carboxylate of 39 (Scheme 10) was generated to undergo O-alkylation with the alkyl methanesulfonate 59 to provide the C23 ester 60 (68%). The third ring within the tricyclic core of batzelladine A was then closed by way of a stereoselective intramolecular iodoamination (I2, Cs2CO3, 72%) to establish the desired C30-R-stereocenter, followed by reductive de-iodination (Et3N, H2, Pd/C, 71%) to afford 61, a fully protected form of batzelladine A. Global deprotection of 61 (TFA) completed the total synthesis of batzelladine A (75%), whose analytical data were found to be identical in all respects to that reported for the natural product.
Scheme 10 a.
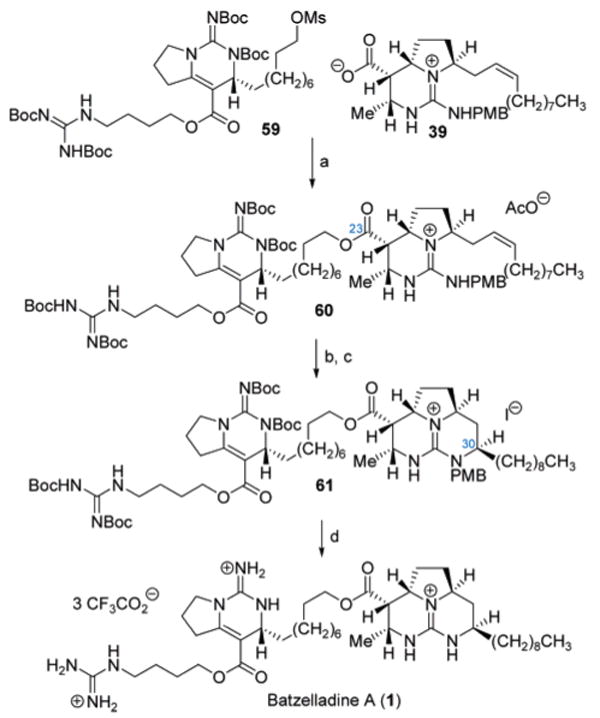
a Reagents and conditions: (a) Cs2CO3, DMF, 50 °C, 68%; (b) I2, Cs2CO3, DME, 23 °C, 72%; (c) 10% Pd/C, TEA, H2 (1 atm), EtOAc, 71%; (d) TFA, 0 °C, 75%.
Conclusions
Through the use of a unified strategy involving the diastereoselective [4 + 2]-annulation of chiral imines with vinyl carbodiimides, the total syntheses of (+)-batzelladine A (1) and (−)-batzelladine D (2) were accomplished in highly convergent and stereoselective sequences. The successful application of this approach to dihydropyrimidine construction in the synthesis of 1 and 2 illustrates its potential for the synthesis of other related alkaloids such as the crambescidin family of natural products, as well as for the construction of complex non-natural N-heterocycles of potential biological utility.
Supplementary Material
Complete ref 1a; experimental details for the preparation and analytical characterization of synthetic intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by the NIH (GM67659), Abbott, Eli Lilly, Johnson & Johnson, Merck, and Pfizer. A Bristol-Myers Squibb predoctoral fellowship to M.A.A. is acknowledged.
References
- 1.(a) 1188 Patil AD, et al. J Org Chem. 1995;60:1182. [Google Scholar]; (b) Patil AD, Freyer AJ, Taylor PB, Carte B, Zuber G, Johnson RK, Faulkner DJ. J Org Chem. 1997;62:1814–1819. [Google Scholar]; (c) Snider BB, Busuyek MV. J Nat Prod. 1999;62:1707–1711. doi: 10.1021/np990312j. [DOI] [PubMed] [Google Scholar]; (d) Braekman JC, Daloze D, Tavares R, Hajdu E, Van Soest RWM. J Nat Prod. 2000;63:193–196. doi: 10.1021/np990403g. [DOI] [PubMed] [Google Scholar]; (e) Structure verification of batzelladines G-I via total synthesis has yet to be accomplished
- 2.(a) Snider BB, Chen J, Patil AD, Freyer AJ. Tetrahedron Lett. 1996;37:6977–6980. [Google Scholar]; (b) Snider BB, Chen J. Tetrahedron Lett. 1998;39:5697–5700. [Google Scholar]
- 3.(a) Franklin AS, Ly SK, Mackin GH, Overman LE, Shaka AJ. J Org Chem. 1999;64:1512–1519. doi: 10.1021/jo981971o. [DOI] [PubMed] [Google Scholar]; (b) Cohen F, Overman LE, Sakata SKL. Org Lett. 1999;1:2169–2172. doi: 10.1021/ol991269u. [DOI] [PubMed] [Google Scholar]; (c) Cohen F, Overman LE. J Am Chem Soc. 2001;123:10782–10783. doi: 10.1021/ja017067m. [DOI] [PubMed] [Google Scholar]; (d) Cohen F, Overman LE. J Am Chem Soc. 2006;128:2594–2603. doi: 10.1021/ja0574320. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cohen F, Overman LE. J Am Chem Soc. 2006;128:2604–2608. doi: 10.1021/ja057433s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Ishiwata T, Hino T, Koshino H, Hashimoto Y, Nakata T, Nagasawa K. Org Lett. 2002;4:2921–2924. doi: 10.1021/ol026303a. [DOI] [PubMed] [Google Scholar]; (b) Shimokawa J, Shirai K, Tanatani A, Hashimoto Y, Nagasawa K. Angew Chem, Int Ed. 2004;43:1559–1562. doi: 10.1002/anie.200353200. [DOI] [PubMed] [Google Scholar]; (c) Shimokawa J, Ishiwata T, Shirai K, Koshino H, Tanatani A, Nakata T, Hashimoto Y, Nagasawa K. Chem–Eur J. 2005;11:6878–6888. doi: 10.1002/chem.200500852. [DOI] [PubMed] [Google Scholar]
- 5.Rao AVR, Gurjar MK, Vasudevan J. J Chem Soc, Chem Commun. 1995:1369–1370. [Google Scholar]
- 6.Louwrier S, Ostendorf M, Tuynman A, Hiemstra H. Tetrahedron Lett. 1996;37:905–908. [Google Scholar]
- 7.(a) Black GP, Murphy PJ, Walshe NDA, Hibbs DE, Hursthouse MB, Malik KMA. Tetrahedron Lett. 1996;37:6943–6946. [Google Scholar]; (b) Black GP, Murphy PJ, Walshe NDA. Tetrahedron. 1998;54:9481–9488. [Google Scholar]; (c) Black GP, Murphy PJ, Thornhill AJ, Walshe NDA, Zanetti C. Tetrahedron. 1999;55:6547–6554. [Google Scholar]
- 8.(a) Elliott MC, Long MS. Tetrahedron Lett. 2002;43:9191–9194. [Google Scholar]; (b) Elliott MC, Long MS. Org Biomol Chem. 2004;2:2003–2011. doi: 10.1039/b404679j. [DOI] [PubMed] [Google Scholar]
- 9.(a) Evans PA, Manangan T. Tetrahedron Lett. 2001;42:6637–6640. [Google Scholar]; (b) Evans PA, Manangan T. Tetrahedron Lett. 2005;46:8811. doi: 10.1016/j.tetlet.2005.06.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Wamhoff H, Richardt G, Stölben S. Adv Heterocycl Chem. 1995;64:159–249. [Google Scholar]; (b) Wamhoff H, Schmidt A. Heterocycles. 1993;35:1055–1066. [Google Scholar]; (c) Saito T, Ohkubo T, Kuboki H, Maeda M, Tsuda K, Karakasa T, Satsumabayashi S. J Chem Soc, Perkin Trans 1. 1998:3065–3080. [Google Scholar]
- 11.For a preliminary report on the use of this strategy for the synthesis of batzelladine D, see Arnold MA, Durón SG, Gin DY. J Am Chem Soc. 2005;127:6924–6925. doi: 10.1021/ja0519029.
- 12.Palacios F, Perez de Heredia I, Rubiales G. J Org Chem. 1995;60:2384–2390. [Google Scholar]
- 13.Schedler DJA, Li J, Ganem B. J Org Chem. 1996;61:4115–4119. doi: 10.1021/jo960286j. [DOI] [PubMed] [Google Scholar]
- 14.Woo K, Jones K. Tetrahedron Lett. 1991;32:6949–6952. [Google Scholar]
- 15.Carbon numbering assignments conform to that of batzelladine D.
- 16.(a) Crabtree RH, Davis MW. Organometallics. 1983;2:681–682. [Google Scholar]; (b) Stork G, Kahne DE. J Am Chem Soc. 1983;105:1072–1073. [Google Scholar]; (c) Crabtree RH, Davis MW. J Org Chem. 1986;51:2655–2661. [Google Scholar]
- 17.Wang Q, Wei H, Schlosser M. Eur J Org Chem. 1999:3263–3268. [Google Scholar]
- 18.Neubert BJ, Snider BB. Org Lett. 2003;5:765–768. doi: 10.1021/ol034042e. [DOI] [PubMed] [Google Scholar]
- 19.Li NS, Piccirilli JA. J Org Chem. 2004;69:4751–4759. doi: 10.1021/jo0495337. [DOI] [PubMed] [Google Scholar]
- 20.Shimokawa J, Ishiwata T, Shirai K, Koshino H, Tanatani A, Nakata T, Hashimoto Y, Nagasawa K. Chem–Eur J. 2005;11:6878–6888. doi: 10.1002/chem.200500852. [DOI] [PubMed] [Google Scholar]
- 21.Carbon numbering assignments conform to that of batzelladine A.
- 22.For the determination of the absolute configuration of C13 in 1, see: Durón SG, Gin DY. Org Lett. 2001;3:1551–1554. doi: 10.1021/ol015848m.
- 23.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 24.Hughes DL. Org Prep Proced Int. 1996;28:127–164. [Google Scholar]
- 25.Diago-Meseguer J, Palomo-Coll AL, Fernandez-Lizarbe JR, Zugaza-Bilbao A. Synthesis. 1980:547–551. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete ref 1a; experimental details for the preparation and analytical characterization of synthetic intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.