

Abstract
Matrix metalloproteinases (MMPs) are induced during inflammatory responses and are important for immune regulation, angiogenesis, wound healing and tissue remodeling. Expression of MMPs needs to be tightly controlled to avoid excessive tissue damage. In this study we investigated the regulation of MMP expression by inflammatory factors in primary human monocytes and macrophages. IFNγ, which augments inflammatory cytokine production in response to macrophage-activating factors such as Toll-like receptor (TLR) ligands, instead broadly suppressed TLR-induced MMP expression. Inhibition of MMP expression was dependent on STAT1 and required de novo protein synthesis. IFNγ strongly enhanced TLR-induced expression of the transcriptional repressor ATF-3 in a STAT1-dependent manner, which correlated with recruitment of ATF-3 to the endogenous MMP-1 promoter as detected by chromatin immunoprecipitation assays. RNA interference experiments further supported a role for ATF-3 in suppression of MMP-1 expression. In addition, IFNγ suppressed DNA binding by AP-1 transcription factors that are known to promote MMP expression and a combination of supershift, RNA interference and overexpression experiments implicated AP-1 family member Fra-1 in the regulation of MMP-1 expression. These results define an IFNγ-mediated homeostatic loop that limits the potential for tissue damage associated with inflammation, and identify transcriptional factors that regulate MMP expression in myeloid cells in inflammatory settings.
Keywords: LPS, IFNγ, STAT1, ATF-3, Macrophages
Introduction
Since the discovery of the first matrix metalloproteinase (MMP) in the regressing tadpole tail by Gross and Lapiere in 1962 (1), the list of functions assigned to various MMPs has been expanding rapidly. MMPs were initially thought to be a group of enzymes with the sole function of regulating extracellular matrix composition. However, intense research in recent years has shown that extracellular matrix (ECM) degradation is not the sole and possibly not the main function of these proteinases. Indeed, plenty of evidence supports roles of MMPs in physiological processes such as embryogenesis and wound healing/tissue remodeling and also in pathological processes that include arthritis, pulmonary diseases, cardiovascular ailments and cancer (2-7). In addition to extracellular matrix remodeling MMPs regulate signal transduction and growth factor function, and recent findings also indicate that MMPs are immunoregulators by acting on inflammatory cytokines, chemokines and other immune proteins (8-18).
Not surprisingly, the activity of MMPs is tightly regulated at multiple levels that include gene expression, compartmentalization, pro-enzyme activation and enzyme inactivation/catabolism/clearance (3,18). Expression of many MMPs is normally low or absent in healthy tissues, but they are induced when tissue remodeling is required, during inflammation, and in disease states such as rheumatoid arthritis and cancer in which high levels of MMPs may signify a poor prognosis in human patients (19). MMP gene expression is mainly regulated at the transcriptional level in response to hormones, growth factors, cytokines and cell-cell/cell-matrix interactions (3,5,20). Though the expression of MMPs in diseased states is intended to restore normal homeostasis, in a setting such as inflammation the up-regulation of various MMPs may lead to more damage than repair (5).
Rheumatoid arthritis (RA) and atherosclerosis are disorders of chronic inflammation, and macrophages are central players in both of these pathological conditions (21-23). Though the importance of macrophage-derived MMPs in cartilage and/or bone destruction in RA has not been fully elucidated, their role in atherosclerosis progression is well-documented (21,24). In atherosclerosis, macrophages accumulate lipid to form foam cells. The accumulation of foam cells in an atheroma is reversible and does not in itself cause clinical consequences. However, the secretion of MMPs by the atheromatous plaque-associated macrophages can cause plaque destabilization, leading to acute coronary events with high morbidity and mortality (22,23,25). Among the MMPs produced by macrophages, MMP-1 (collagenase-1), MMP-2 (gelatinase A), MMP-3 (stromelysin-1), MMP-7 (matrilysin-1), MMP-9 (gelatinase B), MMP-12 (metalloelastase) and MMP-13 (metalloproteinase-13) have been associated with atherosclerotic plaque instability and rupture (22,23,25-27). Therefore, understanding the regulation of MMPs in inflammatory conditions is crucial in formulating a therapeutic intervention to prevent disease progression.
Most studies on regulation of MMP expression have been performed using fibroblasts, chondrocytes, osteoblasts and cell lines such as HEK293, HeLa and NIH-3T3 cells (7,28-45), with very few studies using monocytes/macrophages (46-48). Since MMP expression is regulated differently in the various cell types and cell lines studied to date, we were interested in understanding MMP regulation in primary human monocytes and macrophages that are relevant for inflammatory diseases, and defining mechanisms that inhibit MMP expression. In this study we found that in primary human macrophages IFNγ globally inhibits induction of MMPs by Toll-like receptors, innate immune receptors that play a key role in activation of macrophages by microbial products and endogenous ligands that include ECM degradation products. Further analysis of the regulation of MMP-1 showed that the mechanism of IFNγ-mediated inhibition involves induction of the transcriptional repressor activating transcription factor (ATF)-3 and suppression of DNA binding by AP-1 proteins. Prostaglandin E2 also downregulated LPS-induced MMP-1 expression, and suppression by PGE2 and IFNγ converged on inhibition of AP-1 proteins and regulation of AP-1 family member Fra-1. These results define mechanisms that inhibit MMP expression in primary human macrophages.
Materials and Methods
Biological reagents and cell culture
Peripheral blood mononuclear cells (PBMCs) were obtained from whole blood from disease-free volunteers by density gradient centrifugation with Ficoll (Invitrogen, Carlsbad, CA). CD14+ monocytes were purified from fresh PBMCs with anti-CD14 magnetic beads (Miltenyi Biotec, Auburn, CA), as recommended by the manufacturer. Purity of monocytes was greater than 97% as verified by FACS, and freshly isolated monocytes were used in many experiments, as noted in text and figure legends. Some experiments were performed using macrophages, which were derived by culturing monocytes in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS (Hyclone, Logan, UT) in the presence of 10 ng/ml of human macrophage colony stimulating factor (M-CSF) (Peprotech, Rocky Hill, NJ). THP-1 human monocytic cells were obtained from ATCC and cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS (Hyclone, Logan, UT). LPS (100 ng/ml), PMA (100 ng/ml), LiCl (20 mM), cycloheximide (20 μg/ml) and SB216763 (10 μM) were purchased from Sigma-Aldrich (Milwaukee, WI). PGE2 was obtained from Sigma-Aldrich (Milwaukee, WI) and Calbiochem (San Diego, CA).
Lentiviral gene transduction and RNA interference (RNAi)
A lentivirus-based vector expressing the human STAT1 or Fra-1 cDNA driven by a human phospho-glycerol kinase (hPGK) promoter was used to generate recombinant lentiviral particles as described (49). A construct that contained a transcription cassette encoding enhanced green fluorescent protein (eGFP) driven by the hPGK promoter was used to generate control viral particles for STAT1 and Fra-1 expression experiments. RNAi in the THP-1 monocytic cell line was performed using lentiviral transduction to stably express shRNAs that targeted STAT1 or ATF-3, or DSRed2 as a control. For STAT1 RNA interference, oligonucleotides encoding several different short hairpin RNAs (shRNAs) that target human STAT1 were cloned into the lentivirus-based RNAi vector pLL3.7 that also contains a transcription cassette encoding eGFP driven by a CMV promoter (49). Lentivirus-based RNAi vectors against ATF3 were purchased from Sigma-Aldrich (Milwaukee, WI). Constructs that were effective in suppressing STAT1 or ATF-3 expression were identified using transient co-transfection of HEK 293T cells with expression plasmids encoding the respective genes. The constructs that were most effective in HEK 293T cells (containing the shRNA sequences 5′-GCGTAATCTTCAGGATAAT-3′ or 5′- ACCTTGCAGAACAGAGAAC-3′ for human STAT1 and Sigma's TRC Numbers of 0000013570 and 0000013572 for human ATF-3) were used to generate recombinant lentiviral particles. Lentiviral particles encoding interfering shRNA against red fluorescence protein DSRed2 (5′-GTGGGAGCGCGTGATGAAC-3′) were used as a control for STAT1/ATF3 RNAi experiments. THP-1 cells were incubated overnight with recombinant lentiviral particles at a ratio of 1:50 in the presence of 4 μg/ml polybrene. The efficiency of transduction was evaluated using flow cytometry and fluorescence microscopy to monitor eGFP expression and was typically >90%. For RNAi experiments with primary human monocytes we used nucleofection of short interfering RNA duplexes (siRNA) that targeted GSK3β or Fra-1, or control siRNAs. GSK3β- and Fra-1-specific prevalidated siRNAs and nontargeting control siRNAs were purchased from Dharmacon (Lafayette, CO). siRNAs were transfected into primary human macrophages using the Amaxa Nucleofector device set to program Y-001 with the Human Monocyte Nucleofector kit (Amaxa, Cologne, Germany).
Real time quantitative RT-PCR (qPCR)
For real time PCR, total RNA was extracted using an RNeasy Mini kit and 1 μg of total RNA was reverse transcribed using a First Strand cDNA Synthesis kit (Fermentas, Hanover, MD). Real time, quantitative PCR was performed using iQ™ SYBR-Green Supermix and iCycler iQ™ thermal cycler (Biorad, Hercules, CA) following the manufacturer's protocols. Triplicate reactions were run for each sample and mRNA levels were normalized relative to β-actin. The generation of only the correct size amplification products was confirmed using agarose gel electrophoresis.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed using the ChIP-IT Express kit (Active Motif) according to the manufacturer's instructions. Chromatin was prepared from 2 × 107 primary human monocytes per condition and was sheared to an average size of 500 bp using a Missonics Sonicator 3000. Rabbit polyclonal antibodies against ATF-3 (sc-188) or control rabbit IgG (sc-2027) (Santa Cruz Biotechnologies) were used for immunoprecipitation (IP); each IP contained chromatin derived from 2 ×106 monocytes. Immunoprecipitates were subjected to PCR with primers that span the ATF site located 2017 bp upstream of the transcription start site of the MMP1 gene. Primer sequences are: upstream, 5′- AGGGATTTTGTTTAAGTAAAGG-3′; downstream, 5′-GCATGTGACCATCTGTGATT-3′.
Immunoblotting and Electrophoretic Mobility Shift Assay (EMSA)
Whole cell and nuclear extracts were obtained, and protein levels quantitated using the Bradford assay (Biorad), as previously described (50). For immunoblotting, 30 μg of THP-1 whole cell lysates were fractionated on 7.5% polyacrylamide gels using SDS-PAGE, transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA) and enhanced chemiluminescence was used for detection. For EMSA, 5 μg of nuclear extracts from primary monocytes were incubated for 15 minutes at room temperature with 0.5 ng of 32P-labeled double stranded AP-1 (5′- AAA GCA TGA GTC AGA CAC -3′ and 5′- GTG TCT GAC TCA TGC TTT -3′) or CRE (5′- TTC CAG GGT GAC GTC TTA GGC - 3′ and 5′- GCC TAA GAC GTC ACC CTG GAA -3′) oligonucleotides derived from the human MMP-1 promoter in a 15 μl binding reaction containing 40 mM NaCl and 2 μg of poly-dI-dC (Pharmacia, Piscataway, NJ), and complexes were resolved on nondenaturing 4.5 % polyacrylamide gels. For supershift assays, nuclear extracts were incubated with antibodies on ice for 60 min prior to the addition of radio-labeled oligonucleotides as previously described (51). All supershift antibodies used were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Results
IFNγ suppresses TLR- and PMA-induced MMP expression in primary human monocytes
We wished to identify physiological mechanisms that suppress MMP expression in primary human myeloid cells. We first determined which MMPs are induced in monocytes and macrophages by inflammatory stimuli. Primary monocytes were treated with inflammatory factors for 3 hr or overnight (12 hr) and MMP mRNA levels were measured using real-time PCR (RT-PCR). The TLR4 ligand LPS was the most potent inducer of MMP expression (data not shown), and activated expression of at least 7 MMP genes more than 5-fold at either an early time point (3 hr) (MMP-1, MMP-3, MMP-10 and MMP-12; Figure 1A), or at a later time point after LPS stimulation (12 hr) (MMP-2, MMP-7 and MMP-9; Figure 1B). Expression of many MMPs, with certain exceptions such as MMP-2, is regulated by AP-1 transcription factors and we and others had previously shown that IFN-γ can suppress AP-1-mediated gene expression (52). Therefore, we tested whether IFN-γ could suppress LPS-induced MMP expression. Pre-treatment of monocytes with IFNγ for 3-hr strikingly decreased LPS-induced expression of the MMPs that were studied (Figure 1A and 1B); similar results were obtained with primary M-CSF-differentiated macrophages (data not shown). As IFN-γ typically synergizes with LPS to superinduce expression of inflammatory genes such as COX-2 and IL-6, we measured COX-2 and IL-6 mRNA to rule out nonspecific suppressive effects of IFNγ on gene expression. As expected, IFNγ increased LPS-induced accumulation of COX-2 and IL-6 mRNA (Figure 1C). In addition, IFNγ augmented LPS-induced expression of ADAM-17, a cell surface MMP (Figure 1C). These results show that IFNγ specifically and strongly suppressed LPS-induced expression of multiple secreted MMPs that have AP-1 binding sequences (also known as phorbol ester-responsive elements or TREs) in their promoters and are known to be positively regulated by AP-1 proteins (3,53-55). The phorbol ester PMA is a strong activator of AP-1-mediated transcription, and IFNγ also suppressed PMA-induced expression of MMPs, but not of TNFα (Figure 1D). PMA was a stronger inducer of MMP expression than TLR ligands, which correlated with stronger induction of AP-1 proteins by PMA (data not shown). These results show that IFNγ strongly suppresses MMP expression in primary human monocytes. The dependence of expression of many MMP genes on AP-1, together with the previously reported inhibition of AP-1 proteins by IFNγ, suggested that IFNγ may suppress MMP expression at least in part by inhibiting AP-1. However, IFNγ-mediated inhibition of MMP-2 (Fig. 1B), which is not regulated by AP-1, suggests IFNγ inhibits MMP expression by several mechanisms.
FIGURE 1.
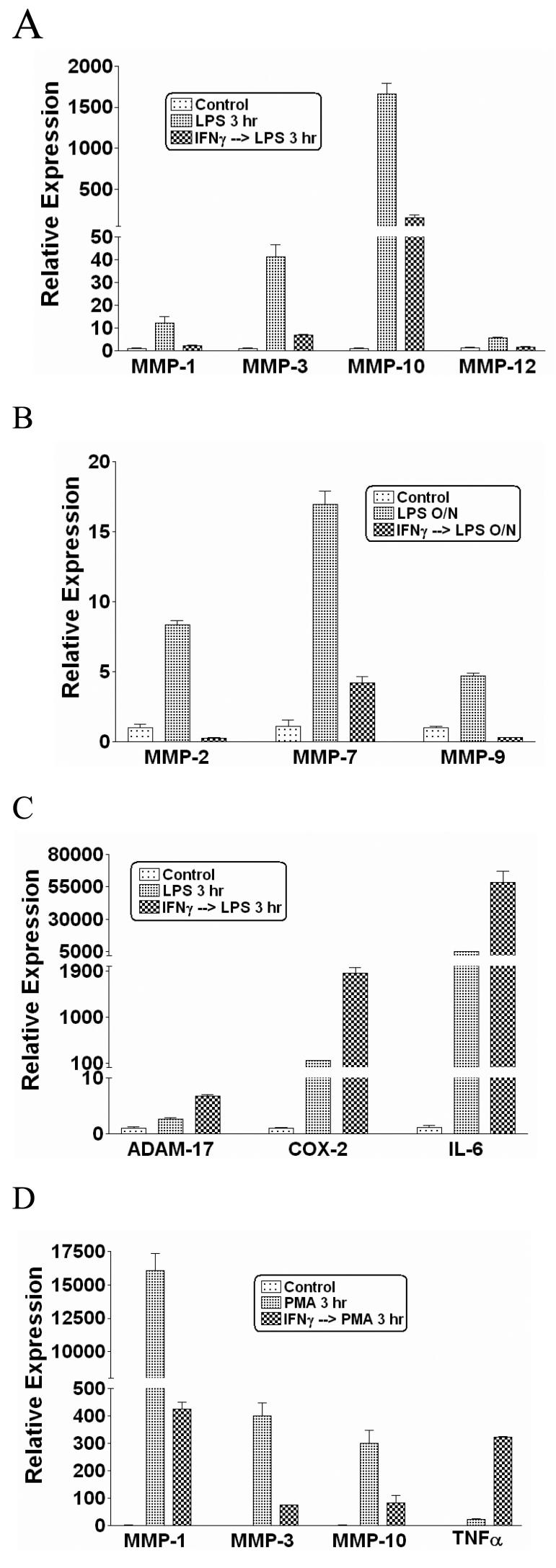
IFNγ inhibits LPS- and PMA-induced MMP expression in primary monocytes. A-C, Primary human monocytes were stimulated with 100 ng/ml of LPS with or without IFNγ pre-treatment (100 U/ml, added 3 hr before LPS) for 3 hr (A and C), or 12 hr (B). D, PMA (100 ng/ml) stimulation of primary monocytes in the absence or presence of IFNγ pre-treatment. mRNA levels were analyzed using real time PCR and normalized relative to β-Actin; results are expressed as mean ± SD of triplicate determinants. Results are shown as expression in stimulated monocytes relative to expression in unstimulated cells (control), which was set at 1.
Previously we (52) and others (56) have shown that IFNγ inhibits TLR-induced IL-10 production by enhancing glycogen synthase kinase 3 (GSK-3) activity and that inhibition of GSK-3 reverses IFNγ suppression of IL-10 production. This prompted us to investigate whether the same mechanism was also applicable to IFNγ regulation of MMPs. However, GSK inhibitors did not have any effect on IFNγ-mediated suppression of MMP expression (Figure 2A); reversal of IFNγ-mediated suppression of IL-10 expression confirmed the efficacy of GSK-3 inhibition (data not shown). In addition, we used siRNA to knock-down GSK-3β expression in primary monocytes to about 30% of control at both mRNA and protein levels (Figure 2B and 2D), a level sufficiently low to attenuate IFNγ-mediated inhibition of IL-10 expression ((52) and data not shown).
FIGURE 2.

IFNγ inhibition of LPS-induced MMP expression is not dependent on GSK-3. A, IFNγ pre-treated primary monocytes were stimulated with LPS for 3 hr in the absence or presence of GSK-3 inhibitors LiCl or SB216763 (added 20 min before IFNγ). B and C, Primary monocytes transfected with control or GSK-3β siRNAs were stimulated with LPS (B) or PMA (C) with or without IFNγ pre-treatment. D, Primary monocytes were transfected with control or GSK-3β siRNAs, and GSK-3β mRNA and protein levels were measured. mRNA levels were measured using real time PCR and normalized relative to β-Actin. Results are expressed as mean ± SD of triplicate determinants. Results are shown as expression in stimulated monocytes relative to expression in unstimulated cells (ctrl), which was set at 1. GSK-3β protein levels were measured using immunoblotting.
However, reduced GSK-3β expression had no discernable effect on IFNγ suppression of TLR4-induced MMP expression (Figure 2B), and similar results were obtained when PMA was used to induce MMP expression (Figure 2C). Our results suggest that the mechanisms by which IFNγ suppresses LPS induction of MMP genes differ from mechanisms used to suppress induction of the potent anti-inflammatory factor IL-10.
Suppression of MMP expression by IFNγ is dependent on STAT1 and de novo protein synthesis
Activation of gene expression by IFNγ is mediated by STAT1, but mechanisms by which IFNγ inhibits gene expression are not well understood (57). Therefore, we investigated whether IFNγ-mediated suppression of MMP expression was dependent on STAT1. STAT1 protein expression was too stable to achieve successful knock down of expression in primary monocytes, so instead we used THP-1 monocytic cells in which STAT1 expression was stably diminished secondary to transduction with lentiviral vectors that express shRNAs targeting STAT1 mRNA (58). IFNγ effectively suppressed TLR4-induced MMP-1 and MMP-10 expression in control THP-1 cells that expressed shRNA that targets the irrelevant DSRed2 mRNA, but IFNγ-mediated suppression of MMP induction was completely abrogated in THP-1 cells expressing low STAT1 levels (Figure 3). Effects of IFNγ alone on MMP expression were not consistently observed (data not shown). Thus, suppression of MMP expression by IFNγ is dependent on STAT1.
FIGURE 3.
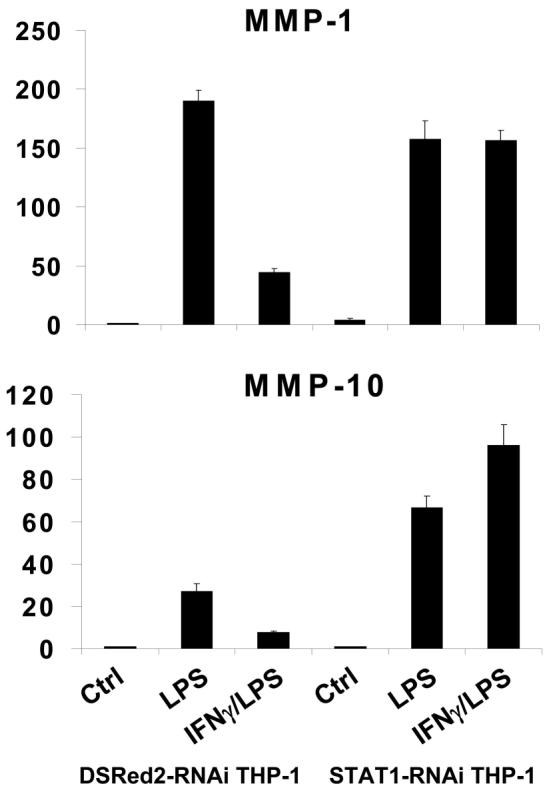
IFNγ inhibition of LPS-induced MMP expression is dependent on STAT1. THP-1 cells transduced with control DSRed2-shRNA- or STAT1-shRNA-encoding lentiviral particles were stimulated with 100 ng/ml of LPS (3 hr) with or without IFNγ pre-treatment (100 U/ml, added 3 hr before LPS) and mRNA levels were measured using real time PCR and normalized relative to β-Actin. Results are expressed as mean ± SD of triplicate determinants. Results are shown as expression in stimulated monocytes relative to expression in unstimulated cells (ctrl), which was set at 1.
STAT1 could suppress MMP expression directly by binding to MMP promoters and recruiting transcriptional corepressors, although there is minimal precedent for such a direct repressive mechanism. Alternatively, STAT1 could activate expression of genes that encode transcriptional repressors that target MMP genes. To differentiate between these two possibilities we tested whether the inhibitory effect of IFNγ on MMP expression was dependent on new protein synthesis. Primary monocytes were stimulated with LPS and IFNγ in the presence of cycloheximide (added 15 min before IFNγ or LPS) to block protein synthesis. As shown in Figure 4A, LPS induced MMP-1, MMP-3 and MMP-10 mRNAs in the absence of protein synthesis, indicating that LPS increases their expression at least in part by inducing post-translational modification of pre-existing proteins. However, cycloheximide abolished the inhibition of MMP-1 and MMP-3 expression by IFNγ, whereas suppression of MMP-10 expression remained intact (Figure 4A). These results suggest that IFNγ induction of a repressor is required for suppression of MMP-1 and MMP-3 expression, whereas inhibition of MMP-10 expression is more direct. This notion was further supported by a kinetic analysis of IFNγ-mediated inhibition. Down-regulation of LPS-induced MMP-3 expression required greater than 1 hr of pre-treatment with IFNγ, consistent with the requirement for synthesis of an inhibitor, whereas MMP-10 expression was suppressed even when IFNγ was added simultaneously with LPS (Figure 4B).
FIGURE 4.
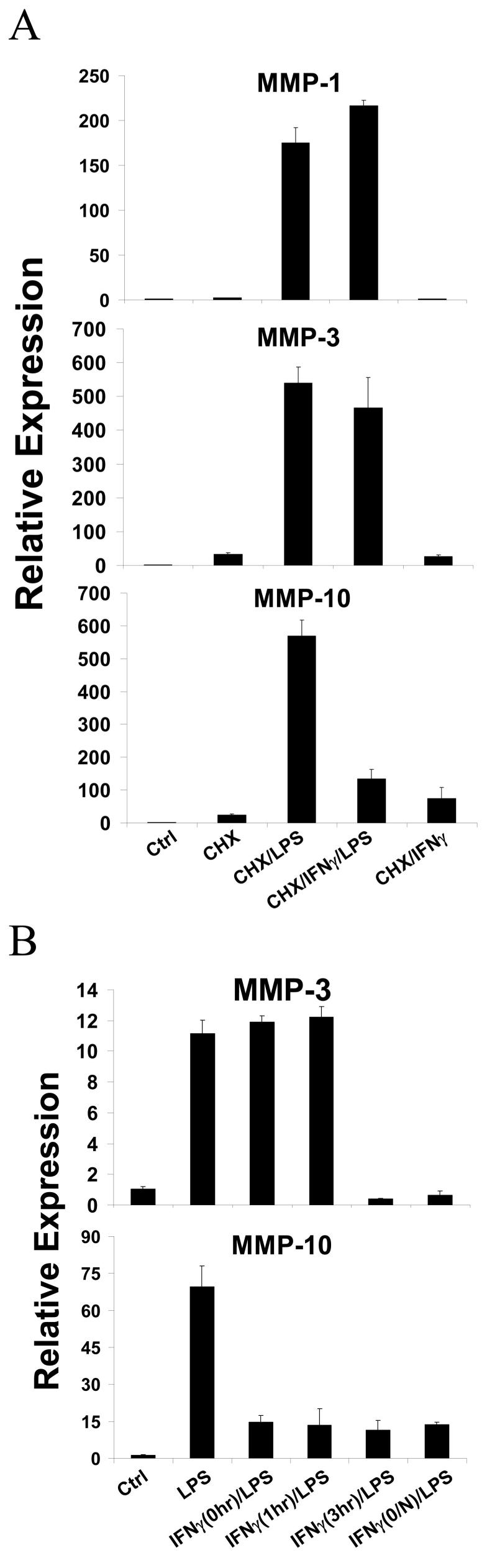
IFNγ inhibition of LPS-induced MMP expression is variably dependent on new protein synthesis. A, IFNγ pre-treated primary monocytes were stimulated with LPS for 3 hr in the absence or presence of cycloheximide (CHX, 20 [.proportional]g/ml, added 20 min before LPS or IFNγ). B, Primary monocytes were pre-treated with IFNγ for different time periods prior to adding LPS. mRNAs levels were measured using real time PCR and normalized relative to β-Actin. Results are expressed as mean ± SD of triplicate determinants. Results are shown as expression in stimulated monocytes relative to expression in unstimulated cells (ctrl), which was set at 1.
Inhibition of MMP-1 expression is mediated by IFNγ-induced ATF-3
The results suggesting that MMP-1 and MMP-3 expression is suppressed by an IFNγ-induced inhibitor prompted us to use microarray analysis to identify transcriptional repressors that were induced by IFNγ in TLR-stimulated monocytes. Interestingly, IFNγ superinduced the expression of the repressor ATF-3 in LPS-stimulated monocytes, and this result was confirmed by real time PCR in monocytes and THP-1 cells (Figure 5A and data not shown). The induction of ATF-3 expression by IFNγ was STAT1-dependent (Figure 5A), consistent with the notion that MMP-1 expression is suppressed by an IFNγ-induced STAT1-dependent protein. ATF-3 has been described as a TLR-induced feedback inhibitor that suppresses TLR-induced gene expression by binding to CRE/ATF promoter elements (59). Because CREs have been implicated in regulation of MMP-1 expression (48), we investigated the role of ATF-3 in regulation of MMP-1 expression in our system. EMSA assays detected an LPS-induced complex that bound to an oligonucleotide corresponding to a functional CRE sequence located upstream of the MMP-1 proximal promoter (−2017 relative to transcription start site) and was further superinduced by IFNγ (Figure 5B and 5C). DNA-binding by this IFNγ-superinduced complex was disrupted by ATF-3 antibodies but not by control antibodies (Figure 5C). Furthermore, treatment of monocytes with LPS plus IFNγ induced recruitment of ATF-3 to the endogenous MMP-1 promoter in the vicinity of the −2017 ATF site (Fig. 5D). Taken together with the real time PCR results (Figure 5A), these results suggest that IFNγ induces DNA binding of the ATF-3 transcriptional repressor that can potentially suppress MMP-1 expression. The role of ATF-3 in regulating MMP-1 expression was investigated by using lentivirus-mediated transduction of THP-1 monocytes to create two stable cell lines that express different shRNAs targeting ATF-3. In those ATF3-RNAi THP-1 cells, the synergistic LPS/IFNγ induction of ATF-3 was reduced to about 40% that of control (Figure 5E), and decreased ATF-3 expression resulted in reversal of IFNγ-mediated suppression of LPS-induced MMP-1 gene expression (Figure 5F). Attempts to test the effects of ATF-3 overexpression were not successful as increased expression of ATF-3 induced cell death (data not shown). These results implicate IFNγ-induced ATF-3 in mediating the suppressive effects of IFNγ on MMP expression.
FIGURE 5.

Inhibition of MMP-1 expression is mediated by IFNγ-induced ATF-3. A, THP-1 cells transduced with control DSRed2-shRNA- or STAT1-shRNA-encoding lentiviral particles were stimulated with LPS (3 hr) with or without IFNγ pre-treatment. B, Nuclear extracts from LPS-stimulated primary monocytes (with or without IFNγ pre-treatment) were subjected to EMSA with a radiolabeled CRE oligonucleotide probe. C, Nuclear extract samples were preincubated for 60 minutes at 4°C with control or ATF-3-specific antibodies as previously described (51) and analyzed by EMSA. D, Primary monocytes were stimulated with LPS (3 hr) with or without IFNγ pre-treatment. Chromatin was immunoprecipitated using control (IgG) or ATF-3 antibodies and analyzed by PCR. E and F, THP-1 cells transduced with control DSRed2-shRNA- or two different (labeled #1 and #2) ATF3-shRNA-encoding lentiviral particles were stimulated with LPS (3 hr) with or without IFNγ pre-treatment and mRNA levels were analyzed using real time PCR.
IFNγ regulates AP-1 and Fra-1 DNA binding
The AP-1 element located between base pairs −72 and −66 of the MMP-1 promoter is crucial for MMP-1 gene expression (41,55,60-62) and we and others have shown that IFNγ suppresses AP-1-dependent gene expression. Thus, despite the negative data on the role of GSK-3 (Figure 2), we further investigated the effects of IFNγ on LPS-induced AP-1 proteins. EMSA experiments using the MMP-1 promoter AP-1 site showed an LPS-induced DNA-binding complex (Figure 6A). Interestingly, LPS-induced binding to the AP-1 oligonucleotide was suppressed by IFNγ (Figure 6A), suggesting suppression of AP-1 proteins as a complementary mechanism by which IFNγ suppresses MMP-1 expression. Supershift experiments showed that the LPS-induced complex was reactive with Fra-1 antibodies, but not with Fos or Jun antibodies (Figure 6B). Because the MMP-1 promoter-derived AP-1 oligonucleotide contains an overlapping Ets site, we also investigated whether the LPS-induced complexes contained Ets proteins, but supershift experiments with PEA-3, PU.1, ETS-1, ETS-2 and ERG 1/2/3 showed no reactivity (Figure 6B and data not shown). These results suggested a role for Fra-1 in LPS and IFNγ regulation of MMP-1 expression, and this was further investigated using RNA interference and overexpression of Fra-1. Indeed, knock down of Fra-1 expression resulted in diminished LPS-induced MMP-1 expression (Figure 6C), and conversely overexpression of Fra-1 increased MMP-1 expression (Figure 6D), thus implicating Fra-1 in induction of MMP-1 expression by LPS. We did not detect regulation of Fra-1 expression by IFNγ (data not shown), but, interestingly, PGE2 which suppressed MMP-1 expression in monocytes (Figure 7A) also suppressed Fra-1 expression (Figure 7B). EMSA using the MMP-1 promoter AP-1 site demonstrated that in the presence of PGE2, the composition of the LPS-induced DNA binding complex was altered such that the complex was no longer shifted by Fra-1 antibodies (Figure 7C). These results support the notion that PGE2 inhibits LPS-induced MMP-1 gene expression by suppressing expression of Fra-1 that contributes to induction of MMP-1 expression through the proximal promoter AP-1 site. Overall, our findings support a role for Fra-1 in the positive and negative regulation of MMP-1 expression, and suggest that IFNγ inhibits MMP-1 transcription in part by suppressing DNA binding by Fra-1. Although IFNγ also suppresses other AP-1 proteins such as Fos and Jun (52), we were not able to link regulation of Fos and Jun with regulation of MMP-1 expression in our system.
FIGURE 6.
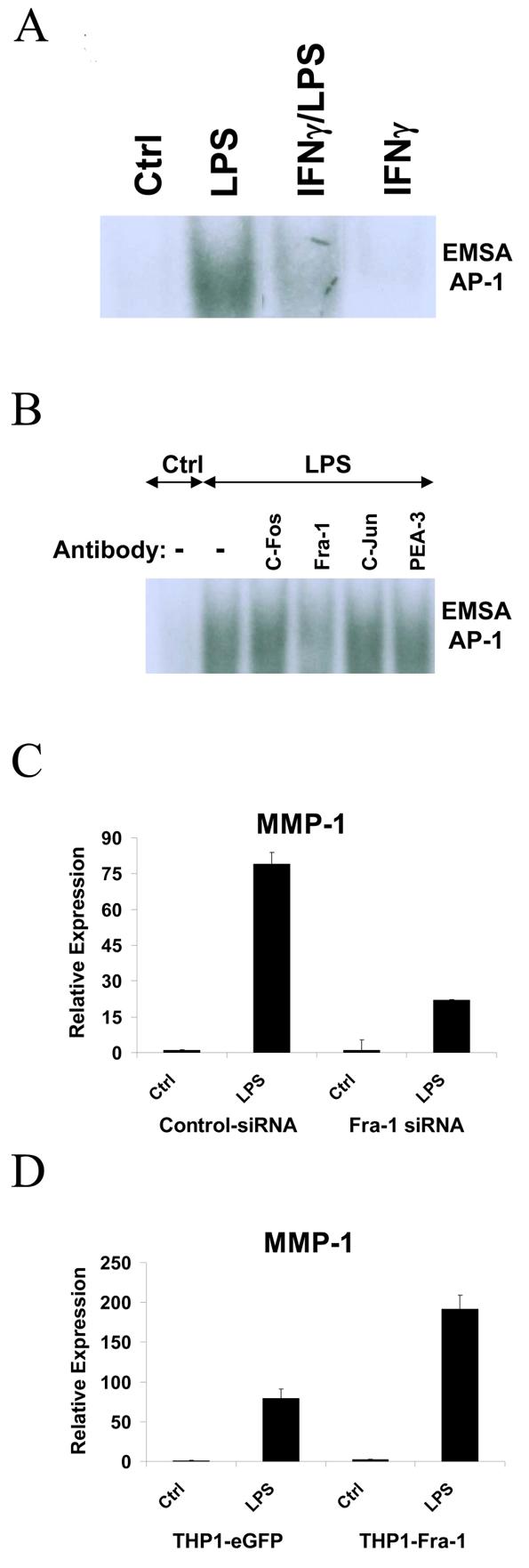
IFNγ inhibits LPS-induced AP-1 DNA-binding complex. A, Nuclear extracts from LPS stimulated primary monocytes (with or without IFNγ pre-treatment) were subjected to EMSA with a radiolabeled AP-1 oligonucleotide probe derived from the MMP-1 promoter. B, Nuclear extracts were preincubated for 60 minutes at 4°C with control or antibodies against various ETS and AP-1 proteins prior to EMSA. C, Primary monocytes transfected with control or Fra-1 siRNAs were stimulated with LPS. D, THP-1 cells transduced with eGFP- or Fra-1-encoding lentiviral particles were stimulated with LPS and RNA levels were measured using real time PCR.
FIGURE 7.
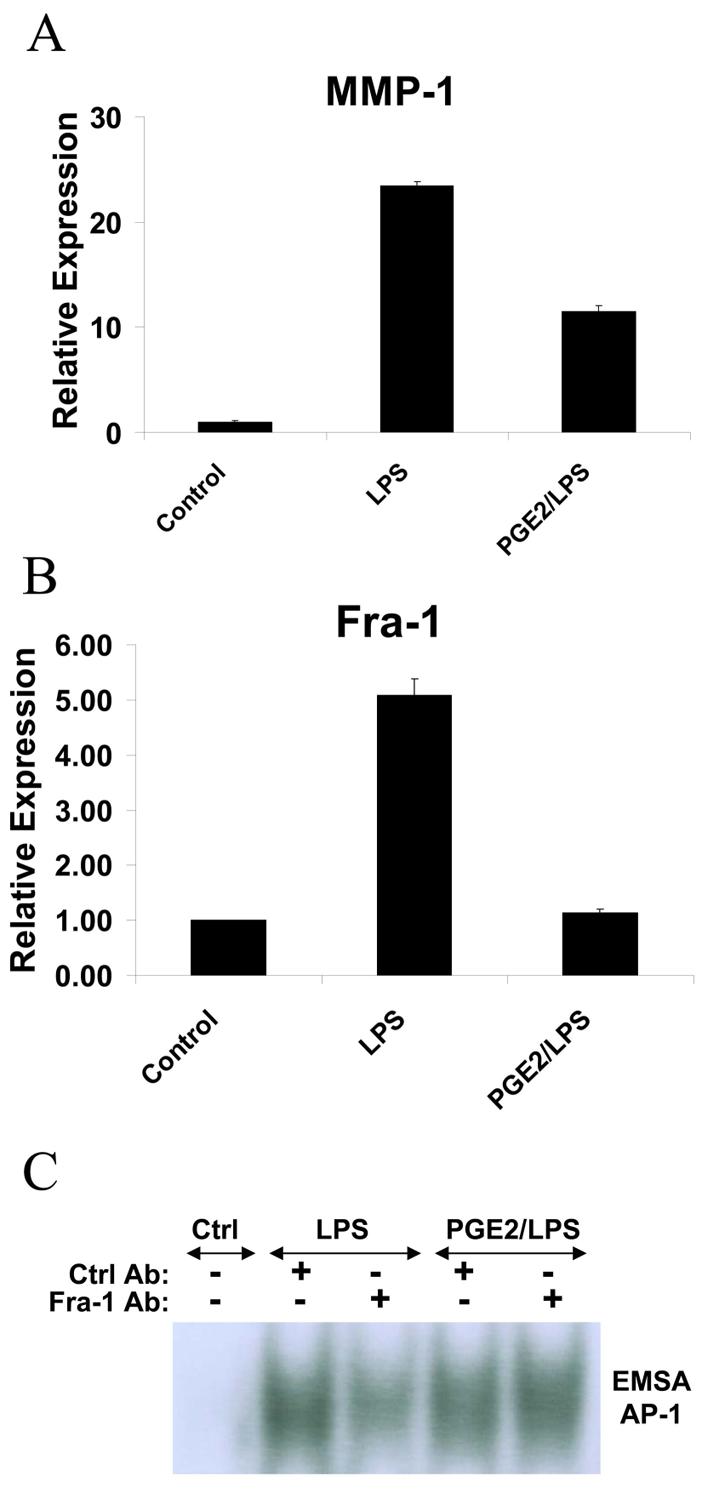
PGE2 inhibits LPS-induced MMP-1 expression and suppresses Fra-1 expression and DNA binding. A and B, Primary monocytes were stimulated with LPS in the absence or presence of PGE2 (1 μM added 20 min before LPS) and RNA was measured using real time PCR. C, Nuclear extracts from primary monocytes stimulated with LPS in the absence or presence of PGE2 were incubated with control or Fra-1 antibodies and subjected to EMSA with a radiolabeled AP-1 oligonucleotide probe.
Discussion
MMP expression needs to be tightly regulated during inflammation to allow necessary angiogenesis and tissue remodeling but to avoid excessive tissue damage. In this study we found that IFNγ, a potent macrophage activating cytokine, globally inhibits MMP expression at the same time that it synergistically enhances the production of proinflammatory cytokines. Thus, as the intensity of inflammation increases in response to IFNγ there is a built-in mechanism to restrain associated tissue damage. We have also identified two mechanisms by which IFNγ inhibits MMP expression, by inducing expression of the transcriptional repressor ATF-3 and simultaneously suppressing the function of AP-1 transcriptional activators.
The regulation of MMP gene expression has been extensively studied, although much of this work has utilized reporter gene approaches in cell lines and nonphysiological activators such as PMA (7,28-45). This previous work has revealed the importance of proximal promoter sequences, especially a composite AP-1/Ets site, in basal and inducible MMP-1 and MMP-3 expression. More recently, the importance of promoter and enhancer sequences outside MMP proximal promoters is becoming increasingly apparent, including upstream CRE sites (48,63). Surprisingly little is known about MMP regulation in primary cells, especially hematopoietic cells such as monocytes and macrophages. In addition, although important MMP promoter elements have been extensively analyzed, less is known about transcription factors that regulate MMP expression. Our work implicates ATF-3 in the regulation of MMP-1 expression. Because ATF-3 is induced by TLR stimulation alone, it participates in feedback inhibition of TLR-induced gene expression, as has been previously reported in other systems (59). In addition, superinduction of ATF-3 by IFNγ contributes to IFNγ-mediated suppression of MMP-1 gene expression. Transfection of primary monocytes or THP-1 cells with MMP promoter-driven reporter plasmids resulted in cell activation, presumably secondary to an innate immune response to exogenous DNA, and rapid extinction of reporter gene expression (H. Ho, unpublished data). Thus, the reporter gene approach was not informative in studying LPS and IFNγ regulation of MMP expression.
A consistent finding in multiple systems and cell types has been the important role of the proximal promoter AP-1 site in the expression of MMP-1 and MMP-3 (3,41,55,60-62). Therefore, given our previous findings that IFNγ can suppress AP-1-mediated IL-10 transcription by a GSK-3 dependent mechanism, we investigated a potential role for regulation of AP-1 proteins by IFNγ in suppression of MMP expression. LPS induced a Fra-1-containing complex that bound to the MMP-1 promoter AP-1 site, and the binding of this complex was suppressed by IFNγ. Fra-1 has been previously implicated in promoting MMP expression (64,65), although its regulation by LPS and IFNγ has not been previously appreciated. We confirmed the importance of Fra-1 in MMP-1 gene expression using both knock down and overexpression approaches and implicated Fra-1 in mediating PGE2 regulation of MMP-1 expression. In contrast to PGE2, IFNγ did not inhibit Fra-1 expression, suggesting that IFNγ may suppress DNA binding of Fra-1 by post-translational modifications or indirectly by regulating other members of the AP-1 family. Overall, the data show that IFNγ regulated MMP-1 expression by at least two mechanisms, induction of ATF-3 and suppression of DNA binding by a Fra-1-containing AP-1 complex. Of the MMPs studied whose expression was inhibited by IFNγ (Figure 1), all contain AP-1 sites with the exception of MMP-2 (66). Thus, although suppression of AP-1 may contribute to diminished expression of several MMPs, it can not explain inhibition of MMP-2 by IFNγ. Interestingly, ATF-3 has been implicated in negative regulation of MMP-2 (59), suggesting a role for ATF-3 in regulation of MMP gene expression beyond the regulation of MMP-1 expression that was investigated in this study. Collectively, our data and previous reports suggest that IFNγ can suppress MMP expression by different mechanisms. This notion is further supported by our results suggesting that MMP-10 expression was suppressed by IFNγ by a mechanism that differed from suppression of MMP-1 because inhibition of MMP-10 did not require new protein synthesis, and IFNγ inhibits MMP-9 expression by modifying chromatin and transcriptional coactivator recruitment (28,30,35,67,68). These findings highlight that IFNγ suppresses MMP expression by several mechanisms and provide insights into how IFNγ downregulates gene expression.
Prior studies from several laboratories including ours have described antagonistic functions for STAT1 and STAT3 in the regulation of cell growth/survival and inflammatory gene expression (58,69). It appears that this opposition of STAT1 and STAT3 also extends to the regulation of MMP expression. STAT3 has been shown to be an important and strong inducer of MMPs such as MMP-1 and MMP-10 (70,71), and was found to bind directly to a STAT-Binding element (SBE) located between base pairs -53 and -45 upstream in the MMP-1 proximal promoter after stimulation with EGF (71). We have shown that inhibition of MMP-1 expression by IFNγ is dependent on STAT1, but likely occurs indirectly via regulation of ATF-3 and AP-1 proteins. Consistent with these results, we were not able to detect binding of STAT1 to the MMP-1 promoter AP-1 site or the previously described SBE that binds STAT3 (H. Ho, unpublished observations), suggesting that STAT1 does not directly compete with STAT3 for binding to the MMP-1 promoter. On the other hand, it is possible that STAT-1 inhibits MMP-10 expression by a direct mechanism, as has been shown for IFNγ suppression of the Perlecan gene (72), because inhibition of MMP-10 expression by IFNγ was STAT1-dependent and rapid and independent of new protein synthesis.
In summary, our results demonstrate that IFNγ differentially regulates gene expression in primary human monoctes and macrophages by augmenting the production of inflammatory cytokines such as IL-6 and TNFα, but at the same time globally suppressing MMP production. The suppression of MMPs by IFNγ may represent a self-protective mechanism to limit undesirable damage to tissues during intense immune and inflammatory reactions that are required to eradicate infectious pathogens. The results also reveal that IFNγ modulates MMP expression by regulating the expression and function of ATF-3, Fra-1 and potentially other AP-1 proteins. Understanding the mechanisms by which IFNγ regulates these genes and transcription factors may yield useful insights into formulating future approaches to enhancing immune clearance of invading pathogens and yet limiting damage to tissues and organ systems.
Acknowledgments
We thank Janice Chen for helpful discussions, Kyung-Kyun Park-Min for review of the manuscript, and Inez Rogatsky and Barbara Nikolajczyk for advice concerning ChIP assays.
1 This work was supported by grants from the NIH (to L.B.I.).
References
- 1.GROSS J, LAPIERE CM. Collagenolytic activity in amphibian tissues: a tissue culture assay. Proc.Natl.Acad.Sci.U.S.A. 1962;48:1014–1022. doi: 10.1073/pnas.48.6.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woessner JF., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5:2145–2154. [PubMed] [Google Scholar]
- 3.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol.Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 4.Sounni NE, Noel A. Membrane type-matrix metalloproteinases and tumor progression. Biochimie. 2005;87:329–342. doi: 10.1016/j.biochi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Lemaitre V, D'Armiento J. Matrix metalloproteinases in development and disease. Birth Defects Res.C.Embryo.Today. 2006;78:1–10. doi: 10.1002/bdrc.20065. [DOI] [PubMed] [Google Scholar]
- 6.Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4:157–164. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huntington JT, Shields JM, Der CJ, Wyatt CA, Benbow U, Slingluff CL, Jr., Brinckerhoff CE. Overexpression of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive melanoma cells: role of BRAF mutation and fibroblast growth factor signaling. J.Biol.Chem. 2004;279:33168–33176. doi: 10.1074/jbc.M405102200. [DOI] [PubMed] [Google Scholar]
- 8.McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289:1202–1206. doi: 10.1126/science.289.5482.1202. [DOI] [PubMed] [Google Scholar]
- 9.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286:113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 10.Powell WC, Fingleton B, Wilson CL, Boothby M, Matrisian LM. The metalloproteinase matrilysin proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr.Biol. 1999;9:1441–1447. doi: 10.1016/s0960-9822(00)80113-x. [DOI] [PubMed] [Google Scholar]
- 11.Hartzell W, Shapiro SD. Macrophage elastase prevents Gemella morbillorum infection and improves outcome following murine bone marrow transplantation. Chest. 1999;116:31S–32S. doi: 10.1378/chest.116.suppl_1.31s-a. [DOI] [PubMed] [Google Scholar]
- 12.Lopez-Boado YS, Wilson CL, Hooper LV, Gordon JI, Hultgren SJ, Parks WC. Bacterial exposure induces and activates matrilysin in mucosal epithelial cells. J.Cell Biol. 2000;148:1305–1315. doi: 10.1083/jcb.148.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corry DB, Rishi K, Kanellis J, Kiss A, Song Lz LZ, Xu J, Feng L, Werb Z, Kheradmand F. Decreased allergic lung inflammatory cell egression and increased susceptibility to asphyxiation in MMP2-deficiency. Nat.Immunol. 2002;3:347–353. doi: 10.1038/ni773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 15.Haro H, Crawford HC, Fingleton B, MacDougall JR, Shinomiya K, Spengler DM, Matrisian LM. Matrix metalloproteinase-3-dependent generation of a macrophage chemoattractant in a model of herniated disc resorption. J.Clin.Invest. 2000;105:133–141. doi: 10.1172/JCI7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, Senior RM, Werb Z. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 17.Haro H, Crawford HC, Fingleton B, Shinomiya K, Spengler DM, Matrisian LM. Matrix metalloproteinase-7-dependent release of tumor necrosis factor-alpha in a model of herniated disc resorption. J.Clin.Invest. 2000;105:143–150. doi: 10.1172/JCI7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat.Rev.Immunol. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 19.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat.Rev.Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 20.Schroen DJ, Brinckerhoff CE. Nuclear hormone receptors inhibit matrix metalloproteinase (MMP) gene expression through diverse mechanisms. Gene Expr. 1996;6:197–207. [PMC free article] [PubMed] [Google Scholar]
- 21.Szekanecz Z, Koch AE. Macrophages and their products in rheumatoid arthritis. Curr.Opin.Rheumatol. 2007;19:289–295. doi: 10.1097/BOR.0b013e32805e87ae. [DOI] [PubMed] [Google Scholar]
- 22.Stoll G, Bendszus M. Inflammation and atherosclerosis: novel insights into plaque formation and destabilization. Stroke. 2006;37:1923–1932. doi: 10.1161/01.STR.0000226901.34927.10. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi K, Takeya M, Sakashita N. Multifunctional roles of macrophages in the development and progression of atherosclerosis in humans and experimental animals. Med.Electron Microsc. 2002;35:179–203. doi: 10.1007/s007950200023. [DOI] [PubMed] [Google Scholar]
- 24.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr.Vasc.Pharmacol. 2005;3:63–68. doi: 10.2174/1570161052773861. [DOI] [PubMed] [Google Scholar]
- 25.Libby P. Changing concepts of atherogenesis. J.Intern.Med. 2000;247:349–358. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 26.Shah PK, Falk E, Badimon JJ, Fernandez-Ortiz A, Mailhac A, Villareal-Levy G, Fallon JT, Regnstrom J, Fuster V. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92:1565–1569. [PubMed] [Google Scholar]
- 27.Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, Libby P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–2509. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Wang L, Buttice G, Sengupta PK, Smith BD. Major histocompatibility class II transactivator (CIITA) mediates repression of collagen (COL1A2) transcription by interferon gamma (IFN-gamma) J.Biol.Chem. 2004;279:41319–41332. doi: 10.1074/jbc.M404174200. [DOI] [PubMed] [Google Scholar]
- 29.Lewis M, Amento EP, Unemori EN. Transcriptional inhibition of stromelysin by interferon-gamma in normal human fibroblasts is mediated by the AP-1 domain. J.Cell Biochem. 1999;72:373–386. doi: 10.1002/(sici)1097-4644(19990301)72:3<373::aid-jcb7>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 30.Ma Z, Chang MJ, Shah RC, Benveniste EN. Interferon-{gamma}-activated STAT-1{alpha} suppresses MMP-9 gene transcription by sequestration of the coactivators CBP/p300. J.Leukoc.Biol. 2005;78:515–523. doi: 10.1189/jlb.0205112. [DOI] [PubMed] [Google Scholar]
- 31.Yokota H, Goldring MB, Sun HB. CITED2-mediated regulation of MMP-1 and MMP-13 in human chondrocytes under flow shear. J.Biol.Chem. 2003;278:47275–47280. doi: 10.1074/jbc.M304652200. [DOI] [PubMed] [Google Scholar]
- 32.Andrews HJ, Bunning RA, Dinarello CA, Russell RG. Modulation of human chondrocyte metabolism by recombinant human interferon gamma: in-vitro effects on basal and IL-1-stimulated proteinase production, cartilage degradation and DNA synthesis. Biochim.Biophys.Acta. 1989;1012:128–134. doi: 10.1016/0167-4889(89)90085-2. [DOI] [PubMed] [Google Scholar]
- 33.Unemori EN, Bair MJ, Bauer EA, Amento EP. Stromelysin expression regulates collagenase activation in human fibroblasts. Dissociable control of two metalloproteinases by interferon-gamma. J.Biol.Chem. 1991;266:23477–23482. [PubMed] [Google Scholar]
- 34.Varga J, Yufit T, Brown RR. Inhibition of collagenase and stromelysin gene expression by interferon-gamma in human dermal fibroblasts is mediated in part via induction of tryptophan degradation. J.Clin.Invest. 1995;96:475–481. doi: 10.1172/JCI118058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nozell S, Ma Z, Wilson C, Shah R, Benveniste EN. Class II major histocompatibility complex transactivator (CIITA) inhibits matrix metalloproteinase-9 gene expression. J.Biol.Chem. 2004;279:38577–38589. doi: 10.1074/jbc.M403738200. [DOI] [PubMed] [Google Scholar]
- 36.Fenrick R, Wang L, Nip J, Amann JM, Rooney RJ, Walker-Daniels J, Crawford HC, Hulboy DL, Kinch MS, Matrisian LM, Hiebert SW. TEL, a putative tumor suppressor, modulates cell growth and cell morphology of ras-transformed cells while repressing the transcription of stromelysin-1. Mol.Cell Biol. 2000;20:5828–5839. doi: 10.1128/mcb.20.16.5828-5839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barchowsky A, Frleta D, Vincenti MP. Integration of the NF-kappaB and mitogen-activated protein kinase/AP-1 pathways at the collagenase-1 promoter: divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine. 2000;12:1469–1479. doi: 10.1006/cyto.2000.0743. [DOI] [PubMed] [Google Scholar]
- 38.Quinones S, Saus J, Otani Y, Harris ED, Jr., Kurkinen M. Transcriptional regulation of human stromelysin. J.Biol.Chem. 1989;264:8339–8344. [PubMed] [Google Scholar]
- 39.Buttice G, Kurkinen M. A polyomavirus enhancer A-binding protein-3 site and Ets-2 protein have a major role in the 12-O-tetradecanoylphorbol-13-acetate response of the human stromelysin gene. J.Biol.Chem. 1993;268:7196–7204. [PubMed] [Google Scholar]
- 40.Buttice G, Quinones S, Kurkinen M. The AP-1 site is required for basal expression but is not necessary for TPA-response of the human stromelysin gene. Nucleic Acids Res. 1991;19:3723–3731. doi: 10.1093/nar/19.13.3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bidder M, Loewy AP, Latifi T, Newberry EP, Ferguson G, Willis DM, Towler DA. Ets domain transcription factor PE1 suppresses human interstitial collagenase promoter activity by antagonizing protein-DNA interactions at a critical AP1 element. Biochemistry. 2000;39:8917–8928. doi: 10.1021/bi000343+. [DOI] [PubMed] [Google Scholar]
- 42.Jinnin M, Ihn H, Mimura Y, Asano Y, Yamane K, Tamaki K. Matrix metalloproteinase-1 up-regulation by hepatocyte growth factor in human dermal fibroblasts via ERK signaling pathway involves Ets1 and Fli1. Nucleic Acids Res. 2005;33:3540–3549. doi: 10.1093/nar/gki648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jayaraman G, Srinivas R, Duggan C, Ferreira E, Swaminathan S, Somasundaram K, Williams J, Hauser C, Kurkinen M, Dhar R, Weitzman S, Buttice G, Thimmapaya B. p300/cAMP-responsive element-binding protein interactions with ets-1 and ets-2 in the transcriptional activation of the human stromelysin promoter. J.Biol.Chem. 1999;274:17342–17352. doi: 10.1074/jbc.274.24.17342. [DOI] [PubMed] [Google Scholar]
- 44.Nakajima H, Hiyama Y, Tsukada W, Warabi H, Uchida S, Hirose S. Effects of interferon gamma on cultured synovial cells from patients with rheumatoid arthritis: inhibition of cell growth, prostaglandin E2, and collagenase release. Ann.Rheum.Dis. 1990;49:512–516. doi: 10.1136/ard.49.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroen DJ, Chen JD, Vincenti MP, Brinckerhoff CE. The nuclear receptor corepressor SMRT inhibits interstitial collagenase (MMP-1) transcription through an HRE-independent mechanism. Biochem.Biophys.Res.Commun. 1997;237:52–58. doi: 10.1006/bbrc.1997.7073. [DOI] [PubMed] [Google Scholar]
- 46.Khan KM, Howe LR, Falcone DJ. Extracellular matrix-induced cyclooxygenase-2 regulates macrophage proteinase expression. J.Biol.Chem. 2004;279:22039–22046. doi: 10.1074/jbc.M312735200. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, McCluskey K, Fujii K, Wahl LM. Differential regulation of monocyte matrix metalloproteinase and TIMP-1 production by TNF-alpha, granulocyte-macrophage CSF, and IL-1 beta through prostaglandin-dependent and -independent mechanisms. J.Immunol. 1998;161:3071–3076. [PubMed] [Google Scholar]
- 48.Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM. Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J.Immunol. 2003;170:6244–6249. doi: 10.4049/jimmunol.170.12.6244. [DOI] [PubMed] [Google Scholar]
- 49.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Zhang M, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat.Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 50.Tassiulas I, Hu X, Ho H, Kashyap Y, Paik P, Hu Y, Lowell CA, Ivashkiv LB. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat.Immunol. 2004;5:1181–1189. doi: 10.1038/ni1126. [DOI] [PubMed] [Google Scholar]
- 51.Sengupta TK, Chen A, Zhong Z, Darnell JE, Jr., Ivashkiv LB. Activation of monocyte effector genes and STAT family transcription factors by inflammatory synovial fluid is independent of interferon gamma. J.Exp.Med. 1995;181:1015–1025. doi: 10.1084/jem.181.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 53.Angel P, Baumann I, Stein B, Delius H, Rahmsdorf HJ, Herrlich P. 12-O-tetradecanoyl-phorbol-13-acetate induction of the human collagenase gene is mediated by an inducible enhancer element located in the 5′-flanking region. Mol.Cell Biol. 1987;7:2256–2266. doi: 10.1128/mcb.7.6.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Westermarck J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999;13:781–792. [PubMed] [Google Scholar]
- 55.Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 1997;15:519–526. doi: 10.1016/s0945-053x(97)90026-3. [DOI] [PubMed] [Google Scholar]
- 56.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat.Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 58.Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J.Biol.Chem. 2006;281:14111–14118. doi: 10.1074/jbc.M511797200. [DOI] [PubMed] [Google Scholar]
- 59.Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Kennedy K, Hai T, Bolouri H, Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 60.Westermarck J, Seth A, Kahari VM. Differential regulation of interstitial collagenase (MMP-1) gene expression by ETS transcription factors. Oncogene. 1997;14:2651–2660. doi: 10.1038/sj.onc.1201111. [DOI] [PubMed] [Google Scholar]
- 61.Oikawa T. ETS transcription factors: possible targets for cancer therapy. Cancer Sci. 2004;95:626–633. doi: 10.1111/j.1349-7006.2004.tb03320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schroen DJ, Brinckerhoff CE. Inhibition of rabbit collagenase (matrix metalloproteinase-1; MMP-1) transcription by retinoid receptors: evidence for binding of RARs/RXRs to the -77 AP-1 site through interactions with c-Jun. J.Cell Physiol. 1996;169:320–332. doi: 10.1002/(SICI)1097-4652(199611)169:2<320::AID-JCP11>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 63.Benbow U, Rutter JL, Lowrey CH, Brinckerhoff CE. Transcriptional repression of the human collagenase-1 (MMP-1) gene in MDA231 breast cancer cells by all-trans-retinoic acid requires distal regions of the promoter. Br.J.Cancer. 1999;79:221–228. doi: 10.1038/sj.bjc.6690037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tower GB, Coon CI, Belguise K, Chalbos D, Brinckerhoff CE. Fra-1 targets the AP-1 site/2G single nucleotide polymorphism (ETS site) in the MMP-1 promoter. Eur.J.Biochem. 2003;270:4216–4225. doi: 10.1046/j.1432-1033.2003.03821.x. [DOI] [PubMed] [Google Scholar]
- 65.Newberry EP, Willis D, Latifi T, Boudreaux JM, Towler DA. Fibroblast growth factor receptor signaling activates the human interstitial collagenase promoter via the bipartite Ets-AP1 element. Mol.Endocrinol. 1997;11:1129–1144. doi: 10.1210/mend.11.8.9958. [DOI] [PubMed] [Google Scholar]
- 66.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J.Cell Physiol. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- 67.Shapiro SD, Campbell EJ, Kobayashi DK, Welgus HG. Immune modulation of metalloproteinase production in human macrophages. Selective pretranslational suppression of interstitial collagenase and stromelysin biosynthesis by interferon-gamma. J.Clin.Invest. 1990;86:1204–1210. doi: 10.1172/JCI114826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nguyen J, Knapnougel P, Lesavre P, Bauvois B. Inhibition of matrix metalloproteinase-9 by interferons and TGF-beta1 through distinct signalings accounts for reduced monocyte invasiveness. FEBS Lett. 2005;579:5487–5493. doi: 10.1016/j.febslet.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 69.Hu X, Park-Min KH, Ho HH, Ivashkiv LB. IFN-gamma-primed macrophages exhibit increased CCR2-dependent migration and altered IFN-gamma responses mediated by Stat1. J.Immunol. 2005;175:3637–3647. doi: 10.4049/jimmunol.175.6.3637. [DOI] [PubMed] [Google Scholar]
- 70.Tsareva SA, Moriggl R, Corvinus FM, Wiederanders B, Schutz A, Kovacic B, Friedrich K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia. 2007;9:279–291. doi: 10.1593/neo.06820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Itoh M, Murata T, Suzuki T, Shindoh M, Nakajima K, Imai K, Yoshida K. Requirement of STAT3 activation for maximal collagenase-1 (MMP-1) induction by epidermal growth factor and malignant characteristics in T24 bladder cancer cells. Oncogene. 2006;25:1195–1204. doi: 10.1038/sj.onc.1209149. [DOI] [PubMed] [Google Scholar]
- 72.Sharma B, Iozzo RV. Transcriptional silencing of perlecan gene expression by interferon-gamma. J.Biol.Chem. 1998;273:4642–4646. doi: 10.1074/jbc.273.8.4642. [DOI] [PubMed] [Google Scholar]