

Abstract
Although mast cells (MCs) often are abundant in the synovial tissues of patients with rheumatoid arthritis (RA), MC’s contribution to joint inflammation and cartilage loss remains poorly understood. MC-restricted tryptase•heparin complexes have pro-inflammatory activity, and significant amounts of hTryptase-β are present in RA synovial fluid. Mouse MC protease-6 (mMCP-6) is the ortholog of hTryptase-β, and this serine protease is abundant in the synovium of arthritic mice. We now report that C57BL/6 (B6) mice lacking their tryptase•heparin complexes have attenuated arthritic responses, with mMCP-6 as the dominant tryptase responsible for augmenting neutrophil infiltration in the K/B×N mouse serum-transfer arthritis model. While inflammation in this experimental arthritis model was not dependent on protease activated receptor-2, it was dependent on the chemokine receptor CXCR2. In support of the latter data, exposure of synovial fibroblasts to hTryptase-β•heparin or mMCP-6•heparin complexes resulted in expression of the neutrophil chemotactic factors CXCL1/KC, CXCL5/LIX, and CXCL8/IL-8. Our proteomics, histochemistry, and immunohistochemistry data also revealed substantial loss of cartilage-derived aggrecan proteoglycans in the arthritic joints of wild-type B6 mice but not mMCP-6-null B6 mice. These observations demonstrate the functional contribution of MC-restricted tryptase•heparin complexes in the K/B×N mouse arthritis model and connect our mouse findings with RA pathophysiology.
Keywords: mast cell, rheumatoid arthritis, inflammation, chemokines, transgenic/knockout mice
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease whose pathophysiology involves multiple cellular lineages and inflammatory processes that conspire to produce synovial inflammation, joint pain, joint swelling, and ultimately destruction of cartilage and bone. Because the synovium in RA patients is often infiltrated with lymphocytes and macrophages, many groups have focused their attention on the roles of these immune cells in the pathology of the autoimmune disease (1). Less widely appreciated is the marked increase in mast cell (MC) numbers in the synovial sublining of RA patients (2-10). Indeed, numerous groups reported that joint tissue isolated from RA patients contains 6- to 25-fold more hTryptase-β-expressing MCs than normal synovial tissue. In some instances, MCs comprise up to 5% of the nucleated cells in the arthritic synovium. This cellular increase in the chronically inflamed joint raises the possibility that MCs have switched from a beneficial physiologic role (e.g., as sentinel cells that protect the vulnerable synovial cavity from infectious organisms) to active participants in the immunopathology of joint inflammation and/or destruction (11). Indeed, MCs tend to localize at the junction of the pannus and cartilage, as well as in areas where the pannus is invading cortical bone (2, 12, 13). Moreover, rapid degradation of cartilage-derived aggrecan proteoglycans occurs when tryptase-expressing MCs are induced to release their granule proteases in MC/chondrocyte and MC/cartilage co-cultures (14). Nevertheless, the definitive contribution of MCs and their varied mediators in human RA remains enigmatic due to the fact that no MC-specific therapeutic intervention has been developed.
Although WBB6F1-KitW/KitW-v (W/Wv) and WCB6F1-KitlSl/KitlSl-d mice possess many non-MC-dependent disorders which greatly complicate data interpretation, numerous studies carried out on these respective Kit- and Kit ligand-defective mice have implicated prominent contributions of MCs to the pathophysiology of autoimmune inflammatory arthritis. For example, we and others noted that MC-deficient W/Wv and WCB6F1-KitlSl/KitlSl-d mice are resistant to arthritis induced by autoantibodies against collagen or glucose-6-phosphate isomerase (15-17). In support of these data, van den Broek and coworkers (18) noted that cartilage erosion in the knee was significantly reduced in W/Wv mice 14 to 35 days after these animals were sensitized with intra-dermal methylated-BSA in the presence of adjuvant followed by intra-articular methylated-BSA challenge. Although inflammation is a prominent feature of the methylated-BSA arthritis model, swelling of knee joints is minimal in contrast to other experimental arthritis models involving distal peripheral joints. In the K/B×N mouse serum-transfer model in which mice are given pathogenic autoantibodies to glucose-6-phosphate isomerase, adoptive transfer of in vitro-differentiated Kit/tryptase-expressing wild-type (WT) MCs into W/Wv mice restored arthritic susceptibility (15). Further studies in this animal model revealed that MCs are activated by IgG Fc-receptors and can contribute to joint inflammation by elaborating IL-1 (19). Although the latter study demonstrated that MC-derived IL-1 was important in the initiation phase of the disease, a mediator(s) that is more restricted to MCs must contribute significantly to the pathogenesis of autoimmune arthritis in order to explain the perceived MC-dependence that occurs in W/Wv mice due to the fact that macrophages and other cell types in the joint also express IL-1.
Tetramer-forming tryptases are selectively expressed in the MCs of every examined mammal, including mice (20, 21) and humans (22-24). The two tetramer-forming tryptases in mice are mouse MC protease (mMCP) 6 and mMCP-7. Their genes reside adjacent to one another on chromosome 17A3.3 (25), and mMCP-6 and mMCP-7 form heterotypic and homotypic tetramers (26). The human ortholog of mMCP-6 is hTryptase-β, and both serine proteases are sequestered in the MC’s granules ionically bound to heparin-containing serglycin proteoglycans. Indeed, the packaging of MC neutral proteases in the cytoplasmic granules of safranin+ MCs is highly dependent on serglycin proteoglycans that contain heparin glycosaminoglycans (27, 28), and the biosynthesis of heparin in these cells is controlled by N-deacetylase/N-sulfotransferase-2 (NDST-2). Heparin stabilizes the tryptase tetramer unit (29, 30) and restricts the enzyme’s substrate specificity (31). These tryptase•heparin complexes are exocytosed into the surrounding microenvironment in a regulated fashion upon MC activation [reviewed in (32, 33)]. MC subpopulations in mice and humans differ in their expression of tryptases and other granule proteases (34). Relevant to our study, human and mouse synovial MCs store abundant amounts of hTryptase-β and mMCP-6, respectively in their secretory granules (7, 8, 13, 35, 36). The observation that IL-33 induces MC-committed progenitors to increase their expression of hTryptase-β (37) is significant considering the recent finding that IL-33 exacerbates collagen-induced arthritis in a MC-dependent manner (38). The accumulated data raise the possibility that MC-restricted tryptases have prominent roles in RA and in experimental arthritis.
Recent studies demonstrated functional roles for MC-restricted tryptases in innate immunity. Of particular relevance to K/B×N mouse experimental arthritis which is dependent on neutrophils (39-41), mMCP-6-/-/mMCP-7-/- (6-/7-) C57BL/6 (B6) mice demonstrated decreased neutrophil recruitment and reduced survival after infection of the peritoneal cavity with Klebsiella pneumoniae (42). In support of these data, administration of recombinant mMCP-6 or hTryptase-β into the peritoneal cavity or lungs of normal mice resulted in a marked influx of neutrophils (30, 31, 43, 44). The contribution of MC-restricted tryptases to the accumulation of peripheral blood neutrophils into the arthritic joint could be due to their ability to stimulate the expression and release of neutrophil-specific chemotactic factors from nearby bystander cells. In this regard, we and others showed that tryptases can induce cultured human epithelial and endothelial cells to increase their expression of CXCL8/IL-8 (31, 45, 46). Nakano and coworkers also reported that cultured human fibroblast-like synoviocytes (FLS) increased their expression of CXCL8 when exposed to a partially purified preparation of synovium-derived hTryptase-β (47). Other possible mechanisms by which MC-restricted tryptases could contribute to inflammatory pathogenesis include activation of matrix metalloproteinases (48-50), cleavage of extracellular matrix components (51), stimulation of fibroblast proliferation and collagen production (52), and proteolysis of fibrinogen (53). Indeed, MC proteases have been demonstrated to contribute to cartilage degradation in vitro in the absence of the inflammatory cells present in the arthritic joint (14).
Having previously demonstrated the presence of mMCP-6 protein in the synovial MCs of healthy and diseased mice (35), we hypothesized that the elaboration of this serine protease and/or its closely related family member mMCP-7 could represent a MC-specific contribution to the pro-inflammatory milieu of the joint in the K/B×N mouse serum-transfer arthritis model. The creation of B6 mice that differ in their expression of mMCP-6, mMCP-7, and NDST-2 allowed us the opportunity to evaluate the potential roles of MC-restricted tryptase•heparin proteoglycan complexes in experimental arthritis. Utilizing the autoimmune K/B×N mouse serum-transfer model of experimental arthritis and our different transgenic mouse strains, we now report that MC-restricted mMCP-6•heparin proteoglycan complexes serve as important intensifiers of inflammation within the joint via enhancing the recruitment of neutrophils in a protease receptor-2 (PAR-2)-independent manner. Further, we show that enzymatically active recombinant mMCP-6 induces cultured mouse FLS to increase their expression of CXCL1/KC/GRO-α and CXCL5/LIX/ENA-78, and we demonstrate that mice lacking CXCR2 develop less joint inflammation. Finally, we show that mMCP-6•heparin proteoglycan complexes contribute to joint destruction by promoting the degradation of the aggrecan proteoglycans in the diseased cartilage.
Materials and Methods
Mice
WT 6+/7- (54) B6 mice and CXCR2-/- BALB/c mice (55) were obtained from the Jackson Labs. PAR-2-/- B6 mice (56) were a kind gift from Dr. Shaun Coughlin (UCSF, San Francisco, CA). These mice and NDST-2-/- (10 generation backcrosses with B6 mice) (27), 6-/7- (generated in B6 ES cells followed by 8 generation backcrosses with B6 mice) (42), and 6-/7+ (10 generation backcrosses with B6 mice) (57) mice were maintained under specific pathogen-free conditions. Although most examined WT mice express mMCP-6 and mMC-7, the MCs in WT B6 mice constitutively lack mMCP-7 (58) due to a splice-site point mutation in this gene that causes rapid catabolism of the defective transcript by the “nonsense” post-transcriptional pathway (54). K/B×N mice were maintained as described (35). All mice were age and gender matched, and experiments were conducted using animal protocols approved by the Dana-Farber Cancer Institute/Brigham and Women’s Hospital Animal Care and Use Committee.
K/B×N mouse serum-transfer arthritis
Arthritogenic K/B×N mouse serum containing autoantibodies to glucose-6-phosphate isomerase was administered to recipient mice using a modification of a previously described experimental approach (59). To induce arthritis, mice received two 150-μl intraperitoneal injections of diluted (50 μl serum + 100 μl sterile PBS) serum on experimental days 0 and 2. Clinical indices were recorded at 24 to 48 h intervals, and indices were graded on a scale of 0 to 12 as described (15). Ankle thickness was measured using a spring loaded dial thickness gauge (Long Island Indicator Service Inc.). Measurements were made axially across the ankle joint, with the ankle in full dorsiflexion.
Histochemistry, enzyme cytochemistry, and immunohistochemistry of the arthritic joint
Ankles were harvested as previously described (35). Briefly, the skin around each ankle was gently removed, and tissue from the distal one-third of the tibia to the midpaw was collected. After fixation with 4% paraformaldehyde, ankles were decalcified in Kristensen’s solution (60), dehydrated, and embedded in paraffin. Tissue was cut into 4-μm sections, and midsagittal ankle sections were stained with hematoxylin and eosin. Histological grading of synovial inflammation and cartilage and bone erosion in the ankle sections were performed as previously described (40, 61). Cell lineages were identified using standard histochemical and enzyme cytochemical approaches (57, 62). For example, toluidine blue stain, Congo red stain, and chloroacetate esterase substrate were used to identify MCs, eosinophils, and neutrophils, respectively. Histomorphometric enumeration of leukocyte lineages were performed by counting stained cells in synovial tissue surrounding ankle and tarsal joints in midsagittal hindpaw sections. An eyepiece reticule (Leica Microsystems) was used to define a unit of 0.04 mm2 restricted to within 200 μm of the synovial lining layer. Observers were blinded to the experimental condition and genotype of the animals in these analyses.
Previously described (35, 57) immunohistochemical approaches were performed on fixed, de-paraffinized sections with a Vectastain® ABC-alkaline phosphatase kit and the relevant rabbit anti-peptide antibodies to identify the presence of mMCP-4, mMCP-5, mMCP-6, and mMCP-7 protein in the synovium samples. Rabbit anti-mouse aggrecan antibody (Millipore) was utilized to analyze aggrecan depletion in cartilage of arthritic ankles. For substrate, FAST™ Fast Red (1 mg/ml) (Sigma) was applied for 20 min. To confirm the presence of neutrophils, after antigen retrieval per manufacturer protocol, sections were stained with the NIMP R14 anti-neutrophil antibody (Abcam) or with control rat IgG, followed by detection with a Vectastain® ABC-HRP kit and DAB substrate. This neutrophil-specific antibody recognizes the surface protein Ly6g/Gr1. Tissue was subsequently counterstained with Gill’s II hematoxylin and mounted with Crystal/Mount (Biomeda) or Cytoseal (Biomeda).
Mass spectrometry analysis of abundant proteins in arthritic joints
Detergent lysates of joint tissues from hindpaws of experimental mice were prepared by mechanical disruption, as previously described (40). Twenty-five μg of protein extract obtained from each mouse ankle joint was fractionated on a 4%-10% Tris-HCl polyacrylamide gradient gel (Bio-Rad) run for 90 min at 130 V with Tris-glycine-SDS buffer (Bio-Rad). All gels were washed and stained with Coomassie Blue G250 (Bio-Rad) for 20 min and then destained in deionized water. A total of 12 gel slices (5 mm2) were excised from each lane in the gel. Individual gel slices were placed in 0.6 ml microfuge tubes (Axygen, Union City, CA) and incubated at room temperature for 10 min in 25 mM NH4HCO3, and then in 50% acetonitrile/25 mM NH4HCO3. This cycle was repeated twice. After the acetonitrile solution was removed, the dried gel slices were covered with 10 mM dithiothreitol and incubated at 56°C for 45 min and then at room temperature for an additional 20 min. Dithiothreitol was discarded and alkylation was performed by adding 55 mM iodoacetamide (MP Biomedicals, Inc., Solon, Ohio) to cover the gel slice. All slices were incubated with iodoacetamide for 45 min at room temperature in the dark. Iodoacetamide was discarded and the gel slices were washed twice with 25 mM NH4HCO3 followed by 50% acetonitrile/25 mM NH4HCO3 as described above. Trypsin (0.150 μg; Promega) was added, along with sufficient 50 mM NH4HCO3 to cover the slice, and the sample incubated overnight at 37°C to generate the tryptic fragments of the unknown protein present in the arthritic joints. Formic acid was added to a final concentration of 1% to stop the reaction. The aqueous solution (peptide fraction) was transferred to a clean microfuge tube. To the gel pieces, 50% acetonitrile/5% formic acid was added; the gel slices were vortexed 30 min and then sonicated for 5 min. The aqueous solution was transferred to the original peptide solution and the above step repeated. The pooled peptide sample was speed vacuum dried to reduce the volume to 10 μl.
The tandem mass spectroscopy (MS/MS) data for the unknown protein of interest in the individual gel slices were obtained using an LTQ mass spectrometer (Thermo Fischer Scientific, Bremen, Germany). The tryptic digests were first run on a pre-column (C18, PepMap100) and separated with a reverse-phase C18 column using mobile phase A (0.1% formic acid in water) and B (84% acetonitrile, 0.1% formic acid in water) with a linear gradient of 2% per min, starting with 100% of A. The peptides were then injected at a flow rate of 300 nl/min. MS/MS spectra was then acquired by the Finnigan™ LTQ. The resulting raw data were analyzed using Mascot (V 2.1.03, Matrix Science) and searched against the NCBI Mouse Protein database. Searches were performed with fixed carbamidomethylation of cysteines and variable oxidation of methionine residues. A fragment tolerance of 0.4 Da was used in searches.
CXCL1, CXCL5, and CXCL8 expression in tryptase-treated FLS
Human and mouse FLS were generated as described (63, 64). In brief, human synovial tissue from RA patients were obtained as discarded specimens from joint arthroplasty surgery with approval of the Partners Institutional Review Board. Synoviocyte cell suspensions were prepared from synovial tissues by mincing, followed by gently rocking for 1 to 2 h at 37°C in DMEM medium (Gibco BRL) containing 1 mg/ml type-IV collagenase (Worthington Biochemicals) and 0.1 mg/ml DNase (Sigma). Mouse FLS isolation was performed similarly under procedures approved by the Dana-Farber Cancer Institute/Brigham and Women’s Hospital Animal Care and Use Committee. After euthanasia, mouse ankle joints were isolated by disarticulation distal to the tibia and at the tarsal/metatarsal junction, taking care not to violate the marrow space. The tissue was infiltrated with collagenase in DMEM and incubated with gentle rocking for 2 h, as described above. The resultant cell suspension was passed through a 100-μm cell strainer (BD Biosciences) and placed in a tissue culture flask (BD Labware) containing DMEM (Gibco BRL) supplemented with 10% heat-inactivated FBS (Hyclone), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol, and 0.1 mM nonessential amino acids (Gibco BRL). Since no bone marrow-derived lineage cells were detected after passage 3 (63, 64), cultured FLS were used in our experiments between passages 4 and 8. For the in vitro stimulation studies, FLS were cultured to 80-90% confluence in 12-well plates (Corning) for 48 to 72 h. The resulting cells were exposed to recombinant hTryptase-β•heparin complexes (Promega) or recombinant pro- and mature mMCP-6•heparin complexes (31) in culture medium containing 1% FCS for 16 h, whereupon supernatants were collected for analysis. As a positive control for chemokine induction, replicate FLS were exposed to TNF-α. Commercial ELISA kits (R&D Systems) were used to quantitate the levels of human CXCL8, mouse CXCL1, and mouse CXCL5 in the supernatants. As assessed by the Limulus Amebocyte lysate kit (Associates of Cape Cod Inc.), the tryptase•heparin preparations used in our study contained less than 0.25 EU LPS/ml.
Statistical analysis
Data were analyzed using Prism software, version 4.00 (GraphPad Software). Cell numbers of respective lineages were described as the mean ± SE. Student’s t-test was performed to compare changes in cell numbers, histological analysis, and ELISA values. Curves of both indices plotted from arthritis experiments were compared using the 2 way-ANOVA test. P values less than 0.05 were considered statistically significant.
Results
Impaired autoimmune inflammatory arthritis in heparin-deficient B6 mice
Since our previous studies demonstrated that the synovium of the arthritic mouse is populated by MCs that presumably contain heparin proteoglycans due to their histochemical properties, we began our assessment of the contribution of MC-restricted tryptase•heparin proteoglycan complexes to joint inflammation in the K/B×N mouse serum-transfer model by examining arthritic responses in NDST-2-/- B6 mice. As expected, we discovered that the amount of total neutral protease stored in the secretory granules of the synovial MCs of NDST-2-/- B6 mice was markedly attenuated relative to that in heparin-sufficient B6 mice (Fig. 1A). Based on these data, we proceeded to quantify joint inflammation induced by K/B×N mouse serum in NDST-2-/- B6 mice. As noted in Fig. 1B, we found an ~50% decrease in clinical measurements of arthritis in these transgenic mice. The day-10 Δ ankle thickness was 0.67 ± 0.05 mm and 0.38 ± 0.06 mm in diseased WT and NDST-2-/- B6 mice, respectively (P = 0.0008; Fig. 1B). Histomorphometric quantification of the arthritic changes in the joint tissues confirmed the clinical assessment, with significant decreases in inflammation and bone erosion scores in the NDST-2-/- B6 mice (Figs. 1, C and D).
FIGURE 1.
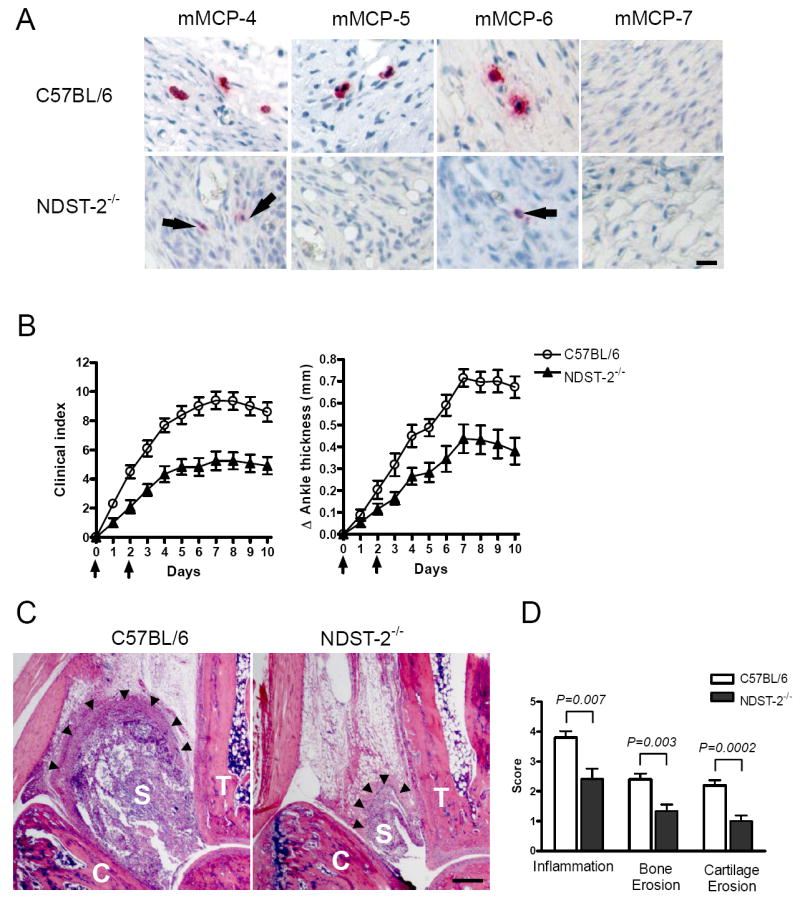
Heparin-deficient NDST-2-/- B6 mice, which fail to store substantial amounts of mMCP-6 and other proteases in their synovial MCs, demonstrate attenuated K/B×N mouse serum-dependent arthritis relative to WT mice. A, Immunohistochemical staining for mMCP-4, mMCP-5, mMCP-6, and mMCP-7 protein in synovial tissue MCs from WT and NDST-2-/- B6 mice. Arrows highlight the marked reduced expression of mMCP-4, mMCP-5, and mMCP-6 in the synovial MCs of NDST-2-/- MCs due to the requirement for heparin-containing serglycin proteoglycans in their granule storage. As expected, the synovial MCs in NDST-2+/+ and NDST-2-/- B6 mice lack mMCP-7 protein due to the exon-2/intron-2 splice-site mutation in the tryptase gene in both mouse strains. Bar, 10 μm. B, Arthritis severity in WT and NDST-2-/- B6 mice. Data were pooled from 20 mice/group in 3 independent experiments (P <0.001). Arrows indicate timing of administration of arthritogenic K/B×N serum. C, Representative mid-sagittal ankle sections demonstrating decreased leukocyte infiltration and synovial hyperplasia in treated NDST-2-/- B6 mice (T, tibia; S, synovial space; C, calcaneus; arrowheads, demarcation of synovial lining). Bar, 200 μm. D, Histomorphometric quantification of arthritic tissue. The data are the mean values ± SEM (N = 20/group).
Impaired arthritis in tryptase-deficient B6 mice
Considering the profound defect in protease expression in the synovial MCs of NDST-2-/- B6 mice (Fig. 1A), we proceeded to assess the contributions of mMCP-6 and mMCP-7 in the arthritis model using WT mMCP-6+/+/mMCP-7-/- (6+/7-) B6 mice and newly created transgenic mMCP-6-/-/mMCP-7+/+ (6-/7+) and 6-/7- mice backcrossed onto the B6 strain (Fig. 2A). Thus, we have mice congenic for mutations in both of the mMCP-6 and mMCP-7 tryptases. In these studies, we observed a significant decrease in clinical joint inflammation in diseased 6-/7- B6 mice (Fig. 2B) comparable to that of diseased NDST-2-/- B6 mice (Fig. 1, B-D). The day-10 Δ ankle thickness was 0.64 ± 0.06 mm and 0.39 ± 0.05 mm for WT 6+/7- B6 mice and transgenic 6-/7- B6 mice, respectively (P = 0.0028; Fig. 2B). Histomorphometric quantification of the tissue arthritic response again confirmed our clinical measurements.
FIGURE 2.
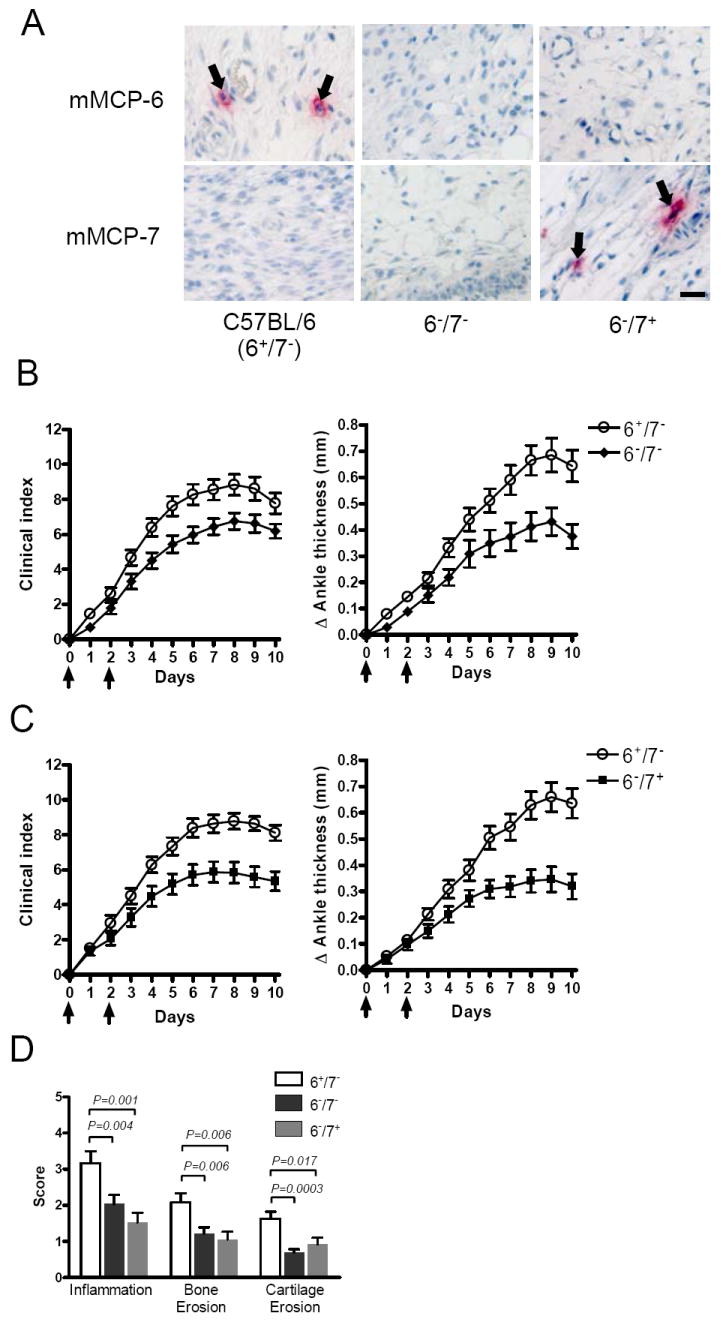
Decreased arthritis in treated mMCP-6-deficient B6 mice. A, Immunohistochemical staining for mMCP-6 and mMCP-7 expression in the synovial tissue MCs of WT 6+/7- and transgenic 6-/7- and 6-/7+ B6 mice. Bar, 10 μm. B-C, Arthritis severity in WT 6+/7- and transgenic 6-/7- and 6-/7+ B6 mice. Arrows indicate when the mice received arthritogenic K/B×N mouse serum. Data were pooled from 3 independent experiments, N = 15 mice/group (P <0.01). D, Histo-morphometric quantification of arthritic tissue. The data are the mean values ± SEM (N = 15/group).
Because the neutrophil accumulation that occurs in the peritoneal cavity of K. pneumoniae-infected mice depends on mMCP-6, we next attempted to define the potential contribution of the tryptase to joint inflammation in the K/B×N mouse serum-transfer model. To address this issue, we compared 6-/7- and 6-/7+ B6 mice. Interestingly, we found that the arthritis severity in 6-/7+ B6 mice was indistinguishable from that in 6-/7- B6 mice (Fig. 2, B and C). The synovium of 6+/7-, 6-/7+, and 6-/7- B6 mice contained 47.2 ± 3.1, 51.6 ± 4.7, and 48.9 ± 3.6 MCs per mm2, respectively. Because no significant difference in synovial MC density was observed among the three populations of B6 mice, the reduced arthritis seen in both mMCP-6-deficient mouse strains was not a consequence of fewer MCs. The accumulated data suggest that mMCP-6 is the dominant pro-inflammatory tryptase in the K/B×N mouse serum-transfer arthritis model.
Characteristics of the cellular infiltrate in the synovium induced by tryptase•heparin complexes
Because large numbers of neutrophils are recruited into the joints of WT mice given proarthritic serum, we examined neutrophil recruitment as a potential mechanism by which MC-restricted mMCP-6•heparin complexes promote synovial inflammation. We quantitated synovial leukocyte populations in the arthritic joints of mMCP-6-deficient and mMCP-6-sufficient B6 mice at day 10 (Fig. 3). These histomorphometric studies revealed that the density of neutrophils in the diseased synovium of NDST-2-null or the tryptase-deficient 6-/7+ and 6-/7- mice was substantially reduced relative to WT B6 mice (Fig. 3, A and C). Since mMCP-6 also can regulate eosinophil infiltration in the skeletal muscle of T. spiralis-infected mice, we next quantified the density of eosinophils in the diseased synovium. We found that the arthritic joints of WT, heparin-deficient, and tryptase-deficient mice contained similar numbers of eosinophils (Fig. 3B). In all arthritic mice, the number of neutrophils greatly exceeded the number of eosinophils. Thus, in the arthritic joint, mMCP-6•heparin complexes preferentially induce the recruitment of neutrophils.
FIGURE 3.
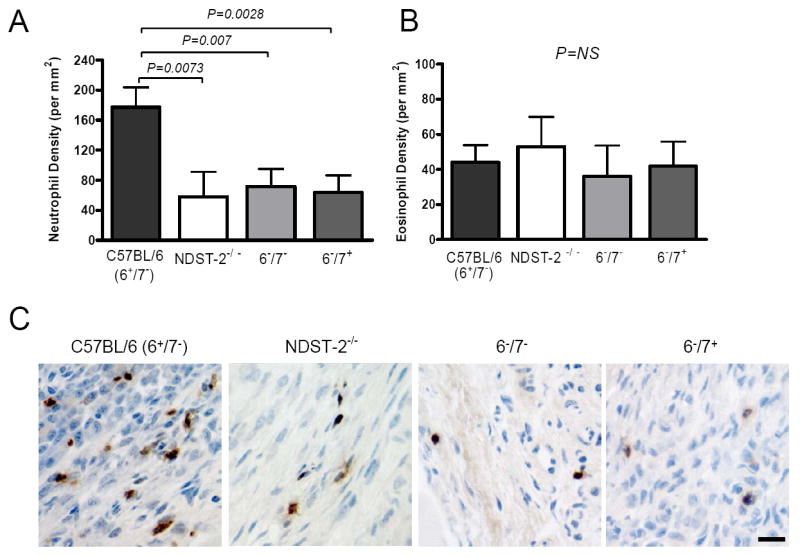
Histomorphometric quantitation of neutrophils and eosinophils in synovial tissue. A-B, Neutrophils (A) and eosinophils (B) were enumerated in arthritic synovial tissue from WT, NDST-2-/-, and tryptase-deficient B6 mice. The data are the mean values ± SEM (N = 15/group). C, Shown is a representative immunohistochemical stain for neutrophils using anti-NIMP-R14 antibody in WT, NDST-2-/-, and tryptase-deficient strains (as labeled). Brown indicates positive staining. Bar, 10 μm.
Development of arthritis is dependent on the chemokine receptor CXCR2 but not on PAR-2/F2RL1
Based on earlier in vitro studies, numerous tryptase substrates of potential relevance to synovial inflammation have been identified. Among these potential substrates, PAR-2 (65) has been most extensively investigated. Although conflicting data have been reported pertaining to the role of this protease-activated receptor in experimental arthritis (66-68), a recent study concluded that PAR-2-null mice developed arthritis when given a high-dose of K/B×N mouse serum (68). Consistent with the findings of Busso and co-workers, we found no difference in arthritic responses when PAR-2-deficient and PAR-2-sufficient B6 mice were given lesser amounts of the pro-arthritic serum (Fig. 4, B and C). These data imply that mMCP-6•heparin complexes regulate neutrophil accumulation in the arthritic joints of our experimental model in a PAR-2-independent manner.
FIGURE 4.
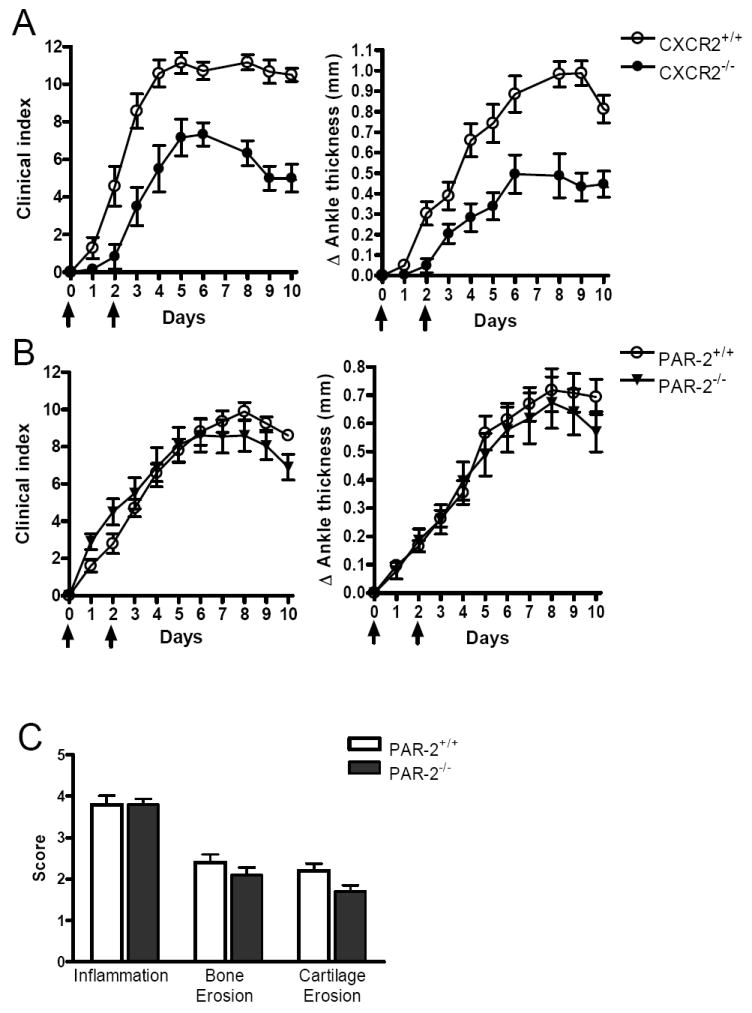
mMCP-6-heparin complexes induce neutrophil accumulation in the arthritic joint in a CXCR2-dependent/PAR-2-independent manner. A, Reduced arthritis severity in CXCR2-/- mice given K/B×N mouse serum relative to CXCR2+/+ mice. The data are the mean values ± SEM (N = 7 mice/group, P <0.05). B-C, PAR-2-/- mice display full intact inflammatory response to K/B×N mouse serum. The data are the mean values ± SEM and data was pooled from 3 independent experiments (N = 10 mice/group, P = NS). Arrows indicate when the mice received arthritogenic K/B×N mouse serum. C, Histomorphometric quantification of arthritic tissue in WT and PAR-2-/- mice. The data are the mean values ± SEM (N = 10 /group, P = NS)
A number of related Glu-Lys-Arg CXCL chemokines (e.g., CXCL1, CXCL2/GRO-β, CXCL3/GRO-γ, CXCL5, CXCL7/NAP-2, and CXCL8) regulate neutrophil accumulation in tissues via the chemokine receptor CXCR2. Because mMCP-6•heparin proteoglycan complexes contribute to the accumulation of neutrophils in the arthritic joint (Fig. 3), we next evaluated arthritis in CXCR2-/- mice. As noted in Fig. 4A, inflammation was markedly reduced in these mice, suggesting that Glu-Lys-Arg CXCL chemokines and their receptors contribute to neutrophil extravasation in this experimental arthritis model.
Tryptase-induced chemokine production in FLS
Tryptase is not directly chemotactic for neutrophils (31) and exocytosed mMCP-6•heparin proteoglycan complexes are retained in connective tissues for hours due to their large size (69). mMCP-6 exocytosed from synovial MCs therefore cannot come in direct contact with peripheral blood neutrophils to induce their extravasation into the diseased joint. Because our experimental arthritis model is partly dependent on CXCR2 (Fig. 4A), we postulated that mMCP-6 and its human ortholog induce granulocyte accumulation in an indirect manner probably by inducing the expression of chemokines that bind to CXCR2. FLS are the primary cells of mesenchymal lineage in synovial tissue, and these cells predominate in rheumatoid pannus tissue. Indeed, stimulation of mouse FLS in vitro with recombinant mMCP-6•heparin complexes resulted in increased expression of the CXCR2 ligands CXCL1 (Fig. 5A) and CXCL5 (Fig. 5B) to levels comparable to that obtained when these mesenchymal cells encountered TNF-α. Interestingly, mMCP-6•heparin complexes elicited CXCL1 production in PAR-2-/- mouse FLS as well (data not shown). To extend our animal work into a human system, we next assessed the ability of recombinant hTryptase-β•heparin complexes to regulate the expression of CXCL8 in cultured human FLS. As noted in Fig. 5C, we found that this neutral protease induced FLS to express significant amounts of CXCL8. The inability of recombinant pro-mMCP-6 (Fig. 5, A and B) or boiled hTryptase-β (Fig. 5C) to induce the expression of significant amounts of CXCL1, CXCL5, or CXCL8 suggests that these serine proteases must be enzymatically active to optimally affect the joint. Given the fact that LPS is heat stable, the chemokine-inducing effects of the hTryptase-β•heparin preparations used in our study were not the result of LPS contamination, thereby supporting our data obtained with the inactive zymogens.
FIGURE 5.
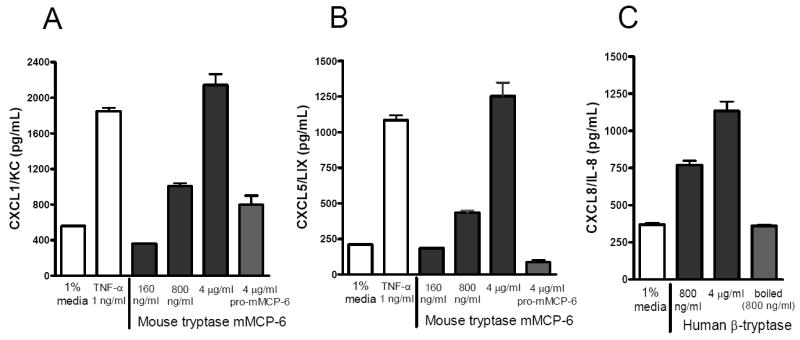
Tryptase•heparin complexes induce human and mouse FLS to express the chemokines CXCL1 (mouse), CXCL5 (mouse), and CXCL8 (human). Primary mouse (A, B) or human (C) FLS (passage 4-8) were exposed to enzymatically active recombinant hTryptase-β•heparin or mMCP-6•heparin complexes. The resulting conditioned media were assayed for their levels of the chemokines via ELISA. The data are the mean values ± SEM. Results shown are representative of 3 independent experiments. As negative controls, replicate human FLS were exposed to boiled hTryptase-β and mouse FLS to pro-mMCP-6. As positive controls, both populations of FLS were exposed to TNF-α.
Tryptase-dependent proteolysis and loss of aggrecan proteoglycans in experimental arthritis
A proteomics approach was next used to identify longitudinal changes in abundant protein constituents between healthy joints and the arthritic joints in the K/B×N mouse experimental model. In these assays, the proteins were extracted from the joints of replicate WT B6 mice at baseline and on days 8 and 15 after arthritis induction. The resulting extracts were subjected to SDS-PAGE and gel slices containing differentially expressed proteins/peptides were subjected to MS/MS.
In this proteomics assay, a large sized protein species was identified in the day-8 and day-15 arthritic samples of WT B6 mice that was not present in the control day-0 sample (data not shown). MS/MS analysis of three trypsin-generated peptides from the unknown protein revealed that it was aggrecan proteoglycan that had lost its protease-susceptible N-terminal hyaluronan (HA)-binding domain. The amino acid sequence of mouse aggrecan and the location of the three MS/MS identified peptides in the isolated aggrecan fragment are noted in Fig. 6. Based on our proteomics data, the proteolytic liberation of soluble aggrecan and concomitant loss of aggrecan proteoglycans from joint cartilage is a major feature of the K/B×N mouse model of arthritis, as occurs in RA patients. We therefore used histochemical and immunohistochemical approaches to compare amounts of aggrecan proteoglycans from the cartilage of WT B6 mice and our transgenic NDST-2-/-, 6-/7+, and 6-/7- B6 mice. As noted in Fig. 7, loss of aggrecan proteoglycans were markedly attenuated in the joints of our tryptase•heparin deficient B6 mouse strains in established arthritis.
FIGURE 6.

Identification of a soluble fragment of aggrecan proteoglycan in the arthritic joints of WT B6 mice. Mouse aggrecan (GenBank accession number AAC37670) is initially translated as a 2132-mer protein that contains a 19-mer signal peptide (77). The proteoglycan’s N-terminal globular domains bind to HA, resulting in macromolecular complexes in normal cartilage that can exceed 1 billon Da. Aggrecan’s HA-binding region is susceptible to numerous proteases. For example, the Glu392→Ala393 peptide bond that is cleaved by aggrecanse-1/ADAMTS4 and aggrecanase-2/ADAMTS5 is highlighted in italic bold. Highlighted in bold are the locations of the three MS/MS identified peptides at residues 595-604, 2003-2026, and 2066-2075 in the isolated aggrecan fragment from the arthritic joints of WT B6 mice at days 8 and 15 after transfer of K/B×N mouse serum.
FIGURE 7.

Loss of aggrecan proteoglycans from arthritic cartilage. A, Proteoglycan depletion of cartilage assessed by toluidine blue staining and immunohistochemistry with anti-aggrecan antibodies 10 day after arthritis induction. Note the significant loss of cartilage proteoglycans in arthritic WT mice relative to arthritic tryptase•heparin deficient mice. Arrowheads delineate depth of proteoglycan depletion. Shown are representative stains from WT and transgenic tryptase-deficient B6 mice at day 10 (N = 12-20 mice/group pooled from 3 experiments). Bar, 10 μm. B, Evaluation of aggrecan loss. The data are the mean values ± SEM (N = 12-20 mice/group).
Discussion
MCs have long been recognized as potential participants in human and mouse inflammatory arthritis. We recently identified an important contribution for MC-derived IL-1 in arthritis initiation in the K/B×N mouse serum transfer model (19). However, because many cell types produce this pro-inflammatory cytokine, it is likely that MCs contribute to synovial inflammation via multiple pathways. We therefore sought to identify a contribution of MCs to arthritis that is more restricted to this cell lineage. Of the diverse array of effector molecules exocytosed from activated MCs, their protease•serglycin proteoglycan complexes are unique to MCs. Our demonstration of a functional role for mMCP-6•heparin complexes in the K/B×N mouse model (Figs. 1-3, 6, and 7) now provides further insight into the roles of MCs and their protease mediators in experimental arthritis.
The identification of tryptase•heparin complexes as contributors to arthritis is consistent with previous functional studies of these macromolecular complexes in mice and humans. Because synovial MCs are positioned near vessels and within the synovial mesenchyme (11, 70), they are ideally situated to amplify arthritogenic stimuli via bulk release of their preformed mediators. Tryptase is present as an active enzyme pre-packaged tetramer in the cytoplasmic granules of human and mouse synovial MCs ionically bound to heparin-containing serglycin proteoglycans (32, 33). Although the safranin+ population of MCs found in the mouse synovial lining preferentially express mMCP-6, all human MCs express its ortholog hTryptase-β, including the MCs in the human synovium. Previous in vivo studies employing either recombinant protein or bacterial infection of tryptase-deficient mice demonstrated that mMCP-6 and hTryptase-β elicit neutrophil accumulation in tissues (30, 31, 35, 42-44). Neutrophils are abundant in the joints of mice given arthritogenic K/B×N serum (Fig. 3) and in the synovial fluid of humans with RA, and these granulocytes contribute to the pathogenesis of arthritis (39-41). We observed a significant decrease in the number of neutrophils in the arthritic synovium of three transgenic mouse strains that lack mMCP-6 (Fig. 3). These findings are consistent with a functional contribution to synovitis via the promotion of neutrophil recruitment into the arthritic joint. Our additional finding that hTryptase-β•heparin and mMCP-6•heparin complexes induce cultured FLS to increase their expression of the neutrophil chemoattractants CXCL1, CXCL5, and CXCL8 (Fig. 5) is mechanistically consistent with an ability of MC-restricted tryptase tetramers to indirectly promote neutrophil accumulation in inflammatory arthritis, in part, by inducing the expression of varied chemokines that bind to CXCR2 on the surface of the granulocytes.
These insights notwithstanding, the mechanism by which mMCP-6•heparin complexes regulate granulocyte accumulation is undoubtedly more complex than just inducing bystander cells in the rheumatoid joint to increase their expression of CXCL chemokines. In this regard, it is now appreciated that mMCP-6 is important in the accumulation of eosinophils in the skeletal muscle of T. spiralis-infected mice (57). It also has been reported that recombinant hTryptase-β can cleave CCL11/eotaxin and CCL5/RANTES, and therefore abrogate their eosinophil chemotactic activities (71). We therefore considered the possibility that large numbers of eosinophils do not accumulate in the rheumatoid joint due to an additional tryptase-mediated inactivation of an induced CCL chemokine that preferentially attracts eosinophils. However, this does not appear to be the case in our experimental arthritis model due to the fact that we did not see increased numbers of eosinophils in the diseased joints of our treated 6-/7- mice (Fig. 3).
Although the safranin+ MCs in most mouse strains express mMCP-7, our studies failed to uncover a significant functional contribution for this tryptase in the K/B×N mouse serum-transfer model of experimental arthritis (Fig. 2). The α chain of fibrinogen is a preferred substrate of mMCP-7, and the injection of this tryptase into the peritoneal cavity of WT mice preferentially results in the recruitment of eosinophils (43, 53). As WT B6 mice are genetically deficient in mMCP-7 (54, 58) but susceptible to arthritis (Figs. 1 and 2), our congenic mouse strains that differed in their mMCP-6 and mMCP-7 expression allowed us to test for possible redundancy of tryptase activity. Our observation that mMCP-7 did not restore inflammation or granulocyte influx into the synovium of the diseased mice (Fig. 2) suggests that mMCP-6 is the dominant pro-inflammatory tryptase in the K/B×N mouse serum-transfer model of arthritis. These observations are consistent with findings in the methylated-BSA/IL-1 (mBSA) mouse model of experimental arthritis of the knee which also displays significant reliance on mast cell tryptase•heparin complexes (72). The latter study confirms a key role for mMCP-6 in a very different type of experimental arthritis. Nevertheless, given redundant mMCP-6 and mMCP-7 function in the mBSA model, it needs to be pointed out that congenic 6-/7- mice might be necessary to uncover the roles for the MC’s tryptase•heparin complexes in other physiologic and/or pathologic situations.
The receptors and/or extracellular matrix proteins in the synovium that are cleaved by enzymatically active mMCP-6 remain to be determined. PAR-2 is a member of the protease-activated receptor family expressed on the surfaces of endothelial cells and synovial fibroblasts, and this receptor is activated by trypsin. Mirza and coworkers (73) reported that recombinant hTryptase-β can weakly activate cells via PAR-2. Nevertheless, using varied combinational approaches, it has been shown that recombinant mMCP-6 (31) and hTryptase-β (43, 74) prefer to cleave amino acid sequences that do not resemble the trypsin-susceptible activation site in PAR-2 (65). The substrate specificity of mMCP-6 becomes even more restricted when this tryptase is ionically bound to heparin (31). Because Compton and coworkers (75) found that naturally occurring hTryptase-β cannot activate PAR-2 if heparin is present, the importance of the proteoglycan cofactor may not have been adequately appreciated in some of the studies that concluded that mMCP-6 and/or hTryptase-β function via PAR-2. Nevertheless, the availability of PAR-2-null mice allowed us to more definitively evaluate the role of this G-protein coupled receptor in the K/B×N mouse serum-transfer model of experimental arthritis. Our results revealed that PAR-2 is dispensable in this arthritis model (Fig. 4). Furthermore, recombinant mMCP-6 stimulated CXCL1 production in PAR-2-null mouse FLS. These observations are consistent with the findings of Busso and coworkers (68) who evaluated the role of PAR-2 in four different mouse models of arthritis.
A major feature of RA is loss of aggrecan proteoglycans due to proteolysis of the N-terminal domain that binds to HA. Using a proteomics approach, we identified a prominent soluble fragment of aggrecan proteoglycan lacking its HA-binding domain in the joints of arthritic WT B6 mice (Fig. 6). Employing histochemistry and immunohistochemistry approaches, we then discovered that the proteolytic loss of cartilage proteoglycans is markedly attenuated in our transgenic B6 mice that lack MC-restricted tryptase•heparin complexes (Fig. 7). While our data could be a consequence of fewer protease-rich neutrophils in the arthritic joints of our tryptase-deficient mice (Fig. 3), we previously noted that an undefined serine protease present in rat peritoneal MCs rapidly cleaves the N-terminal domains of rat and bovine aggrecan proteoglycans in vitro (14), thereby resulting in a large size fragment that cannot recognize HA as identified in Fig. 6. It is now known that the MCs used in that earlier study express the rat ortholog of mMCP-6 and hTryptase-β (76). Because neutrophils and other cell types were not present in the Stevens et al. 1992 aggrecan-susceptibility study (14), it is possible that MC tryptases preferentially cleave the HA-binding domain of aggrecan in a direct manner. In support of this possibility, mouse aggrecan has a number of candidate tryptase-susceptible cleavage sites in its protease-susceptible N-terminal domain (e.g., the Pro-Ile-Val-Ser-Pro-Arg sequence at residues 546-551) (31, 43, 74) (Fig. 6). Alternatively, because pro-aggrecanase-1/ADAMTS4 is activated by tryptic cleavage at a site favorable for cleavage by mMCP-6 and hTryptase-β, MC tryptases could participate in aggrecan loss in our experimental model by activating a metalloproteinase like ADAMTS4.
A number of observations suggest that the contribution to inflammatory arthritis from mouse MCs via their tryptase•heparin complexes may be pertinent in human RA. The numbers of tryptase-expressing MCs are dramatically elevated in the synovial tissue of many patients with RA. Thus, the cellular lineage, localization, and mediators are common between mouse and man. Since abundant neutrophil recruitment to the synovial fluid is a hallmark of RA, modulation of neutrophil recruitment also is an attractive mechanism by which MC-restricted tryptase•heparin complexes could enhance disease activity. Furthermore, the contribution of the synovial mesenchyme to joint inflammation is increasingly appreciated (64). Thus, our observations that arthritis was reduced in CXCR2-/- mice (Fig. 4) and that exposure of human and mouse FLS to tryptase•heparin complexes results in the expression of multiple chemokines (Fig. 5) that regulate neutrophil recruitment provide a plausible and novel inflammatory pathway for disease pathogenesis. Other demonstrated tryptase activities, such as mitogenic activity for fibroblasts, metalloproteinase activation, and proteolysis of extracellular matrix proteins remain attractive additional potential mechanisms by which this MC effector function may contribute to synovial pathophysiology, and remain a worthy subject for further investigation (48-52).
In summary, we demonstrate the functional contribution of MC-restricted mMCP-6•heparin complexes to the pathogenesis of autoantibody-driven inflammatory arthritis in the mouse. While we have identified prominent roles for tryptase•heparin complexes in neutrophil recruitment and aggrecan loss, arthritis was more diminished in our NDST-2/heparin-null C57BL/6 mice than our mMCP-6-null C57BL/6 mice. These data suggest the participation of another MC-restricted protease mediator (e.g., mMCP-5 and/or carboxypeptidase A3) in joint inflammation and/or destruction. We therefore anticipate that the role of MCs and their varied mediators in arthritis will be multi-faceted, and that future investigations will provide further novel insights into their influence in the inflammatory disorder.
Acknowledgments
We wish to acknowledge the expert histotechnical assistance of Nicholas Calderone.
Footnotes
Disclosures The authors have no commercial interests.
This work was supported by the American Arthritis Foundation (KS); Australian Research Council and Arthritis Australia (HPM); University of Texas M. D. Anderson Cancer Center Physician Scientist Program (RA); Cogan Family Foundation (DML); and the National Institutes of Health grants AI054950 (RLS), AI059746 (DML), AI031599 (MFG), AR051321 (PAN), and HL036110 (RLS).
Abbreviations used in this paper: B6, C57BL/6; FLS, fibroblast-like synoviocytes; HA, hyaluronan; MC, mast cell; mMCP, mouse MC protease; 6+/7-, mMCP-6+/+/mMCP-7-/-; 6-/7+, mMCP-6-/-/mMCP-7+/+; 6-/7-, mMCP-6-/-/mMCP-7-/-; MS/MS, tandem mass spectroscopy; NDST-2, N-deacetylase/N-sulfotransferase-2; PAR-2, protease-activated receptor-2; RA, rheumatoid arthritis; WT, wild-type; W/Wv, WBB6F1-KitW/KitW-v.
References
- 1.Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–911. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- 2.Crisp AJ, Chapman CM, Kirkham SE, Schiller AL, Krane SM. Articular mastocytosis in rheumatoid arthritis. Arthritis Rheum. 1984;27:845–851. doi: 10.1002/art.1780270802. [DOI] [PubMed] [Google Scholar]
- 3.Godfrey HP, Ilardi C, Engber W, Graziano FM. Quantitation of human synovial mast cells in rheumatoid arthritis and other rheumatic diseases. Arthritis Rheum. 1984;27:852–856. doi: 10.1002/art.1780270803. [DOI] [PubMed] [Google Scholar]
- 4.Kopicky-Burd JA, Kagey-Sobotka A, Peters SP, Dvorak AM, Lennox DW, Lichtenstein LM, Wigley FM. Characterization of human synovial mast cells. J Rheumatol. 1988;15:1326–1333. [PubMed] [Google Scholar]
- 5.Bridges AJ, Malone DG, Jicinsky J, Chen M, Ory P, Engber W, Graziano FM. Human synovial mast cell involvement in rheumatoid arthritis and osteoarthritis. Relationship to disease type, clinical activity, and antirheumatic therapy. Arthritis Rheum. 1991;34:1116–1124. doi: 10.1002/art.1780340907. [DOI] [PubMed] [Google Scholar]
- 6.Dean G, Hoyland JA, Denton J, Donn RP, Freemont AJ. Mast cells in the synovium and synovial fluid in osteoarthritis. Br J Rheumatol. 1993;32:671–675. doi: 10.1093/rheumatology/32.8.671. [DOI] [PubMed] [Google Scholar]
- 7.Gotis-Graham I, McNeil HP. Mast cell responses in rheumatoid synovium. Association of the MCTC subset with matrix turnover and clinical progression. Arthritis Rheum. 1997;40:479–489. doi: 10.1002/art.1780400314. [DOI] [PubMed] [Google Scholar]
- 8.Kiener HP, Baghestanian M, Dominkus M, Walchshofer S, Ghannadan M, Willheim M, Sillaber C, Graninger WB, Smolen JS, Valent P. Expression of the C5a receptor (CD88) on synovial mast cells in patients with rheumatoid arthritis. Arthritis Rheum. 1998;41:233–245. doi: 10.1002/1529-0131(199802)41:2<233::AID-ART7>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 9.Pu J, Nishida K, Inoue H, Asahara H, Ohtsuka A, Murakami T. Mast cells in osteoarthritic and rheumatoid arthritic synovial tissues of the human knee. Acta Med Okayama. 1998;52:35–39. doi: 10.18926/AMO/31339. [DOI] [PubMed] [Google Scholar]
- 10.Ceponis A, Konttinen YT, Takagi M, Xu JW, Sorsa T, Matucci-Cerinic M, Santavirta S, Bankl HC, Valent P. Expression of stem cell factor (SCF) and SCF receptor (c-kit) in synovial membrane in arthritis: correlation with synovial mast cell hyperplasia and inflammation. J Rheumatol. 1998;25:2304–2314. [PubMed] [Google Scholar]
- 11.Nigrovic PA, Lee DM. Synovial mast cells: role in acute and chronic arthritis. Immunol Rev. 2007;217:19–37. doi: 10.1111/j.1600-065X.2007.00506.x. [DOI] [PubMed] [Google Scholar]
- 12.Bromley M, Woolley DE. Histopathology of the rheumatoid lesion. Identification of cell types at sites of cartilage erosion. Arthritis Rheum. 1984;27:857–863. doi: 10.1002/art.1780270804. [DOI] [PubMed] [Google Scholar]
- 13.Tetlow LC, Woolley DE. Distribution, activation and tryptase/chymase phenotype of mast cells in the rheumatoid lesion. Ann Rheum Dis. 1995;54:549–555. doi: 10.1136/ard.54.7.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens RL, Somerville LL, Sewell D, Swafford JR, Caulfield JP, Levi-Schaffer F, Hubbard JR, Dayton ET. Serosal mast cells maintain their viability and promote the metabolism of cartilage proteoglycans when cocultured with chondrocytes. Arthritis Rheum. 1992;35:325–335. doi: 10.1002/art.1780350312. [DOI] [PubMed] [Google Scholar]
- 15.Lee DM, Friend DS, Gurish MF, Benoist C, Mathis D, Brenner MB. Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science. 2002;297:1689–1692. doi: 10.1126/science.1073176. [DOI] [PubMed] [Google Scholar]
- 16.Corr M, Crain B. The role of FcγR signaling in the K/B × N serum transfer model of arthritis. J Immunol. 2002;169:6604–6609. doi: 10.4049/jimmunol.169.11.6604. [DOI] [PubMed] [Google Scholar]
- 17.Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med. 2007;204:2797–2802. doi: 10.1084/jem.20071391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van den Broek MF, van den Berg WB, van de Putte LB. The role of mast cells in antigen induced arthritis in mice. J Rheumatol. 1988;15:544–551. [PubMed] [Google Scholar]
- 19.Nigrovic PA, Binstadt BA, Monach PA, Johnsen A, Gurish M, Iwakura Y, Benoist C, Mathis D, Lee DM. Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc Natl Acad Sci USA. 2007;104:2325–2330. doi: 10.1073/pnas.0610852103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNeil HP, Reynolds DS, Schiller V, Ghildyal N, Gurley DS, Austen KF, Stevens RL. Isolation, characterization, and transcription of the gene encoding mouse mast cell protease 7. Proc Natl Acad Sci USA. 1992;89:11174–11178. doi: 10.1073/pnas.89.23.11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reynolds DS, Gurley DS, Austen KF, Serafin WE. Cloning of the cDNA and gene of mouse mast cell protease-6: transcription by progenitor mast cells and mast cells of the connective tissue subclass. J Biol Chem. 1991;266:3847–3853. [PubMed] [Google Scholar]
- 22.Miller JS, Westin EH, Schwartz LB. Cloning and characterization of complementary DNA for human tryptase. J Clin Invest. 1989;84:1188–1195. doi: 10.1172/JCI114284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JS, Moxley G, Schwartz LB. Cloning and characterization of a second complementary DNA for human tryptase. J Clin Invest. 1990;86:864–870. doi: 10.1172/JCI114786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanderslice P, Ballinger SM, Tam EK, Goldstein SM, Craik CS, Caughey GH. Human mast cell tryptase: multiple cDNAs and genes reveal a multigene serine protease family. Proc Natl Acad Sci USA. 1990;87:3811–3815. doi: 10.1073/pnas.87.10.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong GW, Yasuda S, Morokawa N, Li L, Stevens RL. Mouse chromosome 17A3.3 contains 13 genes that encode functional tryptic-like serine proteases with distinct tissue and cell expression patterns. J Biol Chem. 2004;279:2438–2452. doi: 10.1074/jbc.M308209200. [DOI] [PubMed] [Google Scholar]
- 26.Huang C, Morales G, Vagi A, Chanasyk K, Ferrazzi M, Burklow C, Qiu WT, Feyfant E, Šali A, Stevens RL. Formation of enzymatically active, homotypic, and heterotypic tetramers of mouse mast cell tryptases: dependence on a conserved Trp-rich domain on the surface. J Biol Chem. 2000;275:351–358. doi: 10.1074/jbc.275.1.351. [DOI] [PubMed] [Google Scholar]
- 27.Humphries DE, Wong GW, Friend DS, Gurish MF, Qiu WT, Huang C, Sharpe AH, Stevens RL. Heparin is essential for the storage of specific granule proteases in mast cells. Nature. 1999;400:769–772. doi: 10.1038/23481. [DOI] [PubMed] [Google Scholar]
- 28.Forsberg E, Pejler G, Ringvall M, Lunderius C, Tomasini-Johansson B, Kusche-Gullberg M, Eriksson I, Ledin J, Hellman L, Kjellén L. Abnormal mast cells in mice deficient in a heparin-synthesizing enzyme. Nature. 1999;400:773–776. doi: 10.1038/23488. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz LB, Bradford TR. Regulation of tryptase from human lung mast cells by heparin: stabilization of the active tetramer. J Biol Chem. 1986;261:7372–7379. [PubMed] [Google Scholar]
- 30.Hallgren J, Karlson U, Poorafshar M, Hellman L, Pejler G. Mechanism for activation of mouse mast cell tryptase: dependence on heparin and acidic pH for formation of active tetramers of mouse mast cell protease 6. Biochemistry (Mosc) 2000;39:13068–13077. doi: 10.1021/bi000973b. [DOI] [PubMed] [Google Scholar]
- 31.Huang C, Friend DS, Qiu WT, Wong GW, Morales G, Hunt J, Stevens RL. Induction of a selective and persistent extravasation of neutrophils into the peritoneal cavity by tryptase mouse mast cell protease 6. J Immunol. 1998;160:1910–1919. [PubMed] [Google Scholar]
- 32.Hallgren J, Pejler G. Biology of mast cell tryptase. An inflammatory mediator. Febs J. 2006;273:1871–1895. doi: 10.1111/j.1742-4658.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 33.Stevens RL, Adachi R. Protease-proteoglycan complexes of mouse and human mast cells and importance of their β-tryptase-heparin complexes in inflammation and innate immunity. Immunol Rev. 2007;217:155–167. doi: 10.1111/j.1600-065X.2007.00525.x. [DOI] [PubMed] [Google Scholar]
- 34.Gurish MF, Austen KF. The diverse roles of mast cells. J Exp Med. 2001;194:F1–5. doi: 10.1084/jem.194.1.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin K, Gurish MF, Friend DS, Pemberton AD, Thornton EM, Miller HR, Lee DM. Lymphocyte-independent connective tissue mast cells populate murine synovium. Arthritis Rheum. 2006;54:2863–2871. doi: 10.1002/art.22058. [DOI] [PubMed] [Google Scholar]
- 36.Gotis-Graham I, Smith MD, Parker A, McNeil HP. Synovial mast cell responses during clinical improvement in early rheumatoid arthritis. Ann Rheum Dis. 1998;57:664–671. doi: 10.1136/ard.57.11.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–2054. doi: 10.4049/jimmunol.179.4.2051. [DOI] [PubMed] [Google Scholar]
- 38.Xu D, Jiang HR, Kewin P, Li Y, Mu R, Fraser AR, Pitman N, Kurowska-Stolarska M, McKenzie AN, McInnes IB, Liew FY. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc Natl Acad Sci USA. 2008;105:10913–10918. doi: 10.1073/pnas.0801898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 40.Chen M, Lam BK, Kanaoka Y, Nigrovic PA, Audoly LP, Austen KF, Lee DM. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J Exp Med. 2006;203:837–842. doi: 10.1084/jem.20052371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J Exp Med. 2006;203:829–835. doi: 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thakurdas SM, Melicoff E, Sansores-Garcia L, Moreira DC, Petrova Y, Stevens RL, Adachi R. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J Biol Chem. 2007;282:20809–20815. doi: 10.1074/jbc.M611842200. [DOI] [PubMed] [Google Scholar]
- 43.Huang C, De Sanctis GT, O’Brien PJ, Mizgerd JP, Friend DS, Drazen JM, Brass LF, Stevens RL. Evaluation of the substrate specificity of human mast cell tryptase βI and demonstration of its importance in bacterial infections of the lung. J Biol Chem. 2001;276:26276–26284. doi: 10.1074/jbc.M102356200. [DOI] [PubMed] [Google Scholar]
- 44.He S, Peng Q, Walls AF. Potent induction of a neutrophil and eosinophil-rich infiltrate in vivo by human mast cell tryptase: selective enhancement of eosinophil recruitment by histamine. J Immunol. 1997;159:6216–6225. [PubMed] [Google Scholar]
- 45.Cairns JA, Walls AF. Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression. J Immunol. 1996;156:275–283. [PubMed] [Google Scholar]
- 46.Compton SJ, Cairns JA, Holgate ST, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1β and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J Immunol. 1998;161:1939–1946. [PubMed] [Google Scholar]
- 47.Nakano S, Mishiro T, Takahara S, Yokoi H, Hamada D, Yukata K, Takata Y, Goto T, Egawa H, Yasuoka S, Furouchi H, Hirasaka K, Nikawa T, Yasui N. Distinct expression of mast cell tryptase and protease activated receptor-2 in synovia of rheumatoid arthritis and osteoarthritis. Clin Rheumatol. 2007;26:1284–1292. doi: 10.1007/s10067-006-0495-8. [DOI] [PubMed] [Google Scholar]
- 48.Gruber BL, Schwartz LB, Ramamurthy NS, Irani AM, Marchese MJ. Activation of latent rheumatoid synovial collagenase by human mast cell tryptase. J Immunol. 1988;140:3936–3942. [PubMed] [Google Scholar]
- 49.Gruber BL, Marchese MJ, Suzuki K, Schwartz LB, Okada Y, Nagase H, Ramamurthy NS. Synovial procollagenase activation by human mast cell tryptase dependence upon matrix metalloproteinase 3 activation. J Clin Invest. 1989;84:1657–1662. doi: 10.1172/JCI114344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lees M, Taylor DJ, Woolley DE. Mast cell proteinases activate precursor forms of collagenase and stromelysin, but not of gelatinases A and B. Eur J Biochem. 1994;223:171–177. doi: 10.1111/j.1432-1033.1994.tb18980.x. [DOI] [PubMed] [Google Scholar]
- 51.Lohi J, Harvima I, Keski-Oja J. Pericellular substrates of human mast cell tryptase: 72,000 dalton gelatinase and fibronectin. J Cell Biochem. 1992;50:337–349. doi: 10.1002/jcb.240500402. [DOI] [PubMed] [Google Scholar]
- 52.Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, Schwartz LB, Korn JH. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol. 1997;158:2310–2317. [PubMed] [Google Scholar]
- 53.Huang C, Wong GW, Ghildyal N, Gurish MF, Sali A, Matsumoto R, Qiu WT, Stevens RL. The tryptase, mouse mast cell protease 7, exhibits anticoagulant activity in vivo and in vitro due to its ability to degrade fibrinogen in the presence of the diverse array of protease inhibitors in plasma. J Biol Chem. 1997;272:31885–31893. doi: 10.1074/jbc.272.50.31885. [DOI] [PubMed] [Google Scholar]
- 54.Hunt JE, Stevens RL, Austen KF, Zhang J, Xia Z, Ghildyal N. Natural disruption of the mouse mast cell protease 7 gene in the C57BL/6 mouse. J Biol Chem. 1996;271:2851–2855. doi: 10.1074/jbc.271.5.2851. [DOI] [PubMed] [Google Scholar]
- 55.Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, Wood WI, Moore MW. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- 56.Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104:397–401. doi: 10.1182/blood-2004-02-0434. [DOI] [PubMed] [Google Scholar]
- 57.Shin K, Watts GF, Oettgen HC, Friend DS, Pemberton AD, Gurish MF, Lee DM. Mouse mast cell tryptase mMCP-6 is a critical link between adaptive and innate immunity in the chronic phase of Trichinella spiralis infection. J Immunol. 2008;180:4885–4891. doi: 10.4049/jimmunol.180.7.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghildyal N, Friend DS, Freelund R, Austen KF, McNeil HP, Schiller V, Stevens RL. Lack of expression of the tryptase mouse mast cell protease 7 in mast cells of the C57BL/6J mouse. J Immunol. 1994;153:2624–2630. [PubMed] [Google Scholar]
- 59.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 60.Humason GL. Animal tissue techniques. W. H. Freeman and Co.; San Francisco: 1967. [Google Scholar]
- 61.Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leder LD. The chloroacetate esterase reaction. A useful means of histological diagnosis of hematological disorders from paraffin sections of skin. Am J Dermatopathol. 1979;1:39–42. [PubMed] [Google Scholar]
- 63.Kiener HP, Lee DM, Agarwal SK, Brenner MB. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. Am J Pathol. 2006;168:1486–1499. doi: 10.2353/ajpath.2006.050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 65.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferrell WR, Lockhart JC, Kelso EB, Dunning L, Plevin R, Meek SE, Smith AJ, Hunter GD, McLean JS, McGarry F, Ramage R, Jiang L, Kanke T, Kawagoe J. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Palmer HS, Kelso EB, Lockhart JC, Sommerhoff CP, Plevin R, Goh FG, Ferrell WR. Protease-activated receptor 2 mediates the proinflammatory effects of synovial mast cells. Arthritis Rheum. 2007;56:3532–3540. doi: 10.1002/art.22936. [DOI] [PubMed] [Google Scholar]
- 68.Busso N, Frasnelli M, Feifel R, Cenni B, Steinhoff M, Hamilton J, So A. Evaluation of protease-activated receptor 2 in murine models of arthritis. Arthritis Rheum. 2007;56:101–107. doi: 10.1002/art.22312. [DOI] [PubMed] [Google Scholar]
- 69.Ghildyal N, Friend DS, Stevens RL, Austen KF, Huang C, Penrose JF, Šali A, Gurish MF. Fate of two mast cell tryptases in V3 mastocytosis and normal BALB/c mice undergoing passive systemic anaphylaxis: prolonged retention of exocytosed mMCP-6 in connective tissues, and rapid accumulation of enzymatically active mMCP-7 in the blood. J Exp Med. 1996;184:1061–1073. doi: 10.1084/jem.184.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castor CW. The microscopic structure of normal human synovial tissue. Arthritis Rheum. 1960;3:140–151. doi: 10.1002/art.1780030205. [DOI] [PubMed] [Google Scholar]
- 71.Pang L, Nie M, Corbett L, Sutcliffe A, Knox AJ. Mast cell β-tryptase selectively cleaves eotaxin and RANTES and abrogates their eosinophil chemotactic activities. J Immunol. 2006;176:3788–3795. doi: 10.4049/jimmunol.176.6.3788. [DOI] [PubMed] [Google Scholar]
- 72.McNeil HP, Shin K, Campbell IK, Wicks IP, Adachi R, Lee DM, Stevens RL. The mouse mast cell-restricted tetramer-forming tryptases mouse mast cell protease 6 and mouse mast cell protease 7 are critical mediators in inflammatory arthritis. Arthritis Rheum. 2008;58:2338–2346. doi: 10.1002/art.23639. [DOI] [PubMed] [Google Scholar]
- 73.Mirza H, Schmidt VA, Derian CK, Jesty J, Bahou WF. Mitogenic responses mediated through the proteinase-activated receptor-2 are induced by expressed forms of mast cell α-or β-tryptases. Blood. 1997;90:3914–3922. [PubMed] [Google Scholar]
- 74.Harris JL, Niles A, Burdick K, Maffitt M, Backes BJ, Ellman JA, Kuntz I, Haak-Frendscho M, Craik CS. Definition of the extended substrate specificity determinants for β tryptases I and II. J Biol Chem. 2001;276:34941–34947. doi: 10.1074/jbc.M102997200. [DOI] [PubMed] [Google Scholar]
- 75.Compton SJ, Renaux B, Wijesuriya SJ, Hollenberg MD. Glycosylation and the activation of proteinase-activated receptor 2 (PAR2) by human mast cell tryptase. Br J Pharmacol. 2001;134:705–718. doi: 10.1038/sj.bjp.0704303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lützelschwab C, Pejler G, Aveskogh M, Hellman L. Secretory granule proteases in rat mast cells: cloning of 10 different serine proteases and a carboxypeptidase A from various rat mast cell populations. J Exp Med. 1997;185:13. doi: 10.1084/jem.185.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walcz E, Deak F, Erhardt P, Coulter SN, Fulop C, Horvath P, Doege KJ, Glant TT. Complete coding sequence, deduced primary structure, chromosomal localization, and structural analysis of murine aggrecan. Genomics. 1994;22:364–371. doi: 10.1006/geno.1994.1396. [DOI] [PubMed] [Google Scholar]