

Abstract
Purpose
Cell cycle progression is governed by the coordinated activities of kinases, phosphatases, and the ubiquitin system. The entire complement of ubiquitin pathway components that mediate this process in retinal pigment epithelial (RPE) cells remains to be identified. This study was undertaken to determine whether the human ubiquitin-conjugating enzyme, UBE2E3, is essential for RPE cell proliferation.
Methods
UBE2E3 expression and localization in telomerase-immortalized, human RPE cells was determined with a UBE2E3-specific antibody. The necessity for UBE2E3 in RPE proliferation was determined using small interfering (si)RNA to target the expression of the enzyme. Cell counts and immunolabeling for the proliferation marker Ki-67 and the cyclin-dependent kinase inhibitor p27Kip1 were performed to assess the consequences of UBE2E3 depletion. A mouse strain harboring a disrupted allele of UbcM2 (the mouse counterpart of UBE2E3) with the coding sequence for β-galactosidase was used to track the developmental expression of the enzyme in murine RPE cells.
Results
UBE2E3 localized in the nucleus of the immortalized RPE cells. Depletion of the enzyme by siRNA resulted in a cell-cycle exit accompanied by a loss of Ki-67, an increase in p27Kip1, and a doubling in cell area. Rescue experiments confirmed the specificity of the RNA interference. In vivo, UbcM2 was transcriptionally downregulated during RPE development in the mouse.
Conclusions
UBE2E3 is essential for the proliferation of RPE-1 cells and is downregulated during RPE layer maturation in the developing mouse eye. These findings indicate that UBE2E3 is a major enzyme in modulating the balance between RPE cell proliferation and differentiation.
Age-related macular degeneration (AMD) is a major cause of vision loss in the elderly. Morphologic changes and disruptions to numerous cell types in the macular region, including retinal pigment epithelial (RPE) cells, contribute to the onset and progression of AMD (reviewed in Ref. 1). Loss of RPE function is widely held to be an early, pathogenic event in AMD. Thus, a comprehensive understanding of the factors that modulate RPE proliferation and differentiation holds promise for the development of novel AMD treatments.
A key regulator of the balance between RPE proliferation and differentiation is p27Kip1. It is a member of the Cip/Kip family of proteins that inhibit the activity of cyclin-dependent kinases (cdks). p27Kip1 has numerous functions,2-7 but is best characterized as an inhibitor of the G1/S transition (reviewed in Ref. 8). In addition, p27Kip1 protein levels increase as RPE cells transition from a proliferative state toward a terminally differentiated state. Studies in mice nullizygous for the p27Kip1gene have revealed that p27Kip1 plays critical roles in the timing and fidelity of RPE differentiation and in the structural integrity of the RPE monolayer.9-11
Ubiquitin-mediated degradation is a primary mechanism by which p27Kip1 levels are regulated. Ubiquitin is a highly conserved, 76-amino-acid polypeptide that is posttranslationally attached to target proteins by the coordinated actions of a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin protein ligase (E3). p27Kip1 degradation by ubiquitin-mediated proteolysis is complex (reviewed in Ref. 12) This complexity is underscored by biochemical and in vivo evidence demonstrating that multiple E2–E3 combinations can mediate p27Kip1 ubiquitylation (e.g., Refs. 2,13-17). Collectively, most of this evidence supports the notion that different E2–E3 complexes function to ubiquitylate p27Kip1 at different times in G1 or as cells transition from G0 to G1. However, the ubiquitin machinery that regulates p27Kip1 in RPE cells has not been comprehensively defined. The identification of this machinery is central to understanding the balance between RPE proliferation and differentiation.
Here, we report a novel link between p27Kip1 and an E2, called UBE2E3, in RPE cells. UBE2E3 is highly conserved among metazoans; the human and mouse counterparts are 100% identical.18 Overexpression of the enzyme reversibly arrests cells in G1, and this effect requires the catalytic activity of the enzyme.19 Furthermore, genetic linkage has implicated loci in the vicinity of the UBE2E3 gene on chromosome 2 to AMD.20 None of these studies, however, demonstrate a requirement for UBE2E3 in cell cycle progression. In the present study, we investigated whether the enzyme is necessary for RPE cell proliferation. Our model system was telomerase-immortalized RPE cells (RPE-1). These are human diploid, non-transformed, contact-inhibited, epithelial cells that display many of the characteristics of parental RPE cells, including expression of RPE-specific proteins and melanin.21 Using siRNA-based knockdown, we found that depletion of UBE2E3 causes a cell cycle exit characterized by a loss of the proliferation marker Ki-67, a dramatic increase in p27Kip1 levels, and an increase in cell size. We corroborated these tissue culture findings by analyzing murine UBE2E3 (called UbcM2) expression in the developing mouse RPE using a heterologous reporter (i.e., β-galactosidase) driven by the endogenous UbcM2 promoter. This mouse model revealed that UbcM2 expression is transcriptionally downregulated during RPE maturation. These findings represent the first report of a specific E2 required for RPE cell proliferation and implicate the enzyme in the developmental transition of RPE cells from a state of proliferation to one of differentiation.
Materials and Methods
Cell Culture
RPE-1 cells were purchased from ATCC (Manassas, VA; CRL-4000) and cultured in Dulbecco's modified Eagle's medium (DMEM, 1 g/L glucose; Mediatech, Manassas, VA) supplemented with 10% heat-inactivated fetal calf serum (FCS; Mediatech) + 100 U/mL penicillin + 100 μg/mL streptomycin (Invitrogen-Gibco, Grand Island, NY). Cells were cultured in a humidified incubator maintained at 37°C and 5% CO2.
Transfections
UBE2E3-specific small-interfering (si)RNA and control siRNA (siCON; Dharmacon; Lafayette, CO) were combined with Interferin (ISC Bio-express, Kaysville, UT) according to the manufacturer's instructions before being added to RPE-1 cells. The final concentration of siRNA added was ≤10 nM. Samples were harvested 24, 48, and 72 hours after transfection, unless otherwise indicated. The UBE2E3-specific siRNA is catalog no. MU-008845 against accession number NM_006357 and siCON is catalog no. D-001220 (Dharmacon). The sense sequences of the UBE2E3 duplexes were: duplex 1, 5′-GCAUAGCCACUCAGUAUUUUU-3′; duplex 2, 5′-GCUAAGUUAUCCACUAGUGUU-3′; duplex 3, 5′-AUAUGAAGGUGGUGUGUUUUU-3′; and duplex 4, 5′-GGAGCUAGCUGAAAUAACCUU-3′. For the rescue experiments, the cells were transfected (Lipofectamine 2000; Invitrogen, Carlsbad, CA) with either a plasmid coding for a fusion of the red fluorescent protein (RFP) to histone H2B (RFP-H2B; also called Mock) or a plasmid encoding an siRNA-impervious UBE2E3 cDNA. The following day, the cells were treated with siRNA. Three days later, the cells were processed for anti-p27Kip1 immunofluorescence. The siRNA-impervious UBE2E3 cDNA was generated by changing the wobble position of each codon of UBE2E3 within the nucleotide stretches complementary to the siRNA oligos. A site-directed mutagenesis kit was used to introduce the mutations (Quickchange; Stratagene, La Jolla, Ca). The backbone plasmid used for transfections is called pRK7 and was kindly provided by Ian Macara (University of Virginia, Charlottesville, VA).
Immunofluorescence and Cell Size Measurement
Immunolabeling was performed on cells that were fixed in 3.7% formaldehyde in phosphate-buffered saline (PBS), permeabilized with methanol, and blocked in 3% bovine serum albumin (BSA)/PBS for an hour at room temperature. Anti-UBE2E3 (α-2E3) was diluted 1:500 and detected with an Alexa488-labeled, goat anti-rabbit secondary antibody diluted 1:4000 in 3% BSA/PBS. α-p27Kip1 (BD Transduction Laboratories; San Jose, CA) was diluted at 1:1000 and detected with an Alexa546-labeled, goat anti-mouse secondary antibody. Anti-Ki-67 (Abcam, Cambridge, MA) was diluted 1:1000 and detected with an Alexa488-labeled, goat anti-mouse secondary antibody. Secondary antibodies were purchased from Invitrogen-Molecular Probes (Eugene, OR). DNA was counterstained with 0.01 μg/mL 4′-6-diamidino-2-phenylindole (DAPI) in PBS. The cells were examined with an inverted, epifluorescence microscope (TE2000; Nikon, Tokyo, Japan) equipped with a 20× objective (NA 0.45). For quantitative immunofluorescence analysis in the rescue experiments, all images were acquired and processed with identical settings, and cells with a saturated fluorescence signal were excluded. Commercial software (Openlab; Improvision, Inc., Coventry, UK) was used to create masks with DAPI-stained nuclei that defined the nucleus of each cell, and the masks were superimposed on the corresponding anti-p27Kip1 images to measure the intensity of the nuclear p27Kip1 immunosignal. To graph the data, the percentage of relative pixel intensity was measured by the following formula (PIcell – PImin)/(PImax – PImin) × 100. PIcell is the p27Kip1 cell immunosignal pixel intensity for a given cell; PImin and PImax are the minimum and maximum p27Kip1 immunosignal pixel intensities, respectively, measured from the 1000 cells analyzed for a given experiment.
For the cell area measurements, images of live cells were captured with a 20× phase-contrast objective (NA 0.45) and imported into ImageJ software (available by ftp at zippy.nimh.nih.gov/ or at http://rsb.info.nih.gov/nih-image; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD). The areas were determined by outlining the edges of the cell with the lasso tool.
Western Blot Analysis
Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), electrotransferred to nitrocellulose, and blocked in 5% nonfat milk/TBST (0.1% Tween-20 in Tris-buffered saline). The following antibodies were used: (1) α-2E3 (affinity purified, rabbit polyclonal, diluted 1:100),22 (2) anti-glutathione S-transferase (GST, rabbit polyclonal diluted 1:1000; Bethyl, Montgomery, TX,), (3) anti-β-tubulin (mouse monoclonal, diluted 1:1000; Sigma-Aldrich, St. Louis, MO), and (4) anti-p27Kip1 (mouse monoclonal, 1:500; BD-Transduction Laboratories, San Jose, CA). Goat anti-rabbit and -mouse horseradish-peroxidase conjugated secondary antibodies were diluted to 1:20,000 (Jackson Laboratories, West Grove, PA), and enhanced chemiluminescence was performed according to the manufacturer's instructions (KPL, Gaithersburg, MD). Blots were exposed to x-ray film (Phenix Research Products, Chandler, NC) and processed using an automated film developer.
Flow Cytometry Analysis
RPE-1 cells (1.25 × 105/well) were seeded in six-well dishes, transfected with either siCON or UBE2E3-specific siRNA, and 2 days later harvested for anti-p27Kip1 immunolabeling and propidium iodide staining. Cells were collected, rinsed in PBS, fixed in 3.7% formaldehyde, and permeabilized with 0.25% Triton X-100. After premeabilization, the cells were pelleted by centrifugation, and resuspended in anti-p27Kip1 diluted 1:250. The anti-p27Kip1 monoclonal antibody was detected with an Alexa488-labeled, goat anti-mouse secondary antibody diluted at 1:1000. DNA was labeled with a propidium iodide solution (20 μg/mL in 0.1% Triton X-100/PBS+200 μg/mL RNase) at 37°C for 1 hour before being subjected to fluorescence-activated cell sorting (FACS) analysis (FACSCalibur; BD Biosciences, Franklin Lanes, NJ).
Proliferation Assay
RPE-1 cells (3.5 × 104/well) were seeded in 12-well dishes, treated with siRNA, and processed for cell counting. For each of the 3 days after transfection, media and detached cells were removed, and adherent cells were collected by trypsinization, pelleted at 1500 rpm for 5 minutes, resuspended in media, and counted with a hemacytometer. Error bars represent SD of data obtained from three independent experiments.
Reverse Transcription–Polymerase Chain Reaction
RPE-1 cells were transfected with siRNA and 2 days later were collected by trypsinization and washed with PBS, and RNA was isolated (RNAqueous; Ambion, Austin, TX) according to the manufacturer's instructions. Five micrograms of each RNA was used for first-strand synthesis (PowerScript; BD Biosciences) and oligo dT primers. Five percent of each first-strand synthesis reaction was subsequently used as the template for semiquantitative polymerase chain reaction (PCR). PCR products were subjected to electrophoresis through 1% agarose gels and stained with ethidium bromide. The sequences of the UBE2E3-specific primers were: 5′-GTGATAGGCAAAGGTCCGATGATG-3′ and 5′-AGTGATAGATTCTGGTGCGGAAAG-3′. The sequences of the importin-11-specific primers were: 5′-CTTTGTTACTACTCCTGTGAGCACCGA-3′ and 5′-CCCCTTTTTATGTACTGCATGTTC-3′. Templates were melted at 95°C for 45 seconds, primers annealed at 50°C for 45 seconds, and extensions performed at 72°C for 1 minute. The UBE2E3 PCR product resulted from 34 cycles and the importin-11 product from 32 cycles.
Mouse Model and β-Galactosidase Staining
All animal studies were performed in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the guidelines of The University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee (IACUC). The retinas were dissected from 6-week-old C57BL6 mice and incubated (Cell Lysis Buffer; Cell Signaling Technology, Inc., Danvers, MA) for 30 minutes on ice. The lysates were clarified by centrifugation (15,000g) for 15 minutes at 4°C and snap frozen. After the lysates were thawed, protein concentration was determined with a BCA protein assay. A strain of mice in the 129 SvEv background that harbors a single disrupted allele of UbcM2 (the mouse counterpart of UBE2E3) was generated by using embryonic stem cells that were created, sequenced verified, and obtained from the International Gene Trap Consortium (www.genetrap.org).23 The disrupted allele contained an insertion encoding a fusion of β-galactosidase and neomycin phosphotransferase II (β-gal-neo). The insertion is between exons 3 and 4 of the UbcM2 gene and results in the expression of the first 86 residues of UbcM2 as a fusion with β-gal-neo. Expression of the fusion is driven by the endogenous UbcM2 promoter. Confirmation that the β-gal-neo cassette inserted into the UbcM2 gene was established using a genetic approach. RNA was isolated from tail snips of newborn mice (RNAqueous; Ambion). The RNA was subjected to RT-PCR (Superscript III; Invitrogen), a UbcM2 exon 5–specific primer (5′-GGTTGCAATCTGTCAAAAGGGAACAAATAGAGA-3′) and a β-gal-neo specific primer (5′-AGTATCGGCCTCAGGAAGATCG-3′). The resulting cDNA templates were used for PCR reactions to amplify the wild-type UbcM2 allele or the disrupted UbcM2 allele. The wild-type allele was amplified with a UbcM2 exon 3–specific primer (5′-AGAAGGAGCTAGCAGAGATAACCCTT-3′) and the above described UbcM2 exon 5–specific primer. The disrupted UbcM2 allele was amplified with the UbcM2 exon 3–specific primer and a nested β-gal-neo–specific primer (5′-ATTCAGGCTGCGCAACTGTTGGG-3′). A schematic of the cDNAs, primer locations and primer sequences is shown in Supplementary Figure S1A and representative data are shown in Supplementary Figure S1B, online at http://www.iovs.org/cgi/content/full/49/12/5611/DC1.
For the data shown in Figure 6, the mice were euthanatized, and cryosections were prepared from embryonic day (E)13 heads and postnatal day (P)17 eyes. Tissues were immediately fixed for 1 hour on ice in 2% paraformaldehyde/0.25%glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) and then rinsed 3 times for 10 minutes in buffer before being cryoprotected overnight at 4°C in 30% sucrose/0.1 M phosphate buffer (pH 7.3). Sections (5–10 μm in thickness) were cut on a microtome (Cryocut 1800; Leica, Bannockburn, IL), placed on slides (Superfrost/Plus; Fisher Scientific, Pittsburgh, PA), air-dried, and washed with rinse buffer (0.1 M sodium phosphate buffer [pH 7.3], 2 mM MgCl2, 0.1% Triton X-100). Sections were reacted at 37°C for 48 hours with X-Gal (5-bromo-4-chloro-3-indolyl galactopyranoside) in staining buffer (rinse buffer+5 mM potassium ferrocyanide+5 mM potassium ferriccyanide+1 mg/mL X-Gal). Samples were postfixed in 10% formaldehyde overnight at 4°C, and the melanin pigment in the RPE and choroid was bleached according to the protocol of Bhutto et al.24 The samples were then counterstained with neutral red for 1 minute and overlaid with mounting medium (Cytoseal; Electron Microscopy Sciences, Fort Washington, PA) and a coverslip.
Images were captured with an upright microscope (80i; Nikon) equipped with a 40× dry objective. X-Gal-positive spots overlying RPE-cells within a specified length of tissue were counted from an average of 10 sections obtained from each group. Blue spots were counted only if they were unambiguously localized to the bleached RPE. Eyes from two different UbcM2βgal and wild-type animals were sectioned and analyzed for each of the E13 and p17 time points.
RESULTS
To characterize the function of UBE2E3, we developed and affinity purified a rabbit polyclonal antibody, α-2E3, directed against the unique amino terminal, 58-amino-acid region of the enzyme. α-2E3 specifically recognized recombinant GST-UBE2E3 by Western blot but not GST fusions of two closely related enzymes, UBE2E2 and UBE2E1 (Fig. 1A). These three enzymes represent the human class III E2s. Their catalytic core domains are 95% identical, and they are distinguished from each other by their unique amino terminal extensions.18 Western blot of RPE-1 cell lysates with α-2E3 detected a primary band of 23 kDa, the predicted size of the enzyme (Fig. 1B, left). A comparable band was also detected in isolated retinas from C57BL6 mice (Fig. 1B, right). The mouse counterpart of UBE2E3 is called UbcM2, and the proteins are 100% identical. Immunolabeling of asynchronously growing RPE-1 cells with α-2E3 revealed that the enzyme localizes to the nucleus of interphase cells and is dispersed throughout mitotic cells (Fig. 1C).
Figure 1.

α-2E3 specifically recognizes UBE2E3, and the enzyme localizes to interphase nuclei in telomerase-immortalized retinal pigment epithelial cells (RPE-1s). (A) α-GST (left) and α-2E3 (right) Western blot of GST and GST-E2 fusion proteins. Ten nanograms of each protein was loaded per lane. (B) Western blot with α-2E3 of solubilized RPE-1 cell lysate (left) and a lysate derived from mouse retina (right). Thirty micrograms of RPE-1 cell lysate and 25 μg of mouse retina lysate were analyzed. The migration of molecular weight markers is shown to the left of blots (A) and (B). UbcM2 is the mouse counterpart of UBE2E3. (C) Representative photomicrographs of RPE-1 cells immunostained with α-2E3 (right). DNA was counterstained with DAPI (left); white arrows: cells in mitosis. Images were captured with a 20× phase objective. Scale bar, 10 μm.
To directly address whether UBE2E3 is essential in RPE cell proliferation, we used a loss-of-function approach with siRNA to knockdown expression of the enzyme. RT-PCR and complementary Western blotting demonstrated that individual siRNA duplexes, as well as pools of duplexes, efficiently knocked down UBE2E3 mRNA (Fig. 2A) and suppressed expression of the enzyme (Fig. 2B). The specificity of the knockdown was further established by showing that the siRNAs did not reduce expression of the closely related enzyme, UBE2E1 (data not shown).
Figure 2.

Knockdown of UBE2E3 by siRNA causes a block of cell proliferation. (A) RNA was isolated from cells treated with duplexes (Du) 1 to 4 or with a mixture of the duplexes (Du 1+2+3+4). RT-PCR products were generated with UBE2E3- and importin-11-specific primers. (B) α-2E3 Western blot demonstrating UBE2E3 knockdown by each of the UBE2E3-specific siRNAs. The cells were harvested 2 days after siRNA treatment and lysates derived from 100,000 cells were loaded in each lane. Left: migration of molecular weight markers. (C) Graph of data obtained from cell counts on the indicated days after siRNA treatment. The graph represents averages obtained from three independent experiments. Error bars SD. NB, The error bar for the day 3 si2E3 bar is not detectable in this graph because of the close grouping of the data points from the three independent experiments (180,900, 189,000, and 182,500 cells).
Next, we assessed the consequences of UBE2E3 depletion on cell proliferation. RPE-1 cells were transfected with siRNA, collected by trypsinization at 1, 2, and 3 days after siRNA treatment and counted. Depletion of UBE2E3 dramatically reduced the number of cells (Fig. 2C) compared with cells treated with siCON, the control siRNA. The reduced proliferation was observed with pools of siRNA, as well as with individual siRNAs (data not shown). Propidium iodide staining and FACS analysis showed that the UBE2E3 knockdown cells had a similar cell cycle distribution profile as control cells (data not shown) indicating that depletion of the enzyme was probably inducing a cell cycle exit. To confirm this, we immunolabeled control and knockdown cells for Ki-67, a marker of proliferating cells.25-27 We predicted that if the knockdown cells were arrested in the cell cycle, their nucleoli would stain positive for Ki-67, whereas if they had exited the cell cycle, they would be negative for the proliferation marker.26 Control siRNA-treated cells were proliferating as indicated by Ki-67 immunolabeling (Fig. 3A, top, right column). Depletion of the enzyme by either of two different siRNA duplexes led to a loss of Ki-67-positive cells (Fig. 3A, middle and bottom, right column). These data show that UBE2E3 depletion causes cells to exit the cell cycle. Consistent with this exit, we also observed that knockdown of UBE2E3 with either duplex 2 or 3 caused an increase in cell spreading and flattening (Fig. 3B, photomicrographs). Cell area measurements showed that depletion of UBE2E3 caused an average twofold increase in area compared with siCON-treated cells (Fig. 3B, graph).
Figure 3.

UBE2E3 knockdown causes RPE-1 cells to exit from the cell cycle and flatten. (A) Immunofluorescence analysis of RPE-1 cells treated with either siCON or UBE2E3-specific siRNA (duplexes 2 and 3). Two days after siRNA treatment, the cells were colabeled with antibodies against p27Kip1 (center column) and Ki-67 (right column). DNA was counterstained with DAPI (left column). (B) Depletion of UBE2E3 caused RPE-1 cells to spread and flatten. The cells were imaged live with a 20× phase objective. Below the photomicrographs is a graph of average cell area (measured in pixels) for the indicated treatments. Data represent the pooled areas of 93 cells (siCon), 95 cells (duplex 2), and 92 cells (duplex 3) from two independent experiments. *Statistically significant, according to a two-tailed distribution, two-sample, equal-variance Student's t-test with P < 0.00001. Scale bar, 32.6 μm.
To further elucidate the mechanism(s) by which UBE2E3 depletion leads to an exit from the cell cycle, we analyzed knockdown cells for levels of the cell cycle inhibitor p27Kip1. p27Kip1 is degraded by the ubiquitin proteolytic system during G1, and this degradation is required for progression from the G1 to the S phase,7 as well as from the G2 to M phase.6 In addition, p27Kip1 accumulation can drive cells out of the cell cycle, and elevated levels of the protein are necessary to maintain a postmitotic state.28 In proliferating RPE-1 cells, p27Kip1 expression is maintained at relatively low levels (e.g., Fig 3A, top row, middle panel). In contrast, cells depleted of UBE2E3 exhibited a robust increase in the percentage of p27Kip1-positive cells as well as in the level of p27Kip1 per cell. This elevation of p27Kip1 was evident by immunofluorescence analysis (Fig. 3A, middle column), FACS analysis (Fig. 4A), and Western blot (Fig. 4B). Furthermore, most of the cells that accumulated p27Kip1 in the nucleus had a corresponding loss of Ki-67 staining (Fig. 3A).
Figure 4.

UBE2E3 knockdown causes a dramatic elevation of p27Kip1 levels. (A) Representative FACS data of cells treated with siCON siRNA (left) or UBE2E3-specific siRNA duplexes 1 to 4 (Si2E3, right). Two days post-siRNA treatment, cells were colabeled with an anti-p27Kip1 antibody and with propidium iodide. The diagonal line across each graph is identically positioned and permits a comparison of the location of the dots between the two graphs. (B) Representative Western blots from the samples used for FACS in (A). Top: increase in p27Kip1 levels in response to UBE2E3 knockdown; middle: si2E3 decreased UBE2E3 levels below the detection limit of α-2E3. The β-tubulin blot illustrates that equal amounts of lysate were loaded in each lane.
The specificity of the p27Kip1 elevation was confirmed by rescue experiments. We first generated a rescue plasmid containing the cDNA sequence for wild-type, untagged, UBE2E3 but harboring silent point mutations such that the mRNA produced would be impervious to UBE2E3-specific siRNA silencing. We used anti-2E3 Western blotting to validate that this plasmid expressed untagged UBE2E3 (Fig. 5A, lanes 1 and 2) and importantly, that this expression was refractory to siRNA-mediated silencing (Fig. 5A, lanes 3 and 4). Control cells were transfected with a construct encoding RFP-H2B. We observed that transfection of RPE-1 cells with the rescue plasmid resulted in an increase in UBE2E3 levels (Fig. 5A, lane 2). UBE2E3 overexpression also led to the production of both a faster-migrating species (e.g., Fig. 5A, lane 2, asterisk) and a high-molecular-weight smear that likely represents auto-ubiquitylated enzyme (e.g., Fig. 5A, lane 2). In cells cotreated with UBE2E3-specific siRNA, UBE2E3 expression from the rescue plasmid was readily apparent, as were the faster-migrating band, and the high-molecular-weight species (Fig. 5A, lane 4).
Figure 5.

The elevation of p27Kip1 levels by UBE2E3 depletion can be rescued by reintroduction of the enzyme. (A) α-2E3 Western blots of lysates derived from RPE-1 cells transfected with plasmids expressing RFP-H2B (Mock) (lane 1) or untagged UBE2E3 (lane 2). The samples in the right blot were also subjected to treatment with UBE2E3-specific siRNA duplexes 1 to 4 (lanes 3 and 4). Asterisk: an α-2E3 reactive band that is produced by overexpression of the enzyme (lanes 2 and 4). The high molecular weight species also result from overexpression of the enzyme (lanes 2 and 4) and likely represent auto-ubiquitylated enzyme. (B) RPE-1 cells were transfected with plasmids encoding either RFP-H2B mRNA (mock; Ba, Bb, Be, Bf) or siRNA-impervious UBE2E3 mRNA (Bc, Bd, Bg, Bh). The following day, the cells were treated with either siCON (Bb, Bd, Bf, Bh) or si2E3 duplexes 1 to 4 (Ba, Bc, Be, Bg). Three days later, the cells were processed for anti-p27Kip1 immunofluorescence and DAPI staining. Representative photomicrographs for each of the conditions tested are shown. (Ba–d) DAPI-stained cells; (Be–h) the corresponding anti-p27Kip1 immunofluorescence. Scale bar, 10 μm. (C) The percent relative pixel intensity of nuclear p27Kip1 per cell was quantitated from a series of photomicrographs and plotted in ascending order for 250 cells/sample. Cells were treated with si2E3 and mock rescue, si2E3 and UBE2E3 rescue, siCON and mock rescue, or siCON and UBE2E3 rescue.
We next performed the rescue experiment by transfecting RPE-1 cells with either the rescue plasmid or a control plasmid encoding RFP-H2B. The following day, the cells were treated with siRNA, and 3 days later, they were fixed, permeabilized, and processed for anti-p27Kip1 immunofluorescence (Fig. 5B). A moderate decrease in p27Kip1 levels was observed in the UBE2E3-rescued cells compared with the mock-rescued cells (Fig. 5B, compare 5E and 5G). Quantitation of nuclear p27Kip1 levels confirmed that expression of siRNA-impervious UBE2E3 mRNA successfully lowered p27Kip1 levels (Fig. 5C, compare open gray boxes to open black boxes). We attribute the intermediate level of rescue to inefficient transfection and/or heterogenous expression from the rescue plasmid among the population of cells. Collectively, these studies that UBE2E3 is essential for the proliferation of RPE-1 cells, and depletion of the enzyme induces a p27Kip1-mediated cell cycle exit.
One prediction of these tissue culture findings is that UBE2E3 promotes RPE cell proliferation during eye development but is then downregulated as the cells transition toward terminal differentiation. To test this prediction, we compared the expression profile of UBE2E3 in the developing and mature mouse RPE. As mentioned, the mouse counterpart of UBE2E3 is called UbcM2, and the proteins are 100% identical. We generated a unique mouse strain to analyze the in vivo expression profile of UbcM2. This 129/SvEv strain harbors one wild-type UbcM2 allele and one allele disrupted with a β-gal-neo insertion under control of the endogenous UbcM2 promoter. This allele expresses the amino-terminal 86 residues of UbcM2 fused in-frame to β-gal-neo. Thus, every cell that expresses UbcM2 expresses the β-gal-neo fusion protein. In addition, both the β-galactosidase and neomycin phosphotransferase II enzymes are functional in the context of the fusion protein (e.g., Fig. 6A and Ref. 29).
Figure 6.
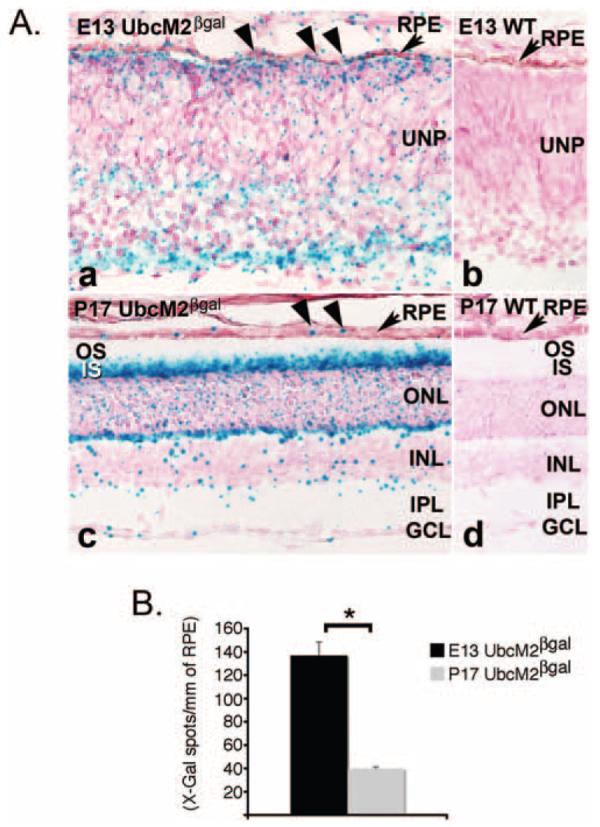
UbcM2, the mouse counterpart of UBE2E3, is downregulated during RPE maturation in the developing mouse eye. (A) Representative photomicrographs of X-Gal-labeled cryosections from E13 embryos (Aa, Ab) and P17 mice (Ac, Ad). (Ab, Ad) Wild-type samples; (Aa, Ac) samples from UbcM2 heterozygous mice. WT, wild type; UNP, undifferentiated retinal precursors; OS, outer segments; IS, inner segments; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. (B) Graph of X-Gal spots counted per mm of RPE from an average of 10 sections from each of two animals for each time point. Error bars, SEM. *Statistically significant, according to a two-tailed distribution, two-sample, equal-variance Student's t-test with P < 0.0002.
We used the β-gal activity as a heterologous reporter to compare the expression of UbcM2 in proliferating RPE cells versus mature RPE cells. This method enabled us to bypass the inability of α-2E3 to specifically detect nuclear enzyme in paraffin-embedded sections and cryosections (data not shown). Cryosections from E13 (representing developing RPE) and P17 (representing mature RPE) mice were prepared and stained in parallel with X-Gal as described in the Materials and Methods section. E13 was chosen as the early time point because RPE cells proliferate at this time, and robust p27Kip1 expression in the developing mouse eye is not observed until E18.30 After an overnight fixation in 10% formaldehyde, the sections were bleached24 to reduce the amount of melanin pigment in the RPE and choroid and thereby facilitate visualization of the X-Gal signal. Representative bright-field photomicrographs from each sample along with sections processed in parallel from wild-type littermates demonstrate the specificity of the reporter system (e.g., Fig. 6A, compare Aa to Ab and Ac to Ad). Quantification of the X-Gal spots per millimeter of RPE revealed that β-gal-neo expression was on average 3.3-fold higher at E13 versus p17 (Fig. 6B). These data demonstrate that in vivo, UbcM2 expression is transcriptionally downregulated during development as RPE cells mature and transition from a proliferative state to one of terminal differentiation. Notably, the X-Gal staining also revealed robust expression from the UbcM2 promoter in several regions of the adult mouse retina including the inner segments of the photoreceptors, the outer plexiform layer, and to a lesser extent, the outer and inner nuclear layers (Fig. 6C). The function(s) of UbcM2 in the retina is currently under investigation.
Discussion
Rigorous genetic and biochemical analyses in a wide range of eukaryotic organisms over the past 25 years have revealed that the ubiquitin system plays a central role in promoting proper progression through each phase of the cell cycle (reviewed in Refs. 31,32). But, despite the importance of this system in cell division and the large number of ubiquitin pathway enzymes in eukaryotic organisms, relatively few of these enzymes have been definitively shown to be essential for cell cycle progression. We have now identified UBE2E3, a ubiquitin-conjugating enzyme highly conserved among vertebrates, as playing an essential role in the proliferation of RPE cells. Specifically, we found that reducing the levels of UBE2E3 by siRNA causes RPE-1 cells to exit the cell cycle (Figs. 2C, 3). This exit is marked by a loss of Ki-67-positive cells (Fig. 3A), a corresponding robust elevation in the levels of the cdk inhibitor, p27Kip1 (Figs. 3A, 4), and a doubling of cell area (Fig. 3B). The specificity of the siRNA-induced phenotypes was confirmed by the observation that two individual siRNAs targeting UBE2E3 expression produced comparable results (e.g., Figs. 3A, 3B) and by rescue experiments (Fig. 5).
In rodent studies, p27Kip1 has been shown to play critical roles in the timing and fidelity of RPE differentiation as well as in the interdigitation of the RPE monolayer with the outer segments.9,33 Mice nullizygous for the p27Kip1 gene have multiple changes in their RPE monolayer compared with their wild-type counterparts, including an increased thickness in the actual monolayer,10 a higher percentage of binucleated cells, and a decreased association with photoreceptor cells.9 These findings reveal that p27Kip1 is essential for the structure and function of the RPE and firmly establish the protein as a primary regulator of the balance between RPE cell proliferation and terminal differentiation. Building on these studies using human RPE-1 cells, we have found that the levels of p27Kip1 are kept relatively low in proliferating cells (e.g., Fig. 3, middle column, top panel), but escalate in response to UBE2E3 depletion (e.g., Figs. 3, 4). Along with the loss of Ki-67 staining and the doubling of cell area, the changes induced by UBE2E3 depletion are consistent with those observed in vivo for RPE cells as they transition from a state of proliferation to one of terminal differentiation.9,33 Of note, we did not observe an upregulation of RPE-specific differentiation markers after UBE2E3 knockdown (data not shown). This finding indicates that, at least in cultured RPE-1 cells, depletion of the enzyme may be necessary to drive cells toward differentiation, but is apparently not sufficient to induce terminal differentiation. Nonetheless, a potential implication of the siRNA findings is that during development of the retina, UBE2E3 levels are sustained in RPE cells until enough cells are generated to form the RPE monolayer. UBE2E3 expression then decreases, which results in p27Kip1 accumulation and triggers cell cycle exit and terminal differentiation. Because of the inherent limitations of using an immortalized tissue culture cell line to test a developmental model, we created a unique mouse strain to evaluate expression of the enzyme in proliferating versus differentiated RPE cells. This mouse strain expresses β-gal-neo driven by the endogenous UbcM2 promoter. A comparison of β-gal activity in proliferating RPE cells from E13 embryos versus differentiated RPE cells from P17 mice revealed a striking, age-dependent decrease in transcription of the UbcM2 gene (Fig. 6). We interpret these findings with the β-gal reporter to indicate that UbcM2 gene expression was decreased by greater than threefold in RPE cells that had exited the cell cycle and undergone terminal differentiation. Together, these studies couple UBE2E3 with p27Kip1 stability and in turn implicate the enzyme as a regulator of the balance between RPE cell proliferation and differentiation. This is the first demonstration that a specific E2 is linked with this process in RPE cells.
It is not yet clear whether the elevation of p27Kip1 directly drives cells out of the cell cycle or occurs secondary to a separate event induced by the depletion of UBE2E3. Further, it remains to be determined whether p27Kip1 is a substrate of UBE2E3 and accumulates because it is no longer being ubiquitylated and marked for degradation when UBE2E3 is depleted. Based on previous work showing that UBE2E3 can enhance the activity of Cul4A,34 we favor the notion that p27Kip1 is ubiquitylated by a complex containing UBE2E3. Cul4A is a multi-subunit E3 ligase that utilizes the DDB1 and Skp2 substrate adaptors to mediate the ubiquitin-dependent degradation of p27Kip1.13 Despite these unresolved issues, this study marks the first report linking a specific E2 to the proliferative capacity of RPE cells. The identification of such factors is an important first step toward manipulating the balance between RPE proliferation and differentiation and may be useful in the development of strategies to replenish the RPE monolayer in diseases such as AMD.
Supplementary Material
Acknowledgments
The authors thank Lindsey Long for technical assistance and Brian Ceresa and Robert E. Anderson for helpful suggestions and encouragement.
Supported by Oklahoma Center for the Advancement of Science and Technology Grants HR05-161S (SMP) and HR06-125 (AFW) and by National Center for Research Resources Grant P20RR024215. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Disclosure: K.S. Plafker, None; K.M. Farjo, None; A.F. Wiechmann, None; S.M. Plafker, None
References
- 1.Nowak JZ. Age-related macular degeneration (AMD): pathogenesis and therapy. Pharmacol Rep. 2006;58(3):353–363. [PubMed] [Google Scholar]
- 2.Hara T, Kamura T, Nakayama K, Oshikawa K, Hatakeyama S, Nakayama K. Degradation of p27(Kip1) at the G(0)-G(1) transition mediated by a Skp2-independent ubiquitination pathway. J Biol Chem. 2001;276(52):48937–48943. doi: 10.1074/jbc.M107274200. [DOI] [PubMed] [Google Scholar]
- 3.Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 2001;413(6853):323–327. doi: 10.1038/35095083. [DOI] [PubMed] [Google Scholar]
- 4.McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23(1):216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakayama K, Nagahama H, Minamishima YA, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000;19(9):2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakayama K, Nagahama H, Minamishima YA, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6(5):661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- 7.Nakayama KI, Hatakeyama S, Nakayama K. Regulation of the cell cycle at the G1-S transition by proteolysis of cyclin E and p27Kip1. Biochem Biophys Res Commun. 2001;282(4):853–860. doi: 10.1006/bbrc.2001.4627. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9(10):1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 9.Defoe DM, Adams LB, Sun J, Wisecarver SN, Levine EM. Defects in retinal pigment epithelium cell proliferation and retinal attachment in mutant mice with p27(Kip1) gene ablation. Mol Vis. 2007;13:273–286. [PMC free article] [PubMed] [Google Scholar]
- 10.Nakayama K, Ishida N, Shirane M, et al. Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85(5):707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida K, Kase S, Nakayama K, et al. Involvement of p27KIP1 in the proliferation of the developing corneal endothelium. Invest Ophthalmol Vis Sci. 2004;45(7):2163–2167. doi: 10.1167/iovs.03-1238. [DOI] [PubMed] [Google Scholar]
- 12.Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol. 2000;183(1):10–17. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 13.Bondar T, Kalinina A, Khair L, et al. Cul4A and DDB1 associate with Skp2 to target p27Kip1 for proteolysis involving the COP9 signalosome. Mol Cell Biol. 2006;26(7):2531–2539. doi: 10.1128/MCB.26.7.2531-2539.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1(4):193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 15.Hara T, Kamura T, Kotoshiba S, et al. Role of the UBL-UBA protein KPC2 in degradation of p27 at G1 phase of the cell cycle. Mol Cell Biol. 2005;25(21):9292–9303. doi: 10.1128/MCB.25.21.9292-9303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamura T, Hara T, Matsumoto M, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6(12):1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 17.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9(12):661–664. doi: 10.1016/s0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 18.Matuschewski K, Hauser HP, Treier M, Jentsch S. Identification of a novel family of ubiquitin-conjugating enzymes with distinct amino-terminal extensions. J Biol Chem. 1996;271(5):2789–2794. doi: 10.1074/jbc.271.5.2789. [DOI] [PubMed] [Google Scholar]
- 19.Pestov DG, Grzeszkiewicz TM, Lau LF. Isolation of growth suppressors from a cDNA expression library. Oncogene. 1998;17(24):3187–3197. doi: 10.1038/sj.onc.1202260. [DOI] [PubMed] [Google Scholar]
- 20.Seddon JM, Santangelo SL, Book K, Chong S, Cote J. A genome-wide scan for age-related macular degeneration provides evidence for linkage to several chromosomal regions. Am J Hum Genet. 2003;73(4):780–790. doi: 10.1086/378505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rambhatla L, Chiu CP, Glickman RD, Rowe-Rendleman C. In vitro differentiation capacity of telomerase immortalized human RPE cells. Invest Ophthalmol Vis Sci. 2002;43(5):1622–1630. [PubMed] [Google Scholar]
- 22.Plafker SM, Plafker KS, Weissman AM, Macara IG. Ubiquitin charging of human class III ubiquitin-conjugating enzymes triggers their nuclear import. J Cell Biol. 2004;167(4):649–659. doi: 10.1083/jcb.200406001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nord AS, Chang PJ, Conklin BR, et al. The International Gene Trap Consortium Website: A portal to all publicly available gene trap cell lines in mouse (database issue) Nucleic Acids Res. 2006 Jan 1;34(suppl):D642–D648. doi: 10.1093/nar/gkj097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhutto IA, Kim SY, McLeod DS, et al. Localization of collagen XVIII and the endostatin portion of collagen XVIII in aged human control eyes and eyes with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2004;45(5):1544–1552. doi: 10.1167/iovs.03-0862. [DOI] [PubMed] [Google Scholar]
- 25.Gerdes J, Schwab U, Lemke H, Stein H. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer. 1983;31(1):13–20. doi: 10.1002/ijc.2910310104. [DOI] [PubMed] [Google Scholar]
- 26.Kill IR. Localisation of the Ki-67 antigen within the nucleolus: evidence for a fibrillarin-deficient region of the dense fibrillar component. J Cell Sci. 1996;109:1253–1263. doi: 10.1242/jcs.109.6.1253. [DOI] [PubMed] [Google Scholar]
- 27.Rawes V, Kipling D, Kill IR, Faragher RG. The kinetics of senescence in retinal pigmented epithelial cells: a test for the telomere hypothesis of ageing? Biochemistry (Mosc) 1997;62(11):1291–1295. [PubMed] [Google Scholar]
- 28.Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16(2):306–317. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 1991;5(9):1513–1523. doi: 10.1101/gad.5.9.1513. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida K, Nakayama K, Kase S, et al. Involvement of p27(KIP1) in proliferation of the retinal pigment epithelium and ciliary body. Anat Embryol (Berl) 2004;208(2):145–150. doi: 10.1007/s00429-004-0382-5. [DOI] [PubMed] [Google Scholar]
- 31.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116(2):221–234. doi: 10.1016/s0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 32.Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4(11):855–864. doi: 10.1038/nrm1246. [DOI] [PubMed] [Google Scholar]
- 33.Levine EM, Close J, Fero M, Ostrovsky A, Reh TA. p27(Kip1) regulates cell cycle withdrawal of late multipotent progenitor cells in the mammalian retina. Dev Biol. 2000;219(2):299–314. doi: 10.1006/dbio.2000.9622. [DOI] [PubMed] [Google Scholar]
- 34.Pick E, Lau OS, Tsuge T, et al. Mammalian DET1 regulates Cul4A activity and forms stable complexes with E2 ubiquitin-conjugating enzymes. Mol Cell Biol. 2007;27(13):4708–4719. doi: 10.1128/MCB.02432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.