

Abstract
Autism is a behaviorally-defined neurodevelopmental disorder usually diagnosed in early childhood that is characterized by impairment in reciprocal communication and speech, repetitive behaviors, and social withdrawal. Although both genetic and environmental factors are thought to be involved, none have been reproducibly identified. The metabolic phenotype of an individual reflects the influence of endogenous and exogenous factors on genotype. As such, it provides a window through which the interactive impact of genes and environment may be viewed and relevant susceptibility factors identified. Although abnormal methionine metabolism has been associated with other neurologic disorders, these pathways and related polymorphisms have not been evaluated in autistic children. Plasma levels of metabolites in methionine transmethylation and transsulfuration pathways were measured in 80 autistic and 73 control children. In addition, common polymorphic variants known to modulate these metabolic pathways were evaluated in 360 autistic children and 205 controls. The metabolic results indicated that plasma methionine and the ratio of S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH), an indicator of methylation capacity, were significantly decreased in the autistic children relative to age-matched controls. In addition, plasma levels of cysteine, glutathione, and the ratio of reduced to oxidized glutathione, an indication of antioxidant capacity and redox homeostasis, were significantly decreased. Differences in allele frequency and/or significant gene-gene interactions were found for relevant genes encoding the reduced folate carrier (RFC 80G>A), transcobalamin II (TCN2 776G>C), catechol-O-methyltransferase (COMT 472G>A), methylenetetrahydrofolate reductase (MTHFR 677C>T and 1298A>C), and GST M1. We propose that an increased vulnerability to oxidative stress (endogenous or environmental) may contribute to the development and clinical manifestations of autism.
Keywords: autism, oxidative stress, genotype, glutathione, methionine
INTRODUCTION
The Centers for Disease Control and the American Academy of Pediatrics have recently released an “Autism Alarm” indicating that the current prevalence of autism is ~1 in 166 children in the US (CDC 2005). The ~10-fold increase in autism diagnosis in the last two decades has generated major public health concern. Nonetheless, research progress has been slow and the biologic basis of this complex disorder remains unknown. Prevailing evidence supports the involvement of both genetic and environmental factors that interact to negatively affect prenatal and postnatal neurologic development (Folstein and Rosen-Sheidley 2001;Keller and Persico 2003). A strong genetic component is widely accepted based on the high concordance among monozygotic twins and the 10-fold increase in risk among siblings of affected children relative to the general population (Bailey et al. 1995). Because monozygotic twin concordance is less than 100%, environmental and epigenetic factors have also been implicated and are thought to be necessary to expose the genetic liability. However, despite intense research effort, no single gene or specific environmental factor has been reproducibly identified (Muhle et al. 2004). It has been estimated that at least 10–15 common and “small effect” susceptibility alleles contribute to the phenotype and that different combinations of mutant alleles may be involved in different individuals. An interacting environmental trigger further complicates the search for susceptibility genes since many unaffected individuals are likely to carry the same genetic risk factors.
The diagnosis of autism is based solely on behavioral criteria that define deficits in social interaction, impairment in verbal and non-verbal receptive/expressive speech, and hyper-focused repetitive behaviors (Lord et al. 2000). The pathophysiology of autism primarily affects three major systems: neurologic, immunologic, and gastrointestinal (Bauman and Kemper 2003;Hornig and Lipkin 2001;Krause et al. 2002;Horvath and Perman 2002;White 2003). An interesting but poorly understood etiologic clue is the fact that four boys are affected for every girl. Further compounding the complexity, autistic behavior encompasses a heterogeneous and variable spectrum of clinical symptoms (Eigsti and Shapiro 2003;Tager-Flusberg and Joseph 2003). Currently, there is no biochemical test for the presence of autism to support the behavioral diagnosis.
Research into the metabolic basis for autism has been relatively underutilized compared to broad scale genomic and proteomic approaches. An integrated metabolic profile reflects the interaction of genetic, epigenetic, environmental, and endogenous factors that perturb the pathway of interest. In addition, the evaluation of an entire metabolic pathway, as opposed to isolated single gene products, provides greater mechanistic insights into disease pathology and can identify new options for targeted intervention strategies. We have used a targeted approach to “metabolomics” by focusing on the dynamics of a single metabolic pathway involving methionine transmethylation and transsulfuration that has been implicated in the pathogenesis of numerous other neurologic disorders (Miller 2003;Serra et al. 2001;Schulz et al. 2000;Muntjewerff et al. 2003;Pogribna et al. 2001).
In a recent case-control study, we measured fasting levels of plasma methionine transmethylation and transsulfuration metabolites in 20 autistic and 33 control children (James et al. 2004). The metabolic profile of children diagnosed with autistic disorder with regressive onset was found to be severely abnormal. The autistic children were found to have significant decreases in methionine levels and in the ratio of plasma S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAM/SAH ratio), an index of methylation capacity. Total glutathione levels (GSH, the major intracellular antioxidant) were decreased and oxidized glutathione disulfide (GSSG) was increased, resulting in a ~3-fold reduction in the redox ratio of reduced (active) GSH to oxidized (inactive) glutathione (GSH/GSSG). Cysteine, the rate-limiting amino acid for glutathione synthesis, was significantly decreased relative to the control children suggesting that GSH synthesis was insufficient to maintain redox homeostasis. The present study was undertaken to confirm and extend these observations in a larger cohort of children and to identify potential polymorphisms in candidate genes known to affect the dynamics of these pathways.
A diagram of methionine transmethylation and transsulfuration pathways is presented in Figure 1. The methionine cycle (transmethylation) involves the regeneration of methionine from homocysteine via the B12-dependent transfer of a methyl group from 5-methyl tetrahydrofolate (5-CH3THF) via the methionine synthase (MS) reaction (Finkelstein 1990). Methionine is then activated to S-adenosylmethionine (SAM), the methyl donor for multiple cellular methyltransferase reactions and the methylation of essential molecules such as DNA, RNA, proteins, phospholipids, creatine, and neurotransmittors (Mato et al. 2002). The transfer of the methyl group from SAM results in the demethylated product S-adenosylhomocysteine (SAH). The reversible hydrolysis of SAH to homocysteine and adenosine by the SAH hydrolase (SAHH) reaction completes the methionine cycle. Homocysteine can then be either remethylated to methionine or irreversibly removed from the methionine cycle by cystathionine beta synthase (CBS). This is a one-way reaction that permanently removes homocysteine from the methionine cycle and initiates the transsulfuration pathway for the synthesis of cysteine and glutathione as indicated in Figure 1 (Finkelstein 1998).
Figure 1.
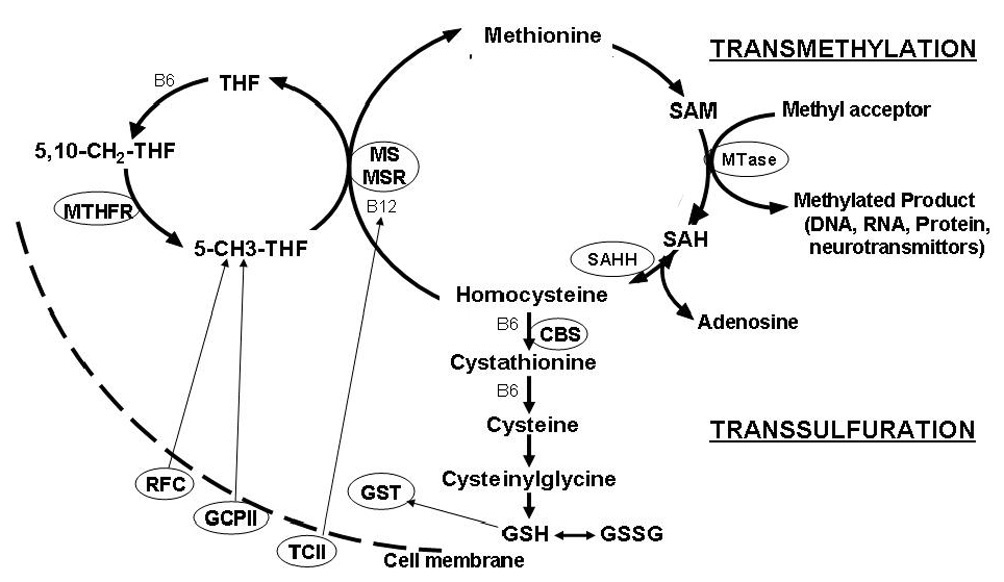
An overview of the pathways involved in folate-dependent methionine transmethylation and transsulfuration. Methylenetetrahydrofolate (MTHFR) catalyzes the synthesis of 5-methyltetrahydrofolate (5-CH3-THF) from 5,10,methylenetetrahydrofolfate (5,10-CH2THF). The methyl group from 5-CH3THF is transferred to homocysteine to regenerate methionine via the folate/B12-dependent methionine synthase (MS) reaction. Methionine synthase reductase (MSR) maintains the B12 cofactor in a reduced state for optimal MS activity. Methionine is then activated to S-adenosylmethionine (SAM), the major methyl donor for multiple cellular methyltransferase (MTase) reactions. After methyl group transfer, SAM is converted to SAH which is further metabolized to homocysteine and adenosine by a reversible reaction catalyzed by SAH hydrolase (SAHH). Homocysteine may be permanently removed from the methionine cycle by irreversible conversion to cystathionine by B6-dependent cystathionine beta synthase (CBS) which initiates the transsulfuration pathway. Cystathionine is subsequently converted to cysteine, the rate limiting amino acid for the synthesis of the tripeptide, glutathione (Glu-Cys-Gly). Reduced active glutathione (GSH) is in dynamic equilibrium with the oxidized disulfide GSSG form of glutathione. Reduced folates are transported from the plasma into the cell by the reduced folate carrier (RFC). Transport of folate into the intestinal mucosa is mediated by glutamate carboxypepsidase II (GCPII). Vitamin B12 is transported into the cell bound to the B12 transport protein transcobalmin II (TCII)
Subtle alterations in gene expression due to multiple polymorphisms and environmental factors that interact to affect the same metabolic pathway can induce chronic metabolic imbalance and alter nutritional requirements (Gueant et al. 2003;Lievers et al. 2003;Bailey and Gregory, III 2000). Using the abnormal metabolic phenotype in autistic children as a guide for the selection of functional candidate genes, we evaluated common SNPs in genes encoding methylenetetrahydrofolate (MTHFR 677C>T, MTHFR 1298A>C), methionine synthase reductase (MTRR 66A>G), transcobalamin II (TCN2 776C>G), catechol-O-methyltransferase (COMT 472G>A), glutathione-S-transferase (GST M1 null, GST T1 null), reduced folate carrier (RFC 80 A>G), glutamate-carboxypepsidase (GCPII 1561C>T). These are among several high frequency low penetrance polymorphisms that have been previously shown to modulate metabolite levels in the methionine transmethylation and transsulfuration pathways.(James et al. 1999;Castel-Dunwoody et al. 2005;Beagle et al. 2005;Stern et al. 2003)
SUBJECTS AND METHODS
Participants
Probands were referral patients recruited from the autism clinics of participating physicians in New York (SMB, KB, MB, PC), and Florida (JJB). The diagnosis of autistic spectrum disorder was made by independent specialists not associated with the study using criteria defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV), the Autism Diagnostic Observation Schedule (ADOS), or the Childhood Autism Rating Scales (CARS). Patients with childhood disintegrative disorder or rare genetic diseases associated with symptoms of autism (e.g., fragile X, Rett syndrome, tuberous sclerosis) were excluded from participation. Patient level of functioning ranged from moderate mental retardation to high functioning based on physician evaluation. The autistic children were between 3 and 14 years of age (mean 7.3 ± 3.2), were 97% Caucasian, 89% male and 11% female. Control children for the metabolic study were healthy Caucasians with no history of chronic disease, autism, or other neurologic disorder who had participated as controls for similar metabolic studies of children with Down syndrome (Pogribna et al. 2001) and cystic fibrosis (Innis et al. 2003). The mean age and SD of the control children was 10.8 ± 4.1 years. Case and control children taking high dose vitamin supplements or medications known to affect methionine metabolism were excluded from the metabolic study. The protocol was approved by the Institutional Review Board of the University of Arkansas for Medical Sciences and parental written informed consent was obtained.
Metabolic Analysis
Fasting blood samples from 80 autistic and 73 unrelated control children were collected into EDTA-Vacutainer tubes and immediately chilled on ice before centrifuging at 4000 × g for 10 minutes at 4°C. Plasma aliquots were transferred into cryostat tubes and stored at −80°C until extraction and HPLC quantification. All samples were analyzed within one month of receipt. For determination of total homocysteine, methionine, total glutathione, cysteinylglycine, and cysteine, 50 µl freshly prepared 1.43 mol/L sodium borohydride solution containing 1.5 µmol/L EDTA, 66 mmol/L NaOH and 10 µl isoamyl alcohol was added to 200 µl plasma to reduce all sulfhydryl bonds and incubated at 40°C in a shaker for 30 minutes. To precipitate proteins, 250 µl ice cold 10% meta-phosphoric acid was added, mixed well, and the sample was incubated for an additional 30 minutes on ice. After centrifugation at 18,000 × g for 15 minutes at 4°C, the supernatant was filtered through a 0.2 µm nylon membrane filter (PGC Scientific, Frederic, MD) and a 20 µl aliquot was injected into the HPLC system.
For determination of SAM, SAH, adenosine, and free reduced glutathione and oxidized disulfide glutathione (GSSG), 100 µl of 10% meta-phosphoric acid was added to 200 µl plasma to precipitate protein; the solution was mixed well and incubated on ice for 30 minutes. After centrifugation for 15 minutes at 18,000 g at 4°C, supernatants were passed through a 0.2 µm nylon membrane filter and 20 µl was injected into the HPLC system.
The separation of metabolites was performed using HPLC with a Shimadzu solvent delivery system (ESA model 580) and a reverse phase C18 column (5 µm; 4.6 × 150 mm, MCM, Inc., Tokyo, Japan) obtained from ESA, Inc. (Chemsford, MA). A 20 µl aliquot of plasma extract was directly injected onto the column using Beckman Autosampler (model 507E). All plasma metabolites were quantified using a model 5200A Coulochem II and CoulArray electrochemical detection systems (ESA, Inc., Chelmsford, MA) equipped with a dual analytical cell (model 5010), a 4-channel analytical cell (model 6210) and a guard cell (model 5020). The concentrations of plasma metabolites were calculated from peak areas and standard calibration curves using HPLC software.
Genetic Analysis
For the genetic analysis, blood samples were obtained from an additional 280 autistic children who did not qualify for the metabolic study because they were not fasting or were taking vitamin supplements (total case n = 360). An additional 132 unaffected controls (total control n = 205) consisted of participants in an ongoing study of congenital heart defect risk described previously (Hobbs et al. 2005). Genomic DNA was extracted using Puregene DNA Purification Kit (Gentra Systems, Inc., Minneapolis, MN). Genotyping was performed with allele-specific fluorescent primer-probe sets supplied by ABI Assays by Design (Applied Biosystems, Foster City, CA) and primer and probe sequences are listed in Table 1. PCR reactions were carried out with ABI PRISM 7700 and 7900 Sequence Detection Systems under the following thermal cycling conditions: one cycle at 95°C for 10 minutes (Taq activation), followed by 40 cycles of 92°C for 15 seconds (denature) and 60°C for 1 minute (anneal/extend). The reaction components were as follows: 900 nM of each primer, 200 nM each probe, 25 mM each dNTP, 1 M Tris-HCL (pH 8.4), 1 M MgCl2, 300 mM KCl, ROX reference dye, 100% glycerol, 0.5 U Taq DNA polymerase (Invitrogen, Carlsbad, CA), and 50 ng genomic DNA. The PCR primers for GST M1 and T1 are listed in Table 1; PCR conditions for GST M1 and GST T1 have been previously reported. (Ye and Parry 2002) The PCR products were visualized on a 2.5% agarose gel with Reliant Fastlane Gel System (Cambrex, Rockland, ME). The presence or absence of a 215 bp band or 480 bp band reflected the GST M1 and GST T1 positive or null genotypes, respectively. [Table 1: Primer-probe sets]
Table I.
Primers and TaqMan allele-specific probes
| Polymorphism | 5’-3’ sequence |
|---|---|
| MTHFR C677T | |
| Forward | TGGCAGGTTACCCCAAAGG |
| Reverse | CACAAAGCGGAAGAATGTGTCA |
| T- Probe 1 | 6FAM-TGATGAAATCGGCTCCCGCA-TAMRA |
| C- Probe 2 | VIC-TGA TGATGAAATCGACTCCCGCA-TAMRA |
| MTHFR A1298C | |
| Forward | GGAGGAGCTGCTGAAGATGTG |
| Reverse | CCCGAGAGGTAAAGAACAAAGACTT |
| A- Probe 1 | VIC-ACCAGTGAAGAAAGTGT |
| C | 6FAM-CAGTGAAGCAAGTGT |
| COMT G472A (Val158Met) | |
| Forward | CCCAGCGGATGGTGGAT |
| Reverse | CAGGCATGCACACCTTGTC |
| A- Probe 1 | VIC- TTCGCTGGCATGAAG |
| G-Probe 2 | 6FAM- TCGCTGGCGTGAAG |
| TCN2 C776G | |
| Forward | ACTCTATCACCAGTTCCTCATGACT |
| Reverse | TTGAGACATGCTGTTCCCAGTT |
| C- Probe 1 | VIC- CTGCCCCAGGCATG |
| G-Probe 2 | 6FAM- CTGCCCCACGCATG |
| MTRR A66G | |
| Forward | AGCAGGGACAGGCAAAGG |
| Reverse | AAGATCTGCAGAAAATCCATGTACCA |
| A-Probe 1 | VIC-TTGCTCACATATTTCTT |
| G-Probe 2 | 6FAM-CTCACACATTTCTT |
| RFC-1 80G>A | |
| Forward | GGCCTGACCCCG AGCT |
| Reverse | AGCCGTAGAAGCAAAGGTAGCA |
| G-Probe 1 | VIC-CACGAG GCGCCGC |
| A-Probe 2 | 6FAM-CGAGGT GCCGCCAG |
| GST M1 | |
| Forward | GAACTCCCTGAAAAGCTAAAGC |
| Reverse | GTTGGGCTCAAATATACGGTGG |
| GCPII C1561T | |
| Forward | GAGTTGATTGTACACCGCTGATG |
| Reverse | CCACCTATGTTTAACATAATACCTCAAG |
| C-Probe 1 | 6FAM-CTTGG TACACAACC TAA |
| T-probe 2 | VIC-AGCTTGGT ATACAACCT |
Allelic discrimination was accomplished by fluorogenic probes with either a 6FAM or VIC reporter dye attached to the 5’ end of the oligonucleotide that is cleaved by the 5’ nuclease activity of Taq DNA polymerase.
Statistical Analysis
Metabolic data are presented as the means ± SD. The data were prospectively collected and analyzed using SigmaStat software. Statistical differences between case and control children were determined using the Student’s t test with significance set at 0.05. For the genotype analysis, odds ratios and 95% confidence intervals were calculated using unconditional logistic regression models and tested using chi square analysis. Cases and controls were tested for Hardy–Weinberg equilibrium using the exact test implemented in STATA GENHW command (Cleves 1999). Gene-gene interactions were tested by including appropriate pair-wise indicator variables into unconditional logistic regression models.
RESULTS
Metabolic Study
Fasting levels of plasma metabolites in the transmethylation and transsulfuration pathways among 73 control and 80 autistic children are presented in Table 2. Within the methionine cycle, levels of methionine, S-adenosylmethionine (SAM), and the SAM/SAH ratio were significantly decreased in the autistic children, whereas the levels of S-adenosylhomocysteine (SAH), and adenosine were significantly elevated. The transsulfuration pathway metabolites, cysteine, total glutathione (free-reduced plus protein-bound), and free reduced glutathione were significantly decreased while cystathionine and the oxidized disulfide form of glutathione, GSSG, were significantly increased. The ratios of total glutathione and free reduced glutathione to oxidized GSSG (redox ratios) were also significantly reduced.
Table II.
Transmethylation and transsulfuration metabolites in autistic cases and controls
| Control* | Autistic* | p value | |
|---|---|---|---|
| (n=73) | (n=80) | ||
| Methionine (µmol/L) | 28.0 ± 6.5 | 20.6 ± 5.2 | <0.0001 |
| SAM (nmol/L) | 93.8 ± 18 | 84.3 ± 11 | <0.0001 |
| SAH (nmol/L) | 18.8 ± 4.5 | 23.3 ± 7.9 | <0.0001 |
| SAM/SAH ratio | 5.5 ± 2.8 | 4.0 ± 1.7 | <0.0001 |
| Adenosine (µmol/L) | 0.19 ± 0.13 | 0.28 ± .13 | 0.001 |
| Homocysteine (µmol/L) | 6.0 ± 1.3 | 5.7 ± 1.2 | 0.03 |
| Cystathionine (µmol/L) | 0.19 ± 0.1 | 0.24 ± 0.1 | <0.0001 |
| Cysteine (µmol/L) | 207 ± 22 | 165 ± 14 | <0.0001 |
| Cysteinylglycine (µmol/L) | 39.4 ± 7.3 | 38.9 ± 11 | 0.78 |
| Total GSH (µmol/L) | 7.53 ± 1.7 | 5.1 ± 1.2 | <0.0001 |
| Free GSH (µmol/L | 2.2 ± 0.9 | 1.4 ± 0.5 | <0.0001 |
| GSSG (µmol/L) | 0.24 ± 0.1 | 0.40 ± 0.2 | <0.0001 |
| Total GSH/GSSG Ratio | 28.2 ± 7.0 | 14.7 ± 6.2 | <0.0001 |
| Free GSH/GSSG Ratio | 7.9 ± 3.5 | 4.9 ± 2.2 | <0.0001 |
Means ± SD
Abbreviations: SAM: S-adenosylmethionine; SAH: S-adenosylhomocysteine; GSH: glutathione; GSSG: glutathione disulfide
Because the severity of clinical symptoms often varies widely between autistic children, the proportion of autistic children with more clinically severe metabolite alterations was also determined. Within the transmethylation pathway, a subset of 50% of autistic children had methionine levels less than 20 µmol/L (mean: 16.6 ± 1.8), 20% of the children had SAM levels <75 µmol/L (mean: 65 ± 8) and 19% had SAH levels >28 nmol/L (mean: 36.6 ± 5.6). A subset of 25% of children had adenosine levels > 0.30 µmol/L (mean: 0.43 ± 0.15). Within the transsulfuration pathway, 65% of the children had cysteine levels <170 µmol/L (mean 155 ±14) and 51% had total glutathione levels < 5.0 µmol/L (mean 4.2 ± 0.5). Free reduced GSH <1.5 µmol/L was present in 68% of the children (mean: 1.15 ± 0.2), whereas oxidized GSSG was above 0.35 µmol/L in 49% (mean: 0.53± 0.16). This subset analysis indicates that there are significant numbers of autistic children whose metabolic profiles are severely abnormal.
Genotyping
All genotype distributions were in Hardy Weinberg equilibrium and all control allele frequencies were consistent with previous reports.(Gueant-Rodriguez et al. 2005;Skibola et al. 2004;Palmatier et al. 1999;Geisler and Olshan 2001) In the univariate analysis, there were no significant differences in allele frequency or genotype distributions at the p < 0.05 level between autistic cases and unaffected controls for MTHFR 677C>T, MTHFR 1298A>C, GST T1 null, GCP 156C>T, or MTRR 66A>G. Significant increases in odds ratios, allele frequencies and genotype distributions among autistic children were found for RFC-1 80A>G, TCN2 776C>G, and COMT 472G>A genes. An increase in the frequencies of MTHFR 677CT and GST M1 null genotypes among autistic cases achieved borderline significance. A decrease in the MTRR homozygous GG genotype and G allele frequency also achieved borderline significance among cases. The odds ratios and 95% confidence intervals for these variant alleles are presented in Table 3.
Table III.
Allele frequencies, genotype distributions, odds ratios, and 95% confidence intervals (CI) in autistic cases and controls. Significant and borderline significant differences are in bold type.
| SNP | Genotype | Cases | Controls | |
|---|---|---|---|---|
| No. (%) | No. (%) | OR (95% CI) | ||
| RFC-1 | A | 290 (42) | 176 (49) | Reference |
| 80 A>G | G | 408 (58) | 182 (51) | 1.36 (1.04,1.7) |
| AA | 55 (16) | 51 (28) | Reference | |
| GA | 180 (52) | 74 (41) | 2.26 (1.37,3.7) | |
| GG | 114 (33) | 54 (26) | 1.96 (1.15,3.3) | |
| GA+GG | 294 (84) | 128 (63) | 2.13 (1.4, 3.4) | |
| COMT | A | 340 (47) | 215 (54%) | Reference |
| 472G>A | G | 376 (53) | 181 (46) | 1.31 (1.02,1.7) |
| AA | 86 (24) | 57 (29) | Reference | |
| AG | 168 (47) | 101 (51) | 1.10 (0.7, 1.7) | |
| GG | 105 (29) | 40 (20) | 1.74 (1.02, 2.9) | |
| TCN2 | C | 375 (52) | 231 (58) | Reference |
| 776 C>G | G | 346 (48) | 169 (42) | 1.25 (0.97, 1.6) |
| CC | 108 (30) | 63 (32) | Reference | |
| CG | 159 (44) | 105 (52) | 0.88 (0.58, 1.3) | |
| GG | 93 (26) | 32 (16) | 1.70 (1.02, 2.8) | |
| CC+CG | 268 (74) | 168 (84) | 0.55 (0.35, 0.8) | |
| GSTM1 | +/+ | 176 (49) | 115 (57) | Reference |
| NULL | 182 (51) | 86 (43) | 1.37 (0.98,1.96) | |
| MTHFR | C | 444 (62) | 276 (67) | Reference |
| 677C>T | T | 268 (38) | 134 (33) | 1.24 (0.96, 1.6) |
| CC | 134 (38) | 93 (45) | Reference | |
| CT | 176 (49) | 90 (44) | 1.36 (.92, 1.99) | |
| TT | 46 (13) | 22 (11) | 1.45 (.79, 2.71) | |
| CT+TT | 222 (62) | 112 (55) | 1.38 (.96, 1.98) | |
| MTRR | A | 348 (49) | 172 (43) | Reference |
| 676A>G | G | 368 (51) | 232 (57) | 0.78 (0.61, 1.02) |
| AA | 91 (25) | 37 (18) | Reference | |
| AG | 166 (46) | 98 (49) | 0.69 (0.42, 1.1) | |
| GG | 101 (28) | 67 (33) | 0.61 (0.36, 1.03) | |
| AG+GG | 267 (75) | 165 (82) | 0.66 (0.42, 1.03) |
Abbreviations: RFC: Reduced Folate Carrier; TCN2: Transcobalamin II; COMT: Catechol-O-methyltransferase; GST: Glutathione-S-transferase; MTHFR: Methylenetetrathydrofolate reductase: MTRR: methionine synthase reductase
The significant gene-gene interactions are presented in Table 4. Three genotype combinations were found to have odds ratios greater than the individual genotypes alone. Among the autistic children, 9.8% inherited the combined homozygous GG genotypes for COMT and TCN2 (4 mutant alleles) compared to 2.5% of control children, raising the odds ratio to 7.0. Homozygous or heterozygous combinations of RFC G allele and the MTHFR 677 T allele (3–4 mutant alleles) also resulted in significant increases in susceptibility to autism: GA/CT, OR 3.2; GA/TT, OR 4.4; and GG/CT, OR 3.1. There was also a significant interaction between the RFC-1 heterozygous GA and homozygous GG alleles and the GST M1 null genotype (3–4 mutant alleles) with odds ratios of 3.78 and 2.67, respectively. An increase in the frequency of compound heterozygous MTHFR 677CT/1298AC reached borderline significance among the autistic children with an OR of 1.78 and also showed an interaction with the RFC 80 G allele.
Table IV.
Gene-gene interactions
| SNP | Genotype | Cases | Controls | |
|---|---|---|---|---|
| No. | No. | OR (95% CI) | ||
| TCN2 776 C>G/COMT 472 G>A | CC/CC | 22 | 22 | Reference |
| GG/GG | 35 | 5 | 7.0 (2.32, 21.2) | |
| RFC-1 80 A>G/MTHFR677C>T | AA/CC | 22 | 28 | Reference |
| GA/CT | 89 | 35 | 3.24 (1.55, 6.78) | |
| GA/TT | 24 | 7 | 4.40 (1.45, 14.0) | |
| GG/CT | 58 | 24 | 3.10 (1.39, 6.84) | |
| RFC-1 80 A>G/GSTM1 Null | AA/++ | 23 | 29 | Reference |
| GA/Null | 90 | 30 | 3.78 (1.80, 7.95) | |
| GG/Null | 53 | 25 | 2.67 (1.22, 5.89) | |
| MTHFR 677 CT/MTHFR 1298AC | CT/AC | 85 | 39 | 1.78 (0.97, 3.26) |
| MTHFR 677CT/ 1298AC/RFC 80G | (CT/AC) / GA | 42 | 26 | 1.33 (1.33, 15.81) |
| (CT/AC) / GG | 25 | 12 | 3.57 (0.97, 13.49) |
DISCUSSION
Autism is recognized to have a complex etiology involving both genetic and environmental factors (Muhle et al. 2004;Keller and Persico 2003). The apparent requirement for an environmental trigger plus the genetic and clinical heterogeneity within autism spectrum disorders greatly complicates the search for candidate genes. The endophenotype represents a reproducible expression of the disease that lies between genes and clinical symptoms and may provide insights into susceptibility alleles (Gottesman and Gould 2003). For example, an endophenotype may be a biochemical, neurologic, hormonal, or immunologic biomarker associated with the disease state. Thus, the abnormal metabolic profile we have discovered in autistic children is an endophenotype that may reflect subtle changes in gene products that regulate flux through methionine transmethylation and transsulfuration pathways. Even small variations in gene expression and enzyme activity, if expressed chronically, could have a significant impact on downstream metabolic dynamics. The correlation between the severity and specificity of autistic symptoms and severity and the specificity of metabolite imbalance is of clinical interest and these studies are currently underway.
The significant decrease in total and free plasma glutathione as well as the GSH/GSSG redox ratio in the autistic children is of particular concern. Glutathione is a tripeptide of cysteine, glycine, and glutamate that is synthesized in every cell of the body. The essential intracellular reducing environment is maintained by the high ratio of reduced glutathione (GSH) to the oxidized disulfide (GSSG) form of glutathione (Schafer and Buettner 2001). The GSH/GSSG redox equilibrium regulates a pleiotropic range of functions that include nitrogen and oxygen free radical scavenger (Dickinson et al. 2003), protein redox status and enzyme activity (Klatt and Lamas 2000), cell membrane integrity and signal transduction (Sagristá et al. 2002;Dickinson and Forman 2002), transcription factor binding and gene expression (Deplancke and Gaskins 2002), phase II detoxification (Pastore et al. 2003), and apoptosis (Hall 1999). Under normal physiologic conditions, glutathione reductase enzyme activity is sufficient to maintain the high GSH/GSSG redox ratio. However, excessive intracellular oxidative stress that exceeds the capacity of GSSG reductase will result in GSSG export to the plasma in attempt to regain intracellular redox homeostasis. Thus, an increase in plasma GSSG is a strong indication of intracellular oxidative stress. Further, GSSG export represents a net loss of glutathione to the cell and increases the requirement for cysteine, the rate-limiting amino acid for glutathione synthesis. Of possible relevance, plasma cysteine levels were severely reduced in over 65% of the autistic children. It is important to note that cysteine is a “conditionally” essential amino acid that is dependent on adequate methionine status; thus, a decrease in methionine precursor levels effectively increases the requirement for preformed cysteine (Griffith 1999). The significant decrease in methionine, cysteine, and glutathione and the increase in plasma GSSG observed in the autistic children suggest that precursor availability is insufficient to maintain glutathione levels and normal redox homeostasis. Consistent with low glutathione levels and increased oxidative stress, autistic children would be expected to have difficulty resisting infection, resolving inflammation, and detoxifying environmental contaminants. Indeed, autistic children have been reported to suffer from recurrent infections (Konstantareas and Homatidis 1987), neuroinflammation (Zimmerman et al. 2005), gastrointestinal inflammation (Horvath and Perman 2002; Jyonouchi et al 2005), and impaired antioxidant and detoxification capacity (Yorbik et al. 2002;Zoroglu et al. 2004;Chauhan et al. 2004)
The abnormalities in the methionine transmethylation pathway in the autistic children are unusual. Reduced plasma methionine and SAM most often reflect a decrease in methionine synthase activity; however, a decrease in methionine synthase activity is most often associated with elevated homocysteine levels (Finkelstein 1998). Similarly, an increase SAH is generally a response to an increase in homocysteine due to the reversibility of the SAH hydrolase enzyme (Figure 1). Despite a significant decrease in methionine and increase in SAH levels, homocysteine levels were not increased in the autistic children. Although an increase in homocysteine would be anticipated, the modest decrease observed is most consistent with an upregulation of transsulfuration pathway in response to insufficient glutathione synthesis (Banerjee and Zou 2005;Mosharov et al. 2000) One explanation for the simultaneous elevation of SAH and adenosine observed in a subset of the children is a downstream defect in adenosine metabolism. An increase in adenosine is well known to bind to the active site of SAH hydrolase as a product inhibitor resulting in an increase SAH levels. Consistent with this possibility, previous studies have reported a decrease in adenosine deaminase activity (Stubbs et al. 1982) and a functional polymorphism in the adenosine deaminase gene in some children with autism (Bottini et al. 2001). The increase in SAH and adenosine in a subset of ~20% of autistic children is of clinical concern because SAH is a potent product inhibitor of most cellular methyltransferases. A low SAM/SAH ratio has been associated with impaired methylation capacity for membrane phosphatidylcholine synthesis and DNA methylation in humans (Innis et al. 2003;Yi et al. 2000). The functional consequences of these metabolic abnormalities on membrane dynamics and gene expression would be of considerable clinical interest especially since we have shown that the metabolic imbalance in autistic children is potentially reversible with targeted nutritional intervention (James et al. 2004). Studies are underway to determine whether treatment to normalize the metabolic imbalance will ameliorate behavioral symptoms.
Because abnormalities in transmethylation and transsulfuration pathways have been associated with heart disease, cancer, birth defects, and neurologic disorders (Saw 1999;Stover 2004;Hobbs et al. 2005;Mattson and Shea 2003), aberrations in these pathways have been well-studied and many enzyme-coding loci in these pathways have now been sequenced for common genetic polymorphisms. It is generally accepted that complex diseases are influenced by genetic alterations at multiple and variable loci that interact together to reach a threshold of toxicity that is critical for the expression of the disease (Jones and Szatmari 2002). Epistasis, or the interaction between genes, is increasingly recognized as an important analytic approach to study genetic contribution to complex disease (Cordell 2002;Jones and Szatmari 2002). Although epistasis is often used to infer biologic meaning from quantitative data, this approach may be tenuous when complex disease risk is the outcome. However, for gene-gene interactions that are involved in the regulation of a common metabolic pathway for which disease-related alterations have previously been demonstrated, a plausible biologic model can be postulated. In this case, epistasis not only contributes to the understanding of biological mechanisms, it also provides insights into genetic factors associated with disease susceptibility (Relton et al. 2004). Based on these considerations, we have initiated a study of candidate genes for proteins that have a functional impact on transmethylation or transsulfuration pathways and oxidative stress. We have used the metabolic endophenotype as a metabolic map for the selection of relevant candidate genes. On an individual level, genotype/metabolic phenotype analysis can provide clues for effective intervention and insights into the basis for individual differences in response to treatment. This is an important future goal that will require a much larger cohort of cases for meaningful correlations.
The reduced folate carrier (RFC) is present on the membrane of every cell and modulates the delivery of reduced folates into the cell (Matherly 2001). The G allele (glutamine>arginine) has been associated with increased risk of birth defects (De Marco et al. 2003) and elevated plasma folate as the result of impaired cell uptake (Yates and Lucock 2005). Relative to controls, autistic children had a significant increase in the frequency of the reduced folate carrier RFC-1 homozygous 80GG (33% vs. 26%) and heterozygous 80GA (52% vs. 41%). Children with either the RFC-1 GA or GG genotypes were approximately 2 times more likely to be autistic (OR: 2.26 and 1.96, respectively). Importantly, a significant interaction between heterozygous RFC-1 80GA genotype and the both the MTHFR 677CT and TT genotypes was observed among in the autistic children with odds ratios of 3.24 and 4.4, respectively. In addition, an interaction between the homozygous RFC-1 80GG and the MTHFR 677CT genotypes conferred a 3-fold increase autism susceptibility. Finally, an interaction between 3–4 loci was found for the compound heterozygous MTHFR 677CT/1298AC and the RFC 80AG and GG genotypes. The RFC-1 80G allele is associated with decreased intracellular folate transport and the MTHFR 677T allele reduces the synthesis of metabolically active folate. Thus, the significant interaction between these MTHFR and RFC genotypes would negatively affect intracellular folate status by two independent mechanisms. Together, common variants in the RFC and MTHFR genes conferred greater susceptibility to autism than either alone and suggest a potential etiologic role for impaired folate-dependent one-carbon metabolism in the susceptibility to autism. Consistent with low intracellular folate availability, methionine levels were decreased among most autistic children. Thus, the metabolic and genetic data support the possibility that the observed alterations in methionine metabolism may be due, in part, to a genetic predisposition for a functional folate deficiency.
Transcobalamin II is the major transport protein required for the cellular uptake of vitamin B12 by receptor-mediated endocytosis (Seetharam 1999). Previous studies indicate that a common 776 C>G transition in the TCN2 gene (proline>arginine) decreases the binding affinity of transcobalamin II for vitamin B12 and reduces the transport of B12 into cells (Afman et al. 2002;Afman et al. 2001;Miller et al. 2002). Vitamin B12 is an essential cofactor for the methionine synthase reaction and accepts the methyl group from 5-methylfolate to generate methionine from homocysteine in the initial step of the methionine transmethylation pathway (Figure 1). The frequency of the homozygous TCN2 776GG variant was significantly increased among the autistic children compared to controls (26% vs. 16%) and the GG variant was associated with a 1.7-fold increased risk of autism. In contrast, the combined wildtype and heterozygous TCN2 genotypes (CC + CG) had an odds ratio of 0.55. Of particular relevance to neurologic disorders, the TCN2 776 GG variant has been associated with lower levels of transcobalamin-bound B12 (holotranscobalamin II) in the cerebral spinal fluid of Alzheimer’s patients (Zetterberg et al. 2003). B12 deficiency is well known to have neuropsychiatric consequences in adults (Zucker et al. 1981) and adversely affect neurodevelopment during infancy (Graham et al. 1992). In toddlers, severe B12 deficiency has been associated with developmental regression similar to that observed in ~33% of autistic children (Grattan-Smith et al. 1997). It is provocative to note that the TCN2 GG variant would be expected to negatively affect B12 cofactor availability for the methionine synthase reaction just as the interaction between RFC-1 80G and MTHFR 677T alleles would be expected to reduce methylfolate availability for the same methionine synthase reaction. Although speculative, the low methionine levels found in many autistic children support the possible contribution of all three variant alleles, independently or combined, to impaired methionine synthesis. In addition, children with a genetic predisposition to impaired methionine synthesis would be especially vulnerable to further reduction in enzyme activity with exposure to endogenous or exogenous oxidative stress (Mosharov et al. 2000).
The third genetic variant found to be significantly more frequent among autistic children was the catechol-O-methyltransferase (COMT) 472G allele. The methylation of dopamine by COMT is an important mechanism for dopamine inactivation and dopaminergic tone in the CNS (Nieoullon 2002). The G>A transition at position 472 (valine>methionine) has been shown to influence protein expression and enzyme activity in an allelic dose/response manner (Chen et al. 2004). The val allele is associated with thermostability and high activity whereas the met allele is associated with low activity and thermolability (Chen et al. 2004). Compared with met carriers, individuals homozygous for the val allele showed poorer attentional control and performance on tests of executive cognition associated with inefficient precortical activity (Blasi et al. 2005). In other studies, the met allele, which encodes the low activity variant, was associated with better performance on tests of prefrontally mediated cognition (Egan et al. 2001;Diamond et al. 2004). The high activity homozygous GG (val/val) genotype was present in 29% of autistic cases and 20% of unaffected controls and was associated with a 1.74-fold increased susceptibility to autism. Unexpectedly, we found an apparent synergistic interaction between homozygous TCN2 GG and homozygous COMT GG genotypes (4 mutant alleles) that increased autism risk 7-fold. Both the TCN2 and COMT allelic variants would be expected to decrease CNS methionine and SAM levels by reducing availability and increasing consumption, respectively. A direct biochemical interaction between dopamine and vitamin B12 deficiencies is not known; however, independent deficiencies in both are well known to negatively affect neurologic function.
Marginal increases in variant allele frequency with borderline significance were found for the GST M1 null genotype (OR: 1.37; CI: 0.98, 1.96) and the combined MTHFR CT+TT genotypes (OR: 1.38; CI: 0.96, 1.98). Despite a modest independent effect, the MTHFR 677 T allele showed significant interactions with the RFC-1 G allele as described above. Similarly, the GST M1 null genotype achieved marginal significance in the univariate analysis, but showed a highly significant interaction with RFC-1 G allele. Children with combined RFC-1 heterozygous 80GA and GST M1 null genotypes had a 3.78-fold increased susceptibility to autism and children with both the RFC homozygous GG and GST M1 null genotypes had a 2.67-fold increase in risk. In contrast, a decrease in MTRR (methionine synthase reductase) homozygous GG genotype among autistic children was suggestive of a protective effect (OR: 0.61). This observation could be interpreted as the A allele representing the risk factor as was concluded for risk of neural tube defects (Relton et al. 2004).
Given the relatively small number of cases and controls in the present study, it encouraging to note that several susceptibility alleles that perturb a common metabolic pathway were increased among the autistic children. This supports the possibility that some forms of autism could be a manifestation of a genetic predisposition to abnormal methionine/glutathione metabolism and oxidative stress. Further, the abnormal metabolic profile observed in a significant proportion of autistic children suggests the provocative possibility that some autistic behaviors could be a neurologic manifestation of a genetically-based systemic metabolic derangement. Such a paradigm shift from a neurodevelopmental disorder to a broader systemic disorder would widen the biologic basis of autism to encompass not only the neurologic manifestations but also the gastrointestinal and immunologic pathology that have received increasing attention in recent years (Horvath and Perman 2002;Jyonouchi et al. 2005). Supporting this possibility, abnormalities in folate-dependent methionine and glutathione metabolism have been associated with gastrointestinal and immunologic dysfunction in addition to impaired CNS function (Bains and Shaw 1997;Martensson et al. 1990;Droge and Breitkreutz 2000). The hypothesis that a genetic component of autism could involve multiple susceptibility alleles that interact to create a fragile, environmentally-sensitive metabolic imbalance is worthy of further pursuit. Moreover, if some children with autism are confirmed to have an abnormal metabolic profile, treatment for this form of autism can be directed toward correcting the metabolic derangements and potentially ameliorating the autistic symptoms.
In summary, we have discovered two key metabolic abnormalities among many autistic children that are indications of significant impairment in methylation capacity (↓SAM/SAH) and in antioxidant capacity and redox homeostasis (↓GSH/GSSG). The significant increase in plasma GSSG levels indicates that these children are under oxidative stress. Preliminary genetic analysis indicates several polymorphic variants affecting methionine and glutathione metabolism are significantly increased among the autistic children supporting the possibility that the metabolic imbalance may be genetically-influenced. Clearly, these new findings should be considered preliminary until confirmed in larger population-based studies.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge and thank all of the autistic children and their families who made this study possible. We thank Laurette Janak (Buffalo, NY) for her help with planning and study logistics. The study was supported by grants from the National Institute of Child Health and Human Development #5R01 HD39054-05, the Autism Research Institute, and the National Center for Research Resources (1C06 RR16517-01 and 3C06 RR16517-01S1)
Reference List
- Afman LA, Lievers KJ, Van der Put NM, Trijbels FJ, Blom HJ. Single nucleotide polymorphisms in the transcobalamin gene: relationship with transcobalamin concentrations and risk for neural tube defects. Eur J Hum Genet. 2002;10:433–438. doi: 10.1038/sj.ejhg.5200830. [DOI] [PubMed] [Google Scholar]
- Afman LA, Van der Put NMJ, Thomas CMG, Trijbels JMF, Blom HJ. Reduced vitamin B12 binding by transcobalamin II increases the risk of neural tube defects. Q J Med. 2001;94:159–166. doi: 10.1093/qjmed/94.3.159. [DOI] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Bailey LB, Gregory JF., III Polymorphisms of methylenetetrahydrofolate reductase and other enzymes; metabolic significance, risks and impact on folate requirement. J Nutr , Supplement: Recent Advances in Nutritional Sciences. 2000;130:919–922. doi: 10.1093/jn/129.5.919. [DOI] [PubMed] [Google Scholar]
- Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res Brain Res Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Zou CG. Redox regulation and reaction mechanism of human cystathionine-β-synthase: a PLP-dependent hemesensor protein. Archives of Biochemistry and Biophysics. 2005;433:144–156. doi: 10.1016/j.abb.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. The neuropathology of the autism spectrum disorders: what have we learned? Novartis Found Symp. 2003;251:112–122. [PubMed] [Google Scholar]
- Beagle B, Yang TL, Hung J, Cogger EA, Moriarty DJ, Caudill MA. The glycine N-methyltransferase (GNMT) 1289 C->T variant influences plasma total homocysteine concentrations in young women after restricting folate intake. J Nutr. 2005;135:2780–2785. doi: 10.1093/jn/135.12.2780. [DOI] [PubMed] [Google Scholar]
- Blasi G, Mattay VS, Bertolino A, Elvevåg B, Callicott JH, Das S, Kolachana BS, Egan MF, Goldberg TE, Weinberger DR. Effect of catechol-O-methyltransferase val158met genotype on attentional control. J Neurosci. 2005;25:5038–5045. doi: 10.1523/JNEUROSCI.0476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottini N, De Luca D, Saccucci P, Fiumara A, Elia M, Porfirio MC, Lucarelli P, Curatolo P. Autism: evidence of association with adenosine deaminase genetic polymorphism. Neurogenetics. 2001;3:111–113. doi: 10.1007/s100480000104. [DOI] [PubMed] [Google Scholar]
- CDC. 2005 http://medicalhomeinfo.org/health/Autism%20downloads/AutismAlarm.pdf.
- Castel-Dunwoody KM, Kauwell GP, Shelnutt KP, Vaughn JD, Griffin ER, Maneval DR, Theriaque DW, Bailey LB. Transcobalamin 776C->G polymorphism negatively affects vitamin B-12 metabolism. Am J Clin Nutr. 2005;81:1436–1441. doi: 10.1093/ajcn/81.6.1436. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Chauhan V, Brown WT, Cohen I. Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin--the antioxidant proteins. Life Sci. 2004;75:2539–2549. doi: 10.1016/j.lfs.2004.04.038. [DOI] [PubMed] [Google Scholar]
- Chen JS, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, Kolachana BS, Hyde TM, Herman MM, Apud J, Egan MF, Kleinman JE, Weinberger DR. Functional analysis of genetic variation in catechol-o-methyltransferase (COMT): Effects on mRNA, protein, and enzyme activity in postmortem human brain. American Journal of Human Genetics. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves MA. Hardy-Weinberg equilibrium tests and allele frequency estimation. STATA Technical Bull. 1999;48:34–37. [Google Scholar]
- Cordell HJ. Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- De Marco P, Calevo MG, Moroni A, Merello E, Raso A, Finnell RH, Zhu HP, Andreussi L, Cama A, Capra V. Reduced folate carrier polymorphism (80A --> G) and neural tube defects. Eur J Hum Genet. 2003;11:245–252. doi: 10.1038/sj.ejhg.5200946. [DOI] [PubMed] [Google Scholar]
- Deplancke B, Gaskins HR. Redox control of the transsulfuration and glutathione biosynthesis pathways. Curr Opin Clin Nutr Metab Care. 2002;5:85–92. doi: 10.1097/00075197-200201000-00015. [DOI] [PubMed] [Google Scholar]
- Diamond A, Briand L, Fossella J, Gehlbach L. Genetic and neurochemical modulation of prefrontal cognitive functions in children. Am J Psychiatry. 2004;161:125–132. doi: 10.1176/appi.ajp.161.1.125. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Moellering DR, Iles KE, Patel RP, Levonen AL, Wigley A, Darley-Usmar VM, Forman HJ. Cytoprotection against oxidative stress and the regulation of glutathione synthesis. Biol Chem. 2003;384:527–537. doi: 10.1515/BC.2003.061. [DOI] [PubMed] [Google Scholar]
- Droge W, Breitkreutz R. Glutathione and immune function. Proc Nutr Soc. 2000;59:595–600. doi: 10.1017/s0029665100000847. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, Straub RE, Goldman D, Weinberger DR. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigsti IM, Shapiro T. A systems neuroscience approach to autism: Biological, cognitive, and clinical perspectives. Ment Retard Dev Disabil Res Rev. 2003;9:205–215. doi: 10.1002/mrdd.10081. [DOI] [PubMed] [Google Scholar]
- Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- Finkelstein JD. The metabolism of homocysteine: pathways and regulation. Eur J Pediatr. 1998;157:S40–S44. doi: 10.1007/pl00014300. [DOI] [PubMed] [Google Scholar]
- Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- Geisler SA, Olshan AF. GSTM1, GSTT1, and the risk of squamous cell carcinoma of the head and neck: a mini-HuGE review. Am J Epidemiol. 2001;154:95–105. doi: 10.1093/aje/154.2.95. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Graham SM, Arvela OM, Wise GA. Long-term neurologic consequences of nutritional vitamin B12 deficiency in infants. J Pediatr. 1992;121:710–714. doi: 10.1016/s0022-3476(05)81897-9. [DOI] [PubMed] [Google Scholar]
- Grattan-Smith PJ, Wilcken B, Procopis PG, Wise GA. The neurological syndrome of infantile cobalamin deficiency: developmental regression and involuntary movements. Mov Disord. 1997;12:39–46. doi: 10.1002/mds.870120108. [DOI] [PubMed] [Google Scholar]
- Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- Gueant JL, Gueant-Rodriguez RM, Anello G, Bosco P, Brunaud L, Romano C, Ferri R, Romano A, Candito M, Namour B. Genetic determinants of folate and vitamin B12 metabolism: a common pathway in neural tube defect and Down syndrome? Clin Chem Lab Med. 2003;41:1473–1477. doi: 10.1515/CCLM.2003.226. [DOI] [PubMed] [Google Scholar]
- Gueant-Rodriguez RM, Juilliere Y, Candito M, Adjalla CE, Gibelin P, Herbeth B, Van Obberghen E, Gueant JL. Association of MTRRA66G polymorphism (but not of MTHFR C677T and A1298C, MTRA2756G, TCN C776G) with homocysteine and coronary artery disease in the French population. Thromb Haemost. 2005;94:510–515. doi: 10.1160/TH05-04-0262. [DOI] [PubMed] [Google Scholar]
- Hall AG. The role of glutathione in the regulation of apoptosis. European Journal of Clinical Investigation. 1999;29:238–245. doi: 10.1046/j.1365-2362.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- Hobbs CA, Cleves JA, Melnyk S, Zhao WZ, James SJ. Congenital heart defects and abnormal maternal biomarkers of methionine and homocysteine metabolism. Am J Clin Nutr. 2005;81:147–153. doi: 10.1093/ajcn/81.1.147. [DOI] [PubMed] [Google Scholar]
- Hornig M, Lipkin WI. Infectious and immune factors in the pathogenesis of neurodevelopmental disorders: epidemiology, hypotheses, and animal models. Ment Retard Dev Disabil Res Rev. 2001;7:200–210. doi: 10.1002/mrdd.1028. [DOI] [PubMed] [Google Scholar]
- Horvath K, Perman JA. Autistic disorder and gastrointestinal disease. Curr Opin Pediatr. 2002;14:583–587. doi: 10.1097/00008480-200210000-00004. [DOI] [PubMed] [Google Scholar]
- Innis SM, Davidson AGF, Chen A, Dyer R, Melnyk S, James SJ. Increased plasma homocysteine and S-adenosylhomocysteine and decreased methionine is associated with altered phosphatidylcholine and phosphatidylethanolamine in cystic fibrosis. J Pediatr. 2003;143:351–356. doi: 10.1067/S0022-3476(03)00326-3. [DOI] [PubMed] [Google Scholar]
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, Neubrander JA. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004;80:1611–1617. doi: 10.1093/ajcn/80.6.1611. [DOI] [PubMed] [Google Scholar]
- James SJ, Pogribna M, Pogribny IP, Melnyk S, Hine RJ, Gibson JB, Yi P, Tafoya DL, Swenson DH, Wilson VL, Gaylor DW. Abnormal folate metabolism and mutation in the methylenetetrahydrofolate reductase gene may be maternal risk factors for Down syndrome. Am J Clin Nutr. 1999;70:495–501. doi: 10.1093/ajcn/70.4.495. [DOI] [PubMed] [Google Scholar]
- Jones MB, Szatmari P. A risk-factor model of epistatic interaction, focusing on autism. American Journal of Medical Genetics. 2002;114:558–565. doi: 10.1002/ajmg.10513. [DOI] [PubMed] [Google Scholar]
- Jyonouchi H, Geng L, Ruby A, Zimmerman-Bier B. Dysregulated innate immune responses in young children with autism spectrum disorders: their relationship to gastrointestinal symptoms and dietary intervention. Neuropsychobiology. 2005;51:77–85. doi: 10.1159/000084164. [DOI] [PubMed] [Google Scholar]
- Keller F, Persico AM. The neurobiological context of autism. Mol Neurobiol. 2003;28:1–22. doi: 10.1385/MN:28:1:1. [DOI] [PubMed] [Google Scholar]
- Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- Konstantareas MM, Homatidis S. Ear infections in autistic and normal children. J Autism Dev Disord. 1987;17:585–594. doi: 10.1007/BF01486973. [DOI] [PubMed] [Google Scholar]
- Krause I, He XS, Gershwin ME, Shoenfeld Y. Brief report: immune factors in autism: a critical review. J Autism Dev Disord. 2002;32:337–345. doi: 10.1023/a:1016391121003. [DOI] [PubMed] [Google Scholar]
- Lievers KJ, Kluijtmans LA, Blom HJ. Genetics of hyperhomocysteinaemia in cardiovascular disease. Ann Clin Biochem. 2003;40:46–59. doi: 10.1258/000456303321016169. [DOI] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH, Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30:205–223. [PubMed] [Google Scholar]
- Martensson J, Jain A, Meister A. Glutathione is required for intestinal function. Proc Natl Acad Sci U S A. 1990;87:1715–1719. doi: 10.1073/pnas.87.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matherly LH. Molecular and cellular biology of the human reduced folate carrier. Prog Nucleic Acid Res Mol Biol. 2001;67:131–162. doi: 10.1016/s0079-6603(01)67027-2. [DOI] [PubMed] [Google Scholar]
- Mato JM, Corrales FJ, Lu SC, Avila MA. S-adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Shea TB. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends in Neurosciences. 2003;26:137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Miller AL. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern Med Rev. 2003;8:7–19. [PubMed] [Google Scholar]
- Miller JW, Ramos MI, Garrod MG, Flynn MA, Green R. Transcobalamin II 775G>C polymorphism and indices of vitamin B12 status in healthy older adults. Blood. 2002;100:718–720. doi: 10.1182/blood-2002-01-0209. [DOI] [PubMed] [Google Scholar]
- Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- Muhle R, Trentacoste SV, Rapin I. The genetics of autism. Pediatr. 2004;113:e472–e486. doi: 10.1542/peds.113.5.e472. [DOI] [PubMed] [Google Scholar]
- Muntjewerff JW, Van der Put N, Eskes T, Ellenbroek B, Steegers E, Blom H, Zitman F. Homocysteine metabolism and B-vitamins in schizophrenic patients: low plasma folate as a possible independent risk factor for schizophrenia. Psychiatry Res. 2003;121:1–9. doi: 10.1016/s0165-1781(03)00200-2. [DOI] [PubMed] [Google Scholar]
- Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67:53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- Palmatier MA, Kang AM, Kidd KK. Global variation in the frequencies of functionally different catechol-O-methyltransferase alleles. Biol Psychiatry. 1999;46:557–567. doi: 10.1016/s0006-3223(99)00098-0. [DOI] [PubMed] [Google Scholar]
- Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta. 2003;333:19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- Pogribna M, Melnyk S, Pogribny I, Chango A, Yi P, James SJ. Homocysteine metabolism in children with Down syndrome: In vitro modulation. American Journal of Human Genetics. 2001;69:88–95. doi: 10.1086/321262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton CL, Wilding CS, Pearce MS, Laffling AJ, Jonas PA, Lynch SA, Tawn EJ, Burn J. Gene-gene interaction in folate-related genes and risk of neural tube defects in a UK population. J Med Genet. 2004;41:256–260. doi: 10.1136/jmg.2003.010694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagristá ML, García AF, De Madariaga MA, Mora M. Antioxidant and pro-oxidant effect of the thiolic compounds N-acetyl-L-cysteine and glutathione against free radical-induced lipid peroxidation. Free Radical Research. 2002;36:329–340. doi: 10.1080/10715760290019354. [DOI] [PubMed] [Google Scholar]
- Saw SM. Homocysteine and atherosclerotic disease: the epidemiologic evidence. Ann Acad Med Singapore. 1999;28:565–568. [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267:4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- Seetharam B. Receptor-mediated endocytosis of cobalamin (vitamin B12) Annual Review of Nutrition. 1999;19:173–195. doi: 10.1146/annurev.nutr.19.1.173. [DOI] [PubMed] [Google Scholar]
- Serra JA, Dominguez RO, de Lustig ES, Guareschi EM, Famulari AL, Bartolome EL, Marschoff ER. Parkinson's disease is associated with oxidative stress: comparison of peripheral antioxidant profiles in living Parkinson's, Alzheimer's and vascular dementia patients. J Neural Transm. 2001;108:1135–1148. doi: 10.1007/s007020170003. [DOI] [PubMed] [Google Scholar]
- Skibola CF, Forrest MS, Coppede F, Agana L, Hubbard A, Smith MT, Bracci PM, Holly EA. Polymorphisms and haplotypes in folate-metabolizing genes and risk of non-Hodgkin lymphoma. Blood. 2004;104:2155–2162. doi: 10.1182/blood-2004-02-0557. [DOI] [PubMed] [Google Scholar]
- Stern LL, Shane B, Bagley PJ, Nadeau M, Shih V, Selhub J. Combined marginal folate and riboflavin status affect homocysteine methylation in cultured immortalized lymphocytes from persons homozygous for the MTHFR C677T mutation. Journal of Nutrition. 2003;133:2716–2720. doi: 10.1093/jn/133.9.2716. [DOI] [PubMed] [Google Scholar]
- Stover PJ. Physiology of folate and vitamin B12 in health and disease. Nutr Rev. 2004;62:S3–S12. doi: 10.1111/j.1753-4887.2004.tb00070.x. [DOI] [PubMed] [Google Scholar]
- Stubbs G, Litt M, Lis E, Jackson R, Voth W, Lindberg A, Litt R. Adenosine deaminase activity decreased in autism. J Am Acad Child Psychiatry. 1982;21:71–74. doi: 10.1097/00004583-198201000-00012. [DOI] [PubMed] [Google Scholar]
- Tager-Flusberg H, Joseph RM. Identifying neurocognitive phenotypes in autism. Philos Trans R Soc Lond B Biol Sci. 2003;358:303–314. doi: 10.1098/rstb.2002.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JF. Intestinal pathophysiology in autism. Exp Biol Med (Maywood ) 2003;228:639–649. doi: 10.1177/153537020322800601. [DOI] [PubMed] [Google Scholar]
- Yates Z, Lucock M. G80A reduced folate carrier SNP modulates cellular uptake of folate and affords protection against thrombosis via a non homocysteine related mechanism. Life Sci. 2005;77:2735–2742. doi: 10.1016/j.lfs.2005.02.029. [DOI] [PubMed] [Google Scholar]
- Ye Z, Parry JM. Genetic polymorphisms in the cytochrome p450 1A1, glutathione S-transferase M1 and T1, and susceptibility to colon cancer. Teratogenesis Carcinog Mutagen. 2002;22:385–392. doi: 10.1002/tcm.10035. [DOI] [PubMed] [Google Scholar]
- Yi P, Melnyk S, Pogribna M, Pogribny IP, Hines RJ, James SJ. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. Journal of Biological Chemistry. 2000;275:29318–29323. doi: 10.1074/jbc.M002725200. [DOI] [PubMed] [Google Scholar]
- Yorbik O, Sayal A, Akay C, Akbiyik DI, Sohmen T. Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot Essent Fatty Acids. 2002;67:341–343. doi: 10.1054/plef.2002.0439. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Nexo E, Regland B, Minthon L, Boson R, Palmer M, Rymo L, Blennow K. The transcobalamin (TC) codon 259 genetic polymorphism influences holo-TC concentration in cerebrospinal fluid from patients with Alzheimer disease. Clin Chem. 2003;49:1195–1198. doi: 10.1373/49.7.1195. [DOI] [PubMed] [Google Scholar]
- Zimmerman AW, Jyonouchi H, Comi AM, Connors SL, Milstien S, Varsou A, Heyes MP. Cerebrospinal fluid and serum markers of inflammation in autism. Pediatr Neurol. 2005;33:195–201. doi: 10.1016/j.pediatrneurol.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Zoroglu SS, Armutcu F, Ozen S, Gurel A, Sivasli E, Yetkin O, Meram I. Increased oxidative stress and altered activities of erythrocyte free radical scavenging enzymes in autism. Eur Arch Psychiatry Clin Neurosci. 2004;254:143–147. doi: 10.1007/s00406-004-0456-7. [DOI] [PubMed] [Google Scholar]
- Zucker DK, Livingston RL, Nakra R, Clayton PJ. B12 deficiency and psychiatric disorders: case report and literature review. Biol Psychiatry. 1981;16:197–205. [PubMed] [Google Scholar]