

Abstract
Vibrio parahaemolyticus harbours two distinct type III secretion systems (T3SS1 and T3SS2). A subset of 10 T3SS1 genes are transcribed when V. parahaemolyticus is grown in tissue culture medium [Dulbecco's modified Eagle's medium (DMEM)], while transcription of these genes (except exsD) is minimal upon growth in Luria–Bertani-Salt (LB-S). Transcription of T3SS1 genes and cytotoxicity towards HeLa cells was prevented by deletion of exsA while complementation with exsA restored these traits. Overexpression of ExsA in the wild-type strain, NY-4, activated the transcription of T3SS1 genes when bacteria were grown in LB-S. Thus, ExsA is necessary and sufficient to induce the transcription of T3SS1 genes. Deletion of the exsD permitted the transcription of T3SS1 genes when bacteria were grown in the LB-S medium and complementation with the wild-type exsD gene-blocked transcription of T3SS1 genes. Overexpression of ExsD in NY-4 prevented the transcription of T3SS1 gene when bacteria were grown in DMEM. A gel mobility shift assay demonstrated that purified ExsA protein binds a novel motif in the upstream region of vp1668 and vp1687, indicating that ExsA interacts directly with the promoter sequences of T3SS1 genes. ExsA positively regulates the expression and secretion of Vp1656 while ExsD negatively regulates the expression and secretion of Vp1656.
Introduction
Vibrio parahaemolyticus is a Gram-negative, halophilic pathogen that is commonly associated with consumption of raw or undercooked seafood (Joseph et al., 1982). Infection with V. parahaemolyticus results in diarrhoea, nausea, vomiting, headache, fever and chills. In addition to typical gastrointestinal infections, c. 5% of V. parahaemolyticus infections advance to septicemia (Hlady and Klontz, 1996) and these infections may be fatal, especially in immunocompromised patients or those with pre-existing medical conditions, such as liver disease or diabetes (Yeung and Boor, 2004).
Thermostable direct haemolysin (TDH) is a major virulence factor of V. parahaemolyticus. TDH forms pores in red blood cells, causes increased short circuit current, increases Cl- secretion in human colonic epithelia cells and enhances Ca2+ entry from the extracellular medium (Takahashi et al., 2001). TDH-related haemolysin is another toxin (Sochard and Colwell, 1977) that is heat-labile and also induces Ca2+-activated Cl- channel leading to altered ion flux and secretory diarrhoea (Takahashi et al., 2001). A tdh deletion mutant has reduced the ability to cause fluid accumulation in ileal loops of a rabbit model (Lin et al., 1993). In contrast, Park et al. (2004) recently demonstrated that a tdh deletion mutant retains the ability to cause fluid accumulation. Furthermore, both V. parahaemolyticus TDH-positive and -negative strains disrupt epithelial tight junctions, possibly resulting in the dissemination of bacteria into the host circulation system (Lynch et al., 2005). These studies indicate that there are factors, in addition to TDH, that contribute to the pathogenesis of V. parahaemolyticus. Some of these include cell-associated haemagglutinin (Nagayama et al., 1994), pili (Nakasone and Iwanaga, 1990, 1992), vibrioferrin (Dai et al., 1992) and ToxR (Lin et al., 1993).
Type III secretion systems (T3SSs) enable bacteria to translocate proteins directly into host cells where they interfere with normal cellular functions (Cornelis and Van Gijsegem, 2000; Cornelis, 2006). T3SSs are divided into three parts: the secretion apparatus, translocators and effectors (Hueck, 1998). The secretion apparatus forms an injector complex in the surface of Gram-negative bacteria. The translocator is a needle structure, which when inserted into the membrane of host cells, allows the effectors to be transported through the cell membrane into the cytosol or other compartments of host cells (Hueck, 1998). While genes encoding the secretion apparatus are conserved among different bacteria, genes encoding effectors can be unique to different organisms (Troisfontaines and Cornelis, 2005).
The genome sequence of V. parahaemolyticus shows that this organism harbours two distinct T3SSs encoded in chromosomes 1 (T3SS1) and 2 (T3SS2) (Makino et al., 2003). T3SS1 is similar to the Ysc secretion system in Yersinia and the T3SS2 is similar to the Inv-Mxi-Spa secretion system in Salmonella and Shigella (Troisfontaines and Cornelis, 2005). The Ysc secretion system is typically associated with cytotoxicity while the Inv-Mxi-Spa secretion system usually contributes to host cell invasion (Troisfontaines and Cornelis, 2005). T3SS1 of V. parahaemolyticus induces host cell death characterized by cell swelling, vacuole formation in the cytosol and pore formation in the membrane of host cells that is caspase-independent (X. Zhou, M.E. Konkel, and D.R. Call, submitted). T3SS2 appears to be involved in the intestinal fluid accumulation (Park et al., 2004).
Secretion of effector proteins is stimulated by contact with host cells, but secretion can also be induced by growing bacteria under suitable conditions (Hueck, 1998). For example, the T3SSs of Pseudomonas and Yersinia can be activated by growing in media containing a chelator, such as nitriloacetate or ethylene glycol tetraacetic acid, which is conventionally referred to as low-calcium media (Straley et al., 1993; Vallis et al., 1999). Activation of the T3SS of Shigella is induced by culturing bacteria in media containing Congo red (Bahrani et al., 1997). The mechanism of transcriptional control of T3SS genes in some bacteria has been described. For example, expression of Pseudomonas T3SS genes in low-calcium media is controlled at the transcriotional level by an AraC-like transcriptional activator, ExsA (Yahr et al., 1995). ExsA binds to a consensus sequence (TNAAANA) approximately 50 bp pairs upstream of the transcriptional start site of the T3SS genes, leading to the transcription of T3SS genes (Hovey and Frank, 1995). ExsA also binds a negative regulator, ExsD, whose overexpression inhibits T3SS gene transcription (McCaw et al., 2002).
The T3SS1 of V. parahaemolyticus is composed of c. 42 genes (vp1656–vp1697) of which 30 genes have sequence similarity with genes encoding the apparatus in Yersinia while the remaining 12 open reading frames are hypothetical genes and may encode effectors proteins. It is unclear how the transcriptional activation of these 42 genes is controlled. At the terminus of the T3SS1 gene cluster, there are two genes, vp1699 and vp1698, that share sequence similarity with the Pseudomonas genes exsA (40%) and exsD (30%) respectively. We hypothesize that the V. parahaemolyticus vp1699 and vp1698, designated from this point forward as exsA and exsD, serve as transcriptional regulators in V. parahaemolyticus. In this study, we examined the conditions that induce the transcription of T3SS genes and demonstrate how ExsA and ExsD regulate transcription of T3SS1 genes.
Results
T3SS1 genes of V. parahaemolyticus are transcribed in Dulbecco's modified Eagle's medium, but not transcribed in Luria–Bertani-salt media
Vibrio parahaemolyticus was grown in Luria–Bertani (LB) medium supplemented with 2.5% sodium chloride [LB-salt (LB-S)] and Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% FBS. We selected 10 T3SS1 genes from the T3SS1 gene cluster to monitor gene expression by RT-PCR. All 10 genes were transcribed when bacterial were grown in DMEM (Fig. 1). Only exsD was clearly transcribed when bacterial were grown in LB-S (Fig. 1). Vp1656, vp1670, vp1687 and vp1696 were very weakly transcribed (faint bands) when bacteria were grown in LB-S (Fig. 1). Transcription of the house-keeping gene, secY, was similar for all culture conditions. In the absence of reverse transcriptase, PCR results were negative, indicating no DNA contamination for these samples. These results demonstrated that T3SS1 genes of V. parahaemolyticus are transcribed when cultured in DMEM. Hereafter we refer to DMEM and LB-S as the inducing condition and non-inducing condition, respectively, for the transcription of T3SS1 genes.
Fig. 1.
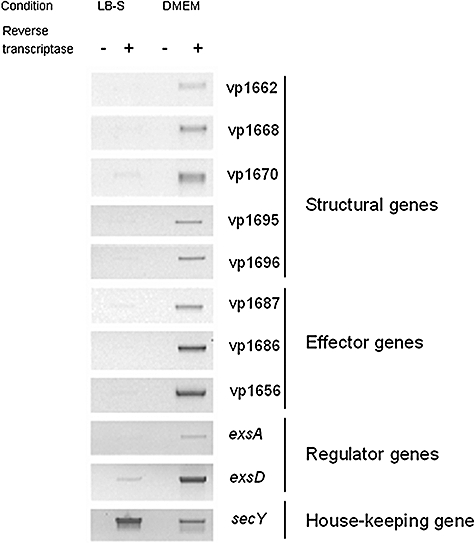
RT-PCR showing expression of several apparatus, effector and hypothetical regulatory genes from T3SS1. cDNA (+) made from total RNA isolated from V. parahaemolyticus strain NY-4 under different conditions (LB-S, DMEM) was used as template for PCR. Mock reactions (−), which did not contain reverse transcriptase in the RT reaction, were used as controls against genomic contamination of the RNA preparations. SecY was a house-keeping gene used as an internal control to ensure that RNA was present in all samples.
ExsA is required for cytotoxicity induced by wild-type V. parahaemolyticus
Because ExsA is a positive regulator of T3SS genes in Pseudomonas, we determined if an exsA knockout is attenuated for cytotoxicity. Cytotoxicity was determined by measuring the lactate dehydrogenase (LDH) release by HeLa cells after infection with V. parahaemolyticus strains. Four hours after infection with the wild-type NY-4 strain, c. 95% of the HeLa cells were lysed, which is consistent with a previous report (X. Zhou, M.E. Konkel, and D.R. Call, submitted), while the ΔexsA strain induced less than 5% of HeLa cell death (Fig. 2A). HeLa cells infected with wild-type strain appeared rounded (Fig. 2C) while HeLa cell infected with ΔexsA strain were similar to uninfected cells (Fig. D and data not shown). To avoid the possibility that deletion of exsA had polar effects, we ectopically expressed ExsA-6xHis in a plasmid and measured if cytotoxicity was restored. Western blot showed that the predicted protein was expressed in ΔexsA complemented with an exsA plasmid (Fig. 2B, lane 3) and the complemented strain recovered the full phenotype with c.100% of the HeLa cells lysed after infection (Fig. 2A). Cell rounding was also restored when HeLa cells were infected with complemented strain (Fig. 2E). These results demonstrated that ExsA is required for V. parahaemolyticus to induce host cell death.
Fig. 2.
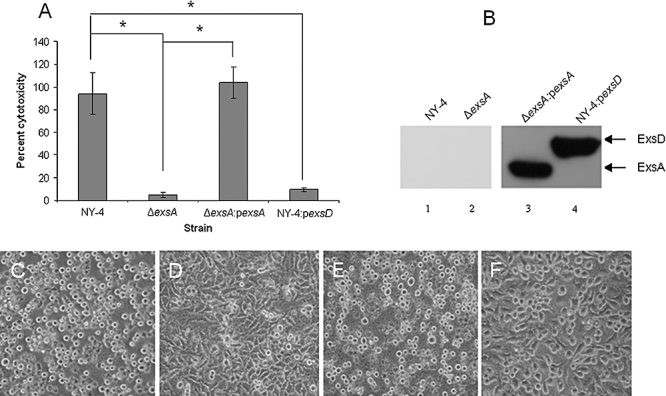
Mutation of exsA gene and overexpression of exsD gene in wild-type strains attenuated the ability of V. parahaemolyticus to induce host cell death.
A. HeLa cells were lysed by wild-type (NY-4) and exsA complement (ΔexsA : pexsA) strains of V. parahaemolyticus while strain ΔexsA and wild-type strain transformed with exsD plasmid (NY-4 : pexsD) had significantly lower levels of cytotoxicity against HeLa cells (bars = standard deviation for three replicates; star represents statistical difference P < 0.05).
B. Expression of exsA and exsD in ΔexsA and wild-type strains respectively. Anti-His antibody detected the ExsA and ExsD in the strains of ΔexsA : pexsA and NY-4 : pexsD respectively (Lanes 3 and 4), but not in the wild-type or ΔexsA controls. Morphological observations under light microscope for HeLa cells infected with (C) NY-4 strain, (D) ΔexsA strain, (E) ΔexsA : pexsA strain and (F) NY-4 : pexsD strain.
ExsA is required for the transcription of T3SS1 genes in DMEM and is sufficient to activate the transcription of T3SS1 genes in LB-S medium
Deletion of the exsA eliminated transcription of T3SS1 genes in the DMEM while expression of ExsA ectopically from a plasmid restored the transcription of T3SS1 genes (Fig. 3), indicating that ExsA is necessary for the transcription of T3SS1 genes in DMEM. Ectopic expression of ExsA in the wild-type strain permitted the transcription of T3SS1 genes even when bacteria were grown in non-inducing medium (i.e. LB-S) (Fig. 3), indicating that ExsA is sufficient to activate the transcription of T3SS1 genes even when bacterial were grown in non-inducing conditions. These results also indicate that T3SS1 transcriptional activation by environmental signals requires transcription of exsA.
Fig. 3.
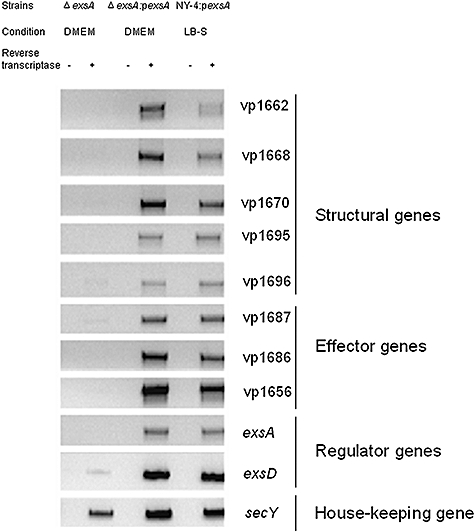
RT-PCR showing expression of T3SS1 genes and secY. cDNA (+) made from total RNA isolated from ΔexsA, ΔexsA : pexsA strains under the inducing (DMEM) growth condition, and NY-4 : pexsA strains under non-inducing (LB-S) growth condition was used as template for the RT reactions. Mock reactions (−), which did not contain reverse transcriptase, were used as controls against genomic contamination of the RNA preparations.
ExsD is a negative regulator of T3SS1 genes of V. parahaemolyticus
Because exsD in Pseudomonas negatively regulates the transcription of T3SS genes, we determined if V. parahaemolyticus vp1698, the putative exsD homologue, has a similar function. Deletion of exsD permitted transcription of T3SS1 genes when bacteria were grown in LB-S, indicating that transcriptional suppression of T3SS1 genes in non-inducing condition is due to the expression of exsD (Fig. 4). To determine if T3SS1 genes of the ΔexsD strain growing in LB-S are transcribed at levels comparable to the wild-type strain under inducing conditions, we diluted the cDNA templates from NY-4 strain growing in DMEM and ΔexsD strain growing in LB-S and tested these dilutions using RT-PCR. The results showed that the abundance of expression of each T3SS1 gene is similar between NY-4 growing in DMEM and ΔexsD growing in LB-S, consistent with T3SS1 genes being transcribed at normal levels for the ΔexsD strain grown in LB-S (Fig. S1). To control for any polar effects of exsD deletion, the ΔexsD strain was provided with a wild-type exsD-6xHis gene in trans and the ectopic expression of ExsD was confirmed by Western blot (Fig. 2B, lane 4). Complementation of ΔexsD strain with a wild-type exsD restored inhibition of T3SS1 gene transcription in non-inducing conditions, indicating that expression of ExsD is sufficient to repress the transcription of T3SS1 genes in non-inducing condition (Fig. 4). Ectopic expression of ExsD in the wild-type strain prevented transcription of T3SS1 genes when the bacteria were grown in DMEM. Furthermore, ectopic expression of ExsD significantly reduced the ability of V. parahaemolyticus wild-type strain to induce host cell death (Fig. 2A and F). These results demonstrated that ExsD is a negative regulator of T3SS1 gene transcription in V. parahaemolyticus.
Fig. 4.
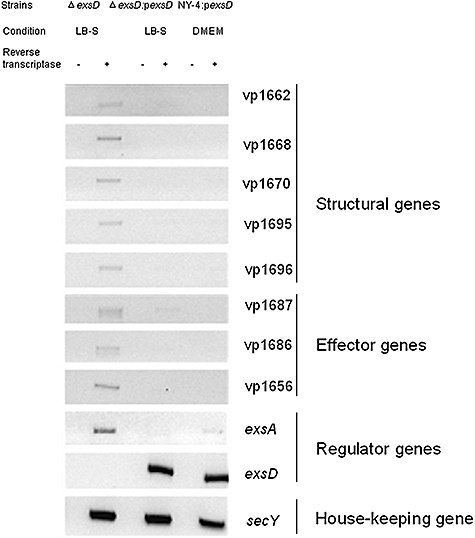
RT-PCR showing expression of T3SS1 genes and secY. cDNA (+) made from total RNA isolated from ΔexsD, ΔexsD : pexsD under the non-inducing (LB-S) growth condition and NY-4 : pexsD under inducing (DMEM) growth condition was used as template for the RT reactions. Mock reactions (−), which did not contain reverse transcriptase, were used as controls against genomic contamination of the RNA preparations.
ExsA but not ExsD binds the promoter sequences of vp1668 and vp1687
We determined if purified proteins ExsA and ExsD were able to bind the promoter sequences of T3SS1 genes. We selected promoter regions upstream of vp1668 and vp1687 because these two regions have hypothetical binding motifs. His-tagged recombinant ExsA, ExsD and Vp1656 proteins were purified to apparent homogeneity as shown by SDS-PAGE and Coomassie brilliant blue staining (Fig. S2). Increasing amounts of protein ExsA and ExsD were mixed with ∼180 bp PCR fragment encompassing the putative promoter region of vp1668 and vp1687. Mixtures were size-fractionated by native acrylamide gel eletrophoresis, followed by ethidium bromide staining and visualization. At lower concentrations of ExsA, a low-molecular-weight shifted band was observed while at higher concentrations of ExsA, another high-molecular-weight complex was detected for the promoters of vp1668 and vp1687 (Fig. 5A and B). The negative control protein, Vp1656, did not bind the promoter of vp1668 (Fig. 5A). Furthermore, when excessive DNA probe was used, a shifted band as well as an unshifted band (Fig. 5A) was detected, indicating that binding can be saturated.
Fig. 5.
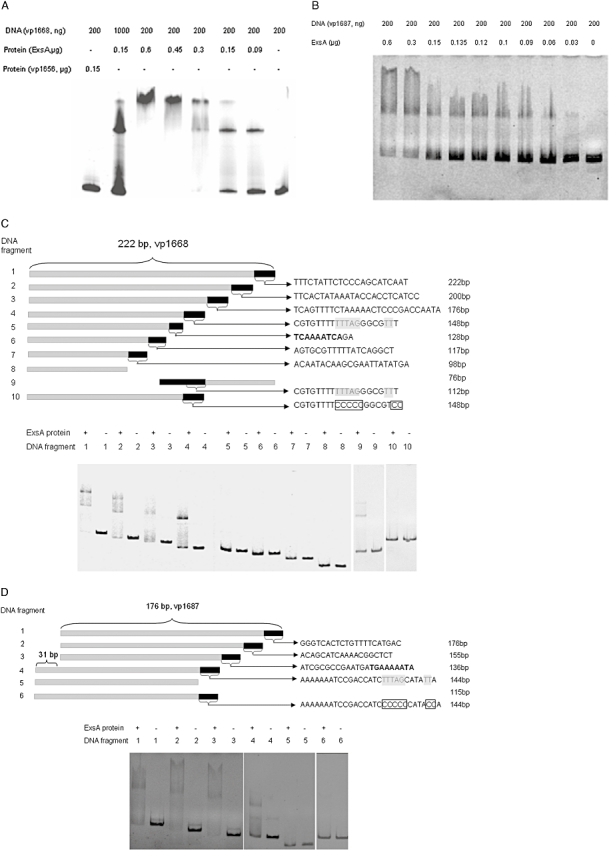
ExsA binds the promoter regions of vp1668 and vp1687.
A. Gel shift analysis using ∼180 bp of DNA sequence from the putative promoter region of vp1668 and purified ExsA.
B. Gel shift analysis using ∼180 bp of DNA sequence from the putative promoter region of vp1687 and purified ExsA.
C. Diagram of the DNA fragments within the promoter region of vp1668 in which the DNA sequences are shown for the black bar; similar sequences to the ExsA binding motif in Pseudomonas are shown in bold and putative ExsA binding motif in V. parahaemolyticus is underlined; potential ExsA binding sequences are replaced with unrelated boxed sequences (upper panel); Gel shift analysis using each DNA fragment and purified ExsA (0.2 μg) (lower panel).
D. Diagram of the DNA fragments within the promoter region of vp1687 in which the DNA sequences are shown for the black bar; similar sequences to the ExsA binding motif in Pseudomonas are shown in bold and a putative ExsA binding motif in V. parahaemolyticus is underlined; potential ExsA binding sequences are replaced with unrelated boxed sequences (upper panel); Gel shift analysis using each DNA fragment and purified ExsA (0.3 μg) (lower panel).
To determine if ExsA binds to specific motif(s) within these two regions, DNA fragments with ∼20 bp truncations at the 3′ end (Fig. 5C and D, upper panel) were amplified and subjected to gel mobility shift assay. For promoter vp1668, DNA fragments 1–4 have the ability to bind ExsA while fragments 5–8 lose the ability to bind ExsA (Fig. 5C), indicating that at least a portion of the last 22 bp segment of fragment 4 is necessary for ExsA binding. For promoter vp1687, DNA fragments 1–4 bind ExsA while fragment 5 does not bind ExsA (Fig. 5D), indicating that at least a portion of the last 29 bp segment of fragment 4 is necessary for binding ExsA. To exclude the possibility that product length affects the ability of ExsA to bind fragment 5 from vp1668 (128 bp), we generated a shorter DNA fragment 9 (112 bp) from 3′ end of the promoter region encompassing the last 22 bp segment of fragment 4. The results showed that fragment 9 is able to bind ExsA (Fig. 5C). Thus, loss of binding is not related to the product length. Because the last 22 bp of fragment 4 in vp1668 and the last 29 bp of fragment 4 in vp1687 share a consensus sequence, TTTAGN4TT, we replaced TTTAGGGCGTTT with CCCCCGGCGTCC (fragment 10) in vp1668 and replaced TTTAGCATATT with CCCCCCATACC (fragment 6) in vp1687. The results showed that both fragment 10 of vp1668 and fragment 6 of vp1687 lost the ability to bind ExsA (Fig. 5C and D), indicating that TTTAGN4TT is the probable ExsA binding motif. In addition, when we used a single concentration of ExsA, two shifted bands appeared in fragments 1 and 2 (possibly 3) of promoter vp1668 while the shifted band with higher molecular weight disappeared for fragments 3–8 (Fig. 5C). When we tested fragment 9, the higher molecular weight band is again evident. These data strongly suggest the presence of a second ExsA binding motif that is independent of the motif described above. ExsD was mixed with promoter vp1668 and vp1687 and no shifted bands were observed (Fig. S3A and S3B), indicating that ExsD does not directly bind the promoter regions of vp1668 and vp1687.
Activity of promoter vp1668 and promoter exsA increases with inducing conditions
We characterized the promoter activity of vp1668 and exsA in different growth conditions and different strains. For unknown reasons, we were unable to generate the single copy of transcriptional lacZ reporter fusion in the bacterial chromosome for promoter vp1687. A single copy of promoters vp1668–lacZ and exsA–lacZ was introduced into wild-type, ΔexsA and ΔexsD strains of V. parahaemolyticus, and β-galactosidase activities were measured. The activity of the promoter vp1668 increased five to six times when the wild-type strain of V. parahaemolyticus was grown in DMEM or after contact with HeLa cells respectively, compared with bacteria grown in LB-S (Fig. 6A). The activity of the promoter exsA increased three to four times when wild-type strain of V. parahaemolyticus was grown in DMEM or contact with HeLa cells respectively, compared with bacteria grown in LB-S (Fig. 6B). These results indicate that the promoter activity of vp1668 and exsA increases when the bacteria are grown in DMEM, and that contact with eukaryotic cells produces an additive effect. Promoter activity of vp1668 in ΔexsA strain was about five times less than the wild-type strain when bacteria were grown in DMEM, suggesting that ExsA is crucial for activating promoter vp1668. Contact with HeLa cells did not increase the promoter activity of vp1668 in the ΔexsA strain (Fig. 6A), indicating that eukaryotic contact does not override the requirement of ExsA for the activation of promoter vp1668. The activity of promoter vp1668 in ΔexsD strain increased approximately eight times compared with the wild-type strain when the bacteria were grown in LB-S (Fig. 6A), verifying that promoter activity of vp1668 is inhibited by ExsD when bacteria are grown in LB-S (Fig. 4). Promoter activity of exsA in the ΔexsA strain is similar to that of the wild-type strain when bacteria were grown in DMEM or contact with HeLa cells (Fig. 6B), indicating that exsA transcription is not affected by ExsA. Finally, promoter activity of exsA in ΔexsD strain increased about three times compared with that of the wild-type strain when bacteria were grown in LB-S, confirming an inhibitory role of ExsD in the expression of ExsA (Fig. 6B).
Fig. 6.
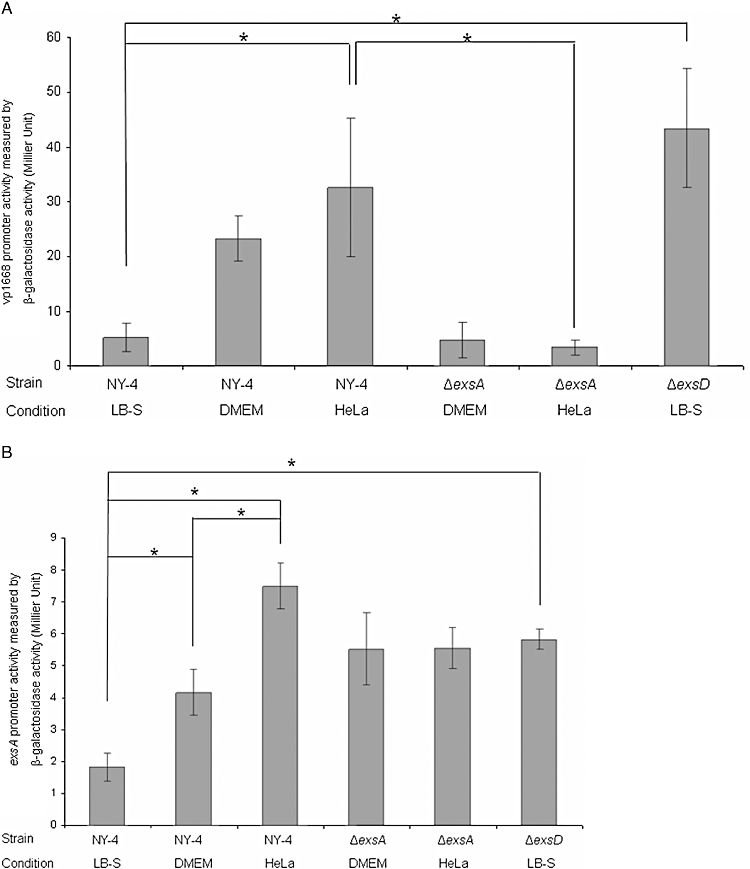
Promoter activity of (A) vp1668 and (B) exsA in different strains and conditions. Bacteria were grown in LB-S, DMEM or contact with HeLa cells for 4 h and levels of β-galactosidase activity were measured (y-axis). The values represent the means ± SD of three replicates. Asterisks represent statistical difference (P < 0.05).
Expression and secretion of Vp1656 is regulated by ExsA and ExsD
Vp1656 was shown previously to be secreted by T3SS1 of V. parahaemolyticus (Park et al., 2004; Ono et al. 2006), so we determined if the expression and secretion of Vp1656 were regulated by ExsA and ExsD. Polyclonal antibody against Vp1656 was generated by immunizing mice with the recombinant protein Vp1656 and the specificity of the polyclonal antibody was analysed by Western blot (Fig. 7A). Two bands were detected in the ΔexsD strain growing in LB-S while the lower band disappeared in the Δvp1656 exsD mutant strain, consistent with the lower band being Vp1656 as specifically recognized by the polyclonal antibody while the upper band is a non-specific protein (Fig. 7A). After the NY-4 strain was grown in DMEM for 2 h, Vp1656 was expressed (Fig. 7B, lane 12) while Vp1656 was not expressed when bacteria were grown in LB-S (Fig. 7B, lane 13). Furthermore, expression of Vp1656 was increased after contact of wild-type strain with HeLa cells for 2 h compared with grown in DMEM (Fig. 7B, lane 11), confirming that eukaryotic cell contact acts in an additive manner on expression of Vp1656. Interestingly, Vp1656 was not detected in the wild-type strain after 4 h of incubation in both DMEM and LB-S (data not shown). We were not able to detect Vp1656 in the supernatant for the wild-type strain (Fig. 7C, lane 7 and 8). This is possibly due to insufficient assay sensitivity for these conditions. Vp1656 was not detected when the ΔexsA strain was grown in DMEM for 2 h (Fig. 7B, lane 6; Fig. 7C, lane 3), or after contact with HeLa cells (Fig. 7B, lane 5). These results indicate that ExsA is required for the expression of Vp1656 in DMEM and eukaryotic cell contact does not override the requirement of ExsA for the expression of Vp1656. Ectopic expression of ExsA in the ΔexsA strain and wild-type strain resulted in significant expression of Vp1656 (Fig. 7B, lanes 1–4; Fig. 7C, lanes 1 and 2). Thus, ExsA is sufficient for the expression and secretion of Vp1656 even when bacteria were grown in the non-inducing condition. Vp1656 was expressed and secreted in ΔexsD strain (Fig. 7B, lane 10; Fig. 7C, lane 6) and complementation of ΔexsD strain with a wild-type exsD gene in trans prevented Vp1656 expression and secretion when bacteria were grown in LB-S (Fig. 7B, lane 9; Fig. 7C, lane 5). The wild-type strain provided with an exsD gene in trans blocked Vp1656 expression and secretion when bacteria were grown in DMEM or in contact with HeLa cells (Fig. 7B, lanes 7 and 8; Fig. 7C, lane 4). Thus, ExsD is sufficient to repress the expression and secretion of Vp1656 and eukaryotic cell contact does not override the inhibitory role of ExsD.
Fig. 7.
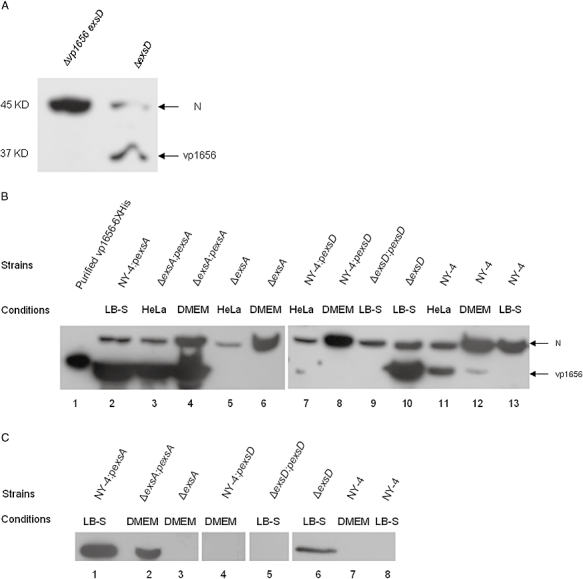
A. Western blot showing the specificity for the polyclonal antibody against Vp1656 for the ΔexsD strain (right lane) and the Δvp1656 exsD strain (left lane). A non-specific protein band (‘N’) was detected with this antisera and serves as a positive detection control in these experiments.
B. Expression of Vp1656 after bacteria were grown for 2 h in different conditions. Lane 1 shows purified protein Vp1656-6xHis.
C. Secretion of Vp1656 after bacteria were grown for 2 h in different conditions.
Expression of T3SS1 genes in the absence of ExsD is ExsA-dependent
ExsA is required for the expression of T3SS1 genes in the wild-type strain grown under inducing conditions or after contact with HeLa cells (Figs 3 and 7). We next determined if ExsA is also required for the exsD mutant strain to express T3SS1 genes when bacteria were grown in non-inducing condition. T3SS1 genes were not transcribed in a ΔexsA exsD double mutant strain growing in LB-S (Fig. 8A), while transcription of T3SS1 genes was detected when ΔexsA exsD double mutant strain was provided with a wild-type exsA gene in trans. As expected, transcription of exsD gene was not detected for both double mutant strain and the complemented strain (Fig. 8A). After the ΔexsA exsD double mutant strain was grown in LB-S, Vp1656 was not expressed (Fig. 8B, upper right panel) or secreted (Fig. 8B, lower right panel), while addition of the double mutant strain with a wild-type exsA gene in trans restored the expression (Fig. 8B, upper left panel) and secretion of Vp1656 (Fig. 8B, lower left panel). These results indicated that expression of T3SS1 genes by ΔexsD strain under non-inducing condition is dependent on ExsA. These results also indicated that transcriptional activity of exsA is independent of ExsD.
Fig. 8.
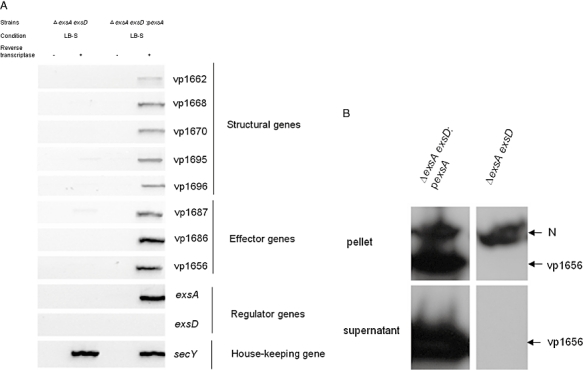
Expression of T3SS1 genes in the absence of ExsD is dependent on ExsA.
A. RT-PCR showing that T3SS1 genes are not transcribed in the ΔexsA exsD strain while transcription of T3SS1 genes is restored in ΔexsA exsD : pexsA strain growing in LB-S. Mock reactions (−), which did not contain reverse transcriptase in the RT reaction, were used as controls against genomic contamination of the RNA preparations. SecY was a house-keeping gene used as an internal control to ensure that RNA was present in all samples.
B. Western blot showing expression and secretion of Vp1656 for the double mutant strain ΔexsA exsD and ΔexsA exsD strain complemented with a wild-type exsA gene in trans when grown in LB-S for 4 h. Vp1656 was detected with polyclonal anti-Vp1656 antibody and ‘N’ in the upper panel represents a non-specific band that serves as a positive detection control.
Discussion
Transcription and expression of T3SS genes are controlled by specific environmental conditions and a subsequent regulatory cascade. Signals that induce transcription include contact between bacteria and eukaryotic cells or other specific growth conditions. For example, transcription and expression of T3SS genes in Pseudomonas and Yersinia are activated in response to low concentration of calcium (Finck-Barbancon et al., 1997; Vallis et al., 1999; Jackson et al., 2004) while in Salmonella, T3SS genes are activated in high-osmolarity and low-oxygen conditions (Lee and Falkow, 1990; Bajaj et al., 1996; Gunn et al., 1996; Arricau et al., 1998). Co-ordinate regulation of T3SS genes ensures that these genes are properly expressed and fully functional during infection; presumably they are not expressed in other environmental conditions to conserve energy. A previous study showed that secretion of protein can be induced when V. parahaemolyticus was cultured in DMEM (Kodama et al., 2007); thus we reasoned that DMEM culture conditions activate the transcription of T3SS1 genes. In this study, we determined transcription patterns for 10 T3SS1 genes in V. parahaemolyticus of which five genes encode apparatus proteins, three genes encode T3SS substrates, and two genes encode regulatory proteins. We showed that when LB-S was used as growth medium, there is minimal transcription of these genes in the wild-type strain NY-4. ExsD is transcribed in LB-S at what appears to be low levels (Fig. 1). When bacteria were grown in tissue culture medium (DMEM), all of the T3SS1 genes tested here were transcribed. For other bacteria, others have shown that tissue culture medium (DMEM) activates a variety of virulence genes including bundle-forming pili (Puente et al., 1996), EspA and EspB proteins (Ebel et al., 1996) and Tir and Intimin (Shin et al., 2001; Abe et al., 2002). In one study, investigators demonstrated that NaHCO3 in DMEM was responsible for the activation of EspA and EspB proteins in Escherichia coli (Abe et al., 2002). The specific factor(s) responsible for the activation of T3SS1 gene transcription in V. parahaemolyticus remain to be determined.
Mutational analysis demonstrated that environmental regulation of T3SS1 genes in V. parahaemolyticus is through the positive regulator, ExsA, and negative regulator, ExsD. ExsA is a member of AraC family of transcriptional activators that are defined by a 100-amino-acid segment of sequence that corresponds to a DNA binding domain of two helix-turn-helix DNA binding motifs (Gallegos et al., 1997). Proteins belonging to AraC family regulate many processes in bacteria, including sugar (Mota et al., 1999; Plumbridge and Pellegrini, 2004; Raposo et al., 2004), amino acid (Zeng and Jin, 2003; Ma et al., 2004) and alcohol degradation (Onaca et al., 2007), stress responses (van der Straaten et al., 2004) and pathogenesis (DiRita, 1992; Gallegos et al., 1997; Egan, 2002). Virulence-associated AraC-type proteins regulate gene expression in response to environmental conditions, such as temperature, osmolarity, pH and metal ions (Gallegos et al., 1993; 1997). Some of the AraC-type regulators control gene expression by direct interaction with specific molecules. For example, TxtR, an AraC regulator in Streptomyces, activates thaxtomin biosynthesis gene expression by binding with cellobiose, an inducer of thaxtomin production (Joshi et al., 2007). Another example is UreR protein of Providencia stuartii that binds urea directly, which significantly increases the affinity of the protein for its DNA binding sites (Thomas and Collins, 1999; Gendlina et al., 2002). Other AraC proteins regulate gene expression by binding directly the promoter region of target genes. In Pseudomonas, ExsA activates the transcription of T3SS genes by directly binding to the promoter sequences of T3SS genes in the calcium-limited condition and the consensus binding site within the promoter sequences are TNAAANA, which is centered ∼15 base pairs upstream of the −35 RNA polymerase binding sites (Yahr and Frank, 1994; Hovey and Frank, 1995; Yahr et al., 1995).
Sequence analysis of the genomic island of T3SS1 genes in V. parahaemolyticus indicated that there were several intergenic spaces that might serve as promoter regions. The first region is between vp1667 and vp1668, and this region may serve as a promoter to initiate the transcription of genes downstream of vp1667 and vp1668 because vp1667 and vp1668 are transcribed in the opposite directions. The second region is between vp1687 and vp1688 and this region may serve as promoter to initiate the transcription of genes downstream of vp1687. The third region is between vp1694 and vp1695 and the fourth region is between vp1698 and vp1699. Further analysis indicated that putative promoter regions for vp1668 and vp1687 contain the DNA sequence of TCAAAATCA and TGAAAAATA respectively, consistent to the ExsA protein binding motif, TNAAANA in Pseudomonas. Electrophoretic mobility shift assay (EMSA) results (Fig. 5A and B) demonstrated that purified ExsA protein is able to directly bind these two DNA regions. Surprisingly, DNA fragment 5 from the vp1668 promoter region, which encompasses a putative binding motif (TCAAAATCA), but does not bind ExsA (Fig. 5C). DNA fragment 4 from the vp1687 promoter does not encompass its putative binding motif (TGAAAAATA), yet this fragment binds ExsA (Fig. 5D). Further analysis between the last 20 residues of fragment 4 within the promoter region of vp1668 (Fig. 5C, upper panel) and the last 29 residues of fragment 4 within the promoter region of vp1687 (Fig. 5D, upper panel) identified a consensus sequence TTTAGN4TT. Mutation of this consensus sequence blocks ExsA binding, and thus the consensus sequence encompasses a unique binding motif. Binding of ExsA within the promoter region results in the transcription and expression of Vp1668, Vp1687 and their downstream genes, because transcription of T3SS1 genes and expression of Vp1656 were blocked in the ΔexsA strain while addition of exsA in trans restored T3SS1 transcription and Vp1656 expression (Figs 3 and 7). Promoter activity of vp1668 in ΔexsA strain decreased to the baseline, indicating that the promoter activity of vp1668 requires the expression of ExsA.
In Pseudomonas, ExsD is a cytosolic protein that directly interacts with ExsA and inhibits ExsA-dependent transcription (McCaw et al., 2002). Deletion of ExsD allows transcription of T3SS genes in Pseudomonas. We found that under non-inducing condition, only exsD was clearly transcribed in V. parahaemolyticus (Fig. 1), consistent with ExsD repression of T3SS1 genes. We demonstrated that transcription of T3SS1 genes (Fig. 4) and expression of Vp1656 (Fig. 7B, lane 10; Fig. 7C, lane 6) occurred in the ΔexsD strain when bacteria were grown in non-inducing conditions. Furthermore, promoter activity of vp1668 and exsA was significantly increased in ΔexsD strain compared with the wild-type strain grown in non-inducing conditions (Fig. 6A and B). These results demonstrated that V. parahaemolyticus ExsD functions as an inhibitor of T3SS1 gene expression. With inducing conditions, overexpression of ExsD in wild-type strain inhibited the transcription of T3SS1 genes (Fig. 4). Furthermore, overexpression of ExsD inhibited the expression of Vp1656 when bacteria were grown in DMEM or while in contact with eukaryotic cells (Fig. 7B, lanes 7 and 8). These results demonstrate that V. parahaemolyticus ExsD alone is sufficient to block transcription and expression of T3SS1 genes. Because ExsD is not able to bind the promoter of vp1668 and vp1687 (Fig. S3A and S3B), it is unlikely that ExsD regulates the transcription of T3SS1 genes directly. Furthermore, when grown in LB-S, the activity of the exsA promoter in the wild-type strain is significantly less than exsA promoter activity in the ΔexsD strain (Fig. 6B), In this situation, there are not environmental signals for transcription of exsA and, therefore, this is clear evidence that ExsD blocks transcription of exsA [probably in conjunction with another interacting protein(s)].
One interesting finding is that transcription of exsD is lower in non-inducing condition than in inducing condition (Fig. 1). In addition, transcription of exsD is lower in the ΔexsA strain compared with the strain complemented with a wild-type exsA gene (Fig. 3). These results indicate that expression of ExsA facilitates the transcription of exsD and expression of ExsD blocks transcription of exsA (Fig. 4). This appears to be a negative feedback system that modulates transcription and expression of T3SS1 genes. Pseudomonas has two additional T3SS gene regulators, exsC and exsE (Yahr and Wolfgang, 2006). ExsC is able to bind ExsD and prevent binding of ExsD with ExsA and thereby permit T3SS gene transcription (Dasgupta et al., 2004). ExsE, a substrate of T3SS, accumulates within the bacterium when bacteria are grown in non-inducing condition and binds ExsC (Urbanowski et al., 2005). Binding of ExsE with ExsC allows ExsD to bind ExsA, leading to the transcriptional repression of T3SS genes (Urbanowski et al., 2005). Sequence analysis showed that vp1701 in V. parahaemolyticus shares 29% similarity with exsC in Pseudomonas, and thus it would be interesting to determine if vp1701 is also involved in the regulation of T3SS1 genes.
We have also found that expression of Vp1656 in wild-type strain was only observed when bacteria were incubated for 2 h while after 4 h of incubation, expression of Vp1656 was not detectable. This is consistent with an earlier report showing that quorum sensing negatively regulates the expression of Vp1656 (Henke and Bassler, 2004). Vp1656 is produced and secreted only when quorum sensing was knocked out (Henke and Bassler, 2004). Our study showed that deletion of exsD resulted in the constitutive expression and secretion of Vp1656, suggesting a possible interaction between exsD with quorum sensing. Furthermore, overexpression of ExsA also led to the constitutive expression and secretion of Vp1656, indicating that ExsA might play a role in the repression of quorum sensing in V. parahaemolyticus. Regulation of quorum sensing by AraC proteins has been observed in Pseudomonas aeruginosa (Dong et al. 2005) and V. parahaemolyticus (Enos-Berlage et al. 2005), and here we suggest another possible mechanism to regulate quorum sensing via the exsA gene in V. parahaemolyticus. We also found that contact with eukaryotic cells increased the expression of Vp1656 in an additive manner compared with growth in DMEM alone.
Based on our results, it appears that when V. parahaemolyticus is grown in non-inducing condition (e.g. LB-S), ExsD is expressed and blocks transcription of exsA. When V. parahaemolyticus is grown under inducing conditions (e.g. DMEM), exsA is transcribed by an environmental signalling pathway and ExsA binds directly to the promoter region of T3SS1 genes, leading to their expression. At the same time, ExsA increases the transcription of exsD (Fig. 3), which in turn provides a negative feedback to downregulate and thereby modulate the expression of T3SS1 genes. Our results suggest that the most parsimonious pathway by which ExsD exerts its effects is by blocking transcription of exsA rather than by blocking the environmental signalling pathway or by blocking transcription of individual T3SS1 genes.
In summary, we identified conditions that induce the expression of T3SS1 genes of V. parahaemolyticus and showed that ExsA activates their expression by directly binding the promoter region of T3SS1 genes. In addition, transcription of T3SS1 genes is blocked by overexpression of ExsD while deletion of exsD gene permitted the transcription of T3SS1 genes under non-inducing conditions. Either deletion of exsA or overexpression of ExsD attenuates the ability of V. parahaemolyticus to induce host cell death.
Experimental procedures
Bacterial strains, plasmids and growth conditions
The strains used in this study are listed in Table 1. All V. parahaemolyticus strains were derived from the wild-type strain NY-4 (X. Zhou, M.E. Konkel, and D.R. Call, submitted). Strains were grown in LB broth or LB agar supplemented with 2.5% sodium chloride at 37°C. E. coli S17 λpir was used in gene deletion experiments and was cultured in LB medium. Plasmid pMMB207 was used in complementation experiments and plasmid pDM4 was used for gene knockout experiments. Other derivative plasmids are listed in Table 1. When appropriate, antibiotics were added at the following concentrations: ampicillin, 100 μg ml−1; chloramphenicol, 25 μg ml−1 for E. coli and 5 μg ml−1 for V. parahaemolyticus.
Table 1.
Strains and plasmids used in this study.
| Strains and plasmids | Descriptions | Sources |
|---|---|---|
| E. coli | ||
| S17-1λpir | thi pro hsdR hsdM+recA RP4-2-Tc::Mu-Km::Tn7λpir | Milton et al. (1992) |
| S17-exsA–lacZ | S17 carrying pNK8-exsA–lacZ | This study |
| S17-1668–lacZ | S17 carrying pNK8-1668–lacZ | This study |
| S17-pMMB207-RBS-exsA | S17 carrying pMMB207-RBS-exsA | This study |
| S17-pMMB207-exsD | S17 carrying pMMB207-exsD | This study |
| S17-pDM4-1699-1F + 2R | S17 carrying pDM4-1699-1F + 2R | This study |
| S17-pDM4-1698-1F + 2R | S17 carrying pDM4-1698-1F + 2R | This study |
| V parahaemolyticus | ||
| NY-4 | Clinical isolate O3 : K6 | |
| ΔexsA | exsA (vp1699) deletion mutant | This study |
| ΔexsA : pexsA | ΔexsA complemented with exsA gene located in the plasmid of pMMB207 | This study |
| NY-4 : pexsA | Wild-type strain containing exsA gene located in the plasmid of pMMB207 | This study |
| vp1668–lacZ::NY-4 | Promoter sequences of vp1668 was inserted in the upstream of lacZ gene in NY-4 strain | This study |
| vp1668–lacZ::ΔexsA | Promoter sequences of vp1668 was inserted in the upstream of lacZ gene in ΔexsA strain | This study |
| vp1668–lacZ::ΔexsD | Promoter sequences of vp1668 was inserted in the upstream of lacZ gene in ΔexsD strain | This study |
| vp1699–lacZ::NY-4 | Promoter sequences of vp1699 was inserted in the upstream of lacZ gene in NY-4 strain | This study |
| vp1699–lacZ::ΔexsA | Promoter sequences of vp1699 was inserted in the upstream of lacZ gene in ΔexsA strain | This study |
| vp1699–lacZ::ΔexsD | Promoter sequences of vp1699 was inserted in the upstream of lacZ gene in ΔexsD strain | This study |
| ΔexsD | exsD (vp1698) deletion mutant | This study |
| ΔexsD : pexsD | ΔexsD complemented with exsD gene located in the plasmid of pMMB207 | This study |
| NY-4 : pexsD | Wild-type strain containing exsD gene located in the plasmid of pMMB207 | This study |
| ΔexsA exsD | exsA and exsD double mutant strain | This study |
| ΔexsA exsD : pexsA | ΔexsA exsD complemented with exsA gene located in the plasmid of pMMB207 | This study |
| Δvp1656 exsD | Insertional mutation of vp1656 on the background of ΔexsD strain | This study |
| Yersenia enterocolitica | ||
| JB580v | Serogroup O : 8, NalrΔyenR (r- m+) | Young and Young (2002) |
| Plasmids | ||
| pMMB207 | RSF1010 derivative, IncQ lacIq Cmr Ptac oriT | Morales et al. (1991) |
| pNK8 | MobRP4 oriR6K, transcriptional reporter vector | Walker and Miller (2004) |
| pDM4 | A suicide vector with ori R6K sacB; Cmr | Milton et al. (1996) |
| pMMB207-RBS-exsA | exsA coding sequences and the sequences for 6 His amino acids at the C-terminus cloned into pMMB207 | This study |
| pMMB207-exsD | exsD coding sequences and the sequences for 6 His amino acids at the C-terminus cloned into pMMB207 | This study |
| pMMB207-1656 | Vp1656 coding sequences and the sequences for 6 His amino acids at the C-terminus cloned into pMMB207 | This study |
| pNK8-exsA–lacZ | Promoter sequences of exsA cloned into pNK8 | This study |
| pNK8-1668–lacZ | Promoter sequences of vp1668 cloned into pNK8 | This study |
| pDM4-1699-1F + 2R | Flanking region sequences of exsA cloned into pDM4 | This study |
| pDM4-1699-1F + 2R | Flanking region sequences of exsD cloned into pDM4 | This study |
| pDM4-1656insertion | Internal region of vp1656 cloned into pDM4 | This study |
Construction of deletion mutants
Deletion mutants were generated by homologous recombination. A chromosomal deletion in the exsA gene wasconstructed by allelic exchange using a suicide vector, pDM4, carrying DNA fragments corresponding to exsA flanking regions (Milton et al., 1996). Two DNA fragments were amplified by PCR with V. parahaemolyticus strain NY-4 chromosomal DNA as a template with primer pairs 1699-1F and 1699-1R and 1699-2F and 1699-2R respectively (Table 2). Fragment 1 (approximately 1000 bp upstream of the start codon) amplified with 1699-1F and 1699-1R was digested with XhoI and BglII and fragment 2 (approximately 1000 bp downstream of the stop codon) amplified with 1699-2F and 1699-2R was digested with BglII and XbaI. These two digested fragments were ligated into the pDM4 suicide vector, which had been digested with XhoI and XbaI. The resultant plasmid was designated pDM4-1699-1F + 2R. This plasmid was transformed into E. coli S17 λpir, resulting in the strain S17-pDM4-1699-1F + 2R. pDM4-1699-1F + 2R was then transferred into the V. parahaemolyticus NY-4 wild-type strain by conjugation, and both ampicillin- and chloramphenicol-resistant transconjugants were selected. Ampicillin was used to select against E. coli and chloramphenicol was used to select for transconjugants. To complete the allelic exchange, the integrated suicide plasmid was induced to excise from the chromosome by growth on LB-S medium containing 5% sucrose. Chloramphenicol-sensitive clones were obtained and screened by PCR with primers 1699-up and 1699-down that lie adjacent to the vp1699. One clone was selected with the exsA gene deletion and designated as ΔexsA. Construction of exsD deletion mutant was performed in the same manner by using the following primers: 16981-F, 1698-1R, 1698-2F, 1698-2R, 1698-up and 1698-down (Table 2). One PCR-confirmed clone was isolated and designated as ΔexsD. Double mutant strain was generated by conjugating the plasmid pDM4-1699-1F + 2R from E. coli S17 λpir to ΔexsD strain and one PCR-confirmed clone was isolated and designated as ΔexsA exsD. For construction of Δvp1656 exsD, primers1656insertion_FW and 1656insertion_RE were used to amplify the internal region of vp1656. PCR fragment was digested with XhoI and BglII and were ligated into pDM4 digested with the same enzyme. The resultant plasmid was designated pDM4-1656insertion and conjugated into ΔexsD strain. One PCR-confirmed clone was isolated and designated as Δvp1656 exsD.
Table 2.
Primers used in this study.
| Primer name | Sequences (5′-3′) |
|---|---|
| 1698-1F | AGGATAAACTCGAGCAAACAGCTCATCCGTCGAG |
| 1698-1R | AGTTAGTCTAGAGGGCAGTATATGTTAGATAAAATG |
| 1698-2F | AGTTAGTCTAGACCCTTTCGAACACTCCAGAAT |
| 1698-2R | AGTTAGAGATCTGTAGAAAATGGATGTGTCAGG |
| 1698-up | TTTTCTCCGGCAATAAAAGGCC |
| 1698-down | TTTTGAAAGTCACCGACGGAC |
| 1698-His-up | AGGATAGAATTCTAAGGAGGTAGGATAATAATGCGGAGAAGAACACAAATG |
| 1698-His-down | AGTTAGTCTAGATTAATGGTGATGGTGATGGTGAATCTGGCTGAGATGGTTACAAG |
| 1699-1F | AGGATAAACTCGAGTCGTTATAAACCACGTCACATGC |
| 1699-1R | AGTTAGTCTAGAAATGACTTATGACTAAAATTTTC |
| 1699-2F | AGTTAGTCTAGATTTCTACCCTTCATAATTTTTAATTTC |
| 1699-2R | AGTTAGAGATCTTGTTATTGACTGGATGCGCTC |
| 1699-up | TTCATTTGTGTTCTTCTCCGC |
| 1699-down | CTTTGCTTCTTTAATTGAAATTG |
| 1699-His-up | AGGATAGGATCCTAAGGAGGTAGGATAATAATGGATGTGTCAGGCCAACT |
| 1699-His-down | AGTTAGTCTAGATTAATGGTGATGGTGATGGTGATTCGCGATGGCGACTTG |
| lacZ-exsA-FW | AGGATATCTAGAGTGATTTATGATTATATCCTACGC |
| lacZ-exsA-RE | AGTTAGAGATCTTTTCTACCCTTCATAATTTTTAAT |
| lacZ-1668-FW | AGGATATCTAGAAATACTCAT TCACTTGCACTC |
| lacZ-1668-RE | AGTTAGAGATCTAATGTAAAAAATATGCGCAATG |
| Promoter1668-FW | AATACTCATTCACTTGCACTC |
| Promoter1668-RE | AATGTAAAAAATATGCGCAATG |
| Promoter1687-FW | CAGAGTAGGGCATCACCG |
| Promoter1687-RE | TTGAATGAATAATCCTATAGTG |
| 1668FW_motif | CGGACCGATGCATCAAAC |
| 1668RE_motif1 | ATTGATGCTGGGAGAATAG |
| 1668RE_motif2 | GGATGAGGTGGTATTTATAG |
| 1668RE_motif3 | TATTGGTCGGGAGTTTTTAG |
| 1668RE_motif4 | AAACGCCCTAAAAAAACACG |
| 1668RE_motif5 | GTCTGATTTTGACAGCCTG |
| 1668RE_motif6 | CAGCCTGATAAAAACGCAC |
| 1668RE_motif7 | TCATATAATTCGCTTGTATTG |
| 1668RE_motif8 | ACGAAAGGAGTGCAAGTGA |
| 1668FW_motif9 | CAGGCTGTCAAAATCAGACG |
| 1668RE_motif10 | GGACGCCGGGGGAAAACACGTCTGATTTTGAC |
| EMSA1687motif_FW | AAACAACTCAGTGAGCGG |
| EMSA1687motif1_RE | GTCATGAAAACAGAGTGACC |
| EMSA1687motif2_RE | AGAGCCGTTTTGATGCTG |
| EMSA1687motif3_RE | TATTTTTCATCATTCGGCGC |
| EMSA1687motif4_RE | TAATATGCTAAAGATGGTCGG |
| EMSA1687motif5_RE | CGTTTTAAGACGTTTGAAAAC |
| EMSA1687motif6_RE | TGGTATGGGGGGGATGGTCGGATTTTTTTCG |
| 1656insertion_FW | AGGATAAACTCGAGATTAGATGGGCCGAAAGCTC |
| 1656insertion_RE | AGTTAGAGATCTCGAATGTGCACGTTTTACTTG |
| vp1656-up | AGGATAGAATTCAGGAGATATACCATGTTGGATAAAATTGGTGGAAC |
| vp1656-down | AGTTAGTCTAGATTAATGGTGATGGTGATGGTGCACTGTCGGGATAGATGCGC |
| 1662–600-up | GTCACGCAAAAAGGCATGAGC |
| 1662–600-down | AAAACGCAGGTGAATTCCCGG |
| 1668–600-up | CAAGAGCCAAGTGCTTGGTATC |
| 1668–600-down | CATGTCGTCACCTTCGACCA |
| 1670–600-up | AGAGCGCTCTCTTGCGTCAC, |
| 1670–600-down | TTTGGGATGTTCATCATTCGGG |
| 1686–600-up | ATTCTAAATGAAGGCAAACTCAGC |
| 1686–600-down | GTTTAAATCCGTACTTGCGAGC |
| 1687–600-up | ATGGCTAATGGATTTATTACCG |
| 1687–600-down | TTACACCCTTAATGTAGAGAATTG |
| 1695–600-up | GGATGTCAGGACGTAACCATC |
| 1695–600-down | CCGTCAGCAGCAACGTGC |
| 1696–600-up | ATGTGGTACTTCGATGGCG |
| 1696–600-down | TTGTGGCTGATCGAGTTGAG |
| 1698-600-up | GTTGTCCACGTGAATCACCGA |
| 1698-600-down | GTAGTCCGCCAGTTCTTTCAA |
| 1699-600-up | GTCGTTCACAATGGTCAATTACG |
| 1699-600-down | CGTCAGCAAAAGCTTGTGTG |
| secY1 | AGGATAAACTCGAGTGGTGCTCTGGAGCGTGCATC |
| secY2 | AGTTAGTCTAGACCTTGTTGACGCTTCGCGTAG |
Complementation
Primers 1699-His-up and 1699-His-down were used to amplify the complete gene of exsA with a 6xHis tag added at the C-terminus of the gene. Amplified PCR product was digested with BamHI and XbaI and ligated into the plasmid pMMB207 (digested with the same enzymes), resulting in the plasmid pMMB207-RBS-exsA. Plasmid pMMB207-RBS-exsA was transformed into E. coli S17 λpir, resulting in the strain S17-pMMB207-RBS-exsA, and then conjugated from E. coli S17 λpir into ΔexsA, ΔexsA exsD and the V. parahaemolyticus NY-4 wild-type strain, resulting in ΔexsA : pexsA, ΔexsA exsD : pexsA and NY-4 : pexsA respectively. Primers 1698-His-up and 1698-His-down were used to amplify the complete gene of exsD with a 6xHis tag added at the C-terminus of the gene. Amplified PCR product was digested with EcoRI and XbaI and ligated into the plasmid pMMB207 digested with the same enzymes, resulting in the plasmid pMMB207-exsD. Plasmid pMMB207-exsD was transformed into E. coli S17 λpir by electroporation and then conjugated from E. coli S17 λpir into ΔexsD and wild-type NY-4 strain, resulting in ΔexsD : pexsD and NY-4 : pexsD respectively (Table 1).
Construction of single-copy lacZ reporter fusions in the bacterial chromosom
Transcriptional lacZ reporter fusions were constructed by cloning the upstream region of exsA and vp1668 into a suicide vector, pKN8. For the exsA promoter, approximately 500 bp DNA fragment upstream of exsA gene was amplified by PCR using primers lacZ-exsA-FW and lacZ-exsA-RE (Table 2). PCR product was digested with XbaI and BglII and cloned into pNK8 (digested with the same enzymes) resulting in the plasmid pNK8-exsA–lacZ. For vp1668 promoter, approximately 160 bp DNA fragment upstream of vp1668 was amplified by PCR using primers lacZ-1668-FW and lacZ-1668-RE. PCR product was digested with XbaI and BglII and cloned into pNK8 (digested with the same enzymes), resulting in the plasmid pNK8-1668–lacZ. These two plasmids were transformed into E. coli S17 λpir by electroporation, resulting in the strains S17-exsA–lacZ and S17-1668–lacZ respectively. pNK8-1668–lacZ and pNK8-exsA–lacZ were integrated into V. parahaemolyticus strains of NY-4, ΔexsA and ΔexsD by conjugation, resulting in the lacZ transcriptional fusion strains: vp1668–lacZ::NY-4, vp1668–lacZ::ΔexsA, vp1668–lacZ::ΔexsD, vp1699–lacZ::NY-4, vp1699–lacZ::ΔexsA, vp1699–lacZ::ΔexsD (Table 1).
Infection and LDH assay
Vibrio parahaemolyticus was harvested from an overnight broth culture and pelleted by centrifugation at ∼6000 g r.p.m. at room temperature. The bacterial pellets were re-suspended in DMEM (Invitrogen, Carlsbad, CA) containing 1% (v/v) Fetal Bovine Serum (HyClone, Logan, Utah). Bacterial suspensions (10 μl) were added to the wells of a 12-well plate, where each well contained HeLa cells (106) (CCL-2), to achieve a multiplicity of infection (m.o.i.) of ∼100 cfu per cell. Plates were centrifuged at ∼600 g to synchronize bacteria–HeLa host cell contact. For LDH release assay, the FBS concentration in the medium was reduced from 10% to 1% to reduce background LDH activity. Supernatants were collected at 4 h post infection and LDH activity was measured with Cytotoxicity Detection Kit (Promega, Madison, WI) according to manufacturer's instructions. Maximum LDH release was achieved by lysis of cells with lysis buffer provided in the kit. Spontaneous LDH release in the supernatant of uninfected cells was also measured as described. Percentage cytotoxicity was calculated with the formula:
 |
RNA isolation and RT-PCR
For bacterial growth in DMEM, an overnight culture (1.5 ml) was centrifuged and the pellet was re-suspended in 15 ml of DMEM supplemented with 1% FBS. For bacterial growth in LB-S, overnight culture (0.15 ml) was directly inoculated into 15 ml of LB-S medium. After 3 h incubation, total RNA was isolated by RNA Easy kit (Qiagen, Valencia, CA). Isolated RNA was treated with DNase I for 30 min to remove DNA and was reverse-transcribed into cDNA using 2 μg of RNA, 200 ng of random hexamers and Superscript III (Invitrogen, Carlsbad, CA). PCR was performed according to the manufacturer's instructions (1 U of Master Taq polymerase, 200 μM each of the four dNTPs and 1 μM each primer). Primer pairs (the last 22 primers listed in Table 2) amplifying internal fragments were used for semi-quantitative analysis of gene expression. Cycling parameters were identical for all primer sets (Table 2): one cycle of 94°C for 4 min; 30 cycles of 95°C for 1 min, 52°C for 1 min and 72°C for 1 min; and a final incubation at 72°C for 5 min.
Cloning and expression of exsA, exsD and vp1656
For cloning of vp1656, primers vp1656-up and vp1656-down were used to amplify the entire coding sequences along with sequences for 6 His amino acids at the C-terminus. PCR product digested with EcoRI and XbaI was cloned into pMMB207 (digested with the same enzymes), resulting in the plasmid pMMB207-1656. Plasmid pMMB207-1656 was transformed into E. coli S17 λpir, resulting in the strain S17-pMMB207-1656, which was conjugated into Yersinia enterocolitica for expression. For expression of exsA and exsD, the plasmids pMMB207-RBS-exsA and pMMB207-exsD were conjugated from E. coli S17 λpir into Y. enterocolitica. We used Y. enterocolitica to express these proteins because ExsA and ExsD were insoluble when expressed in the heterologous host strain E. coli BL21. An overnight culture (20 ml) of Y. enterocolitica containing each plasmid was diluted into 500 ml of LB medium and incubated for 2 h with shaking at 26°C before adding 1 mM IPTG. Protein expression was induced for 12 h in 26°C with vigorous shaking before pelletting cells. Pellets were re-suspended in 15 ml of binding buffer (50 mM NaH2PO4, pH 8.0, 0.5 M NaCl, 10 mM imidazole) and sonicated for 15 min. Insoluble cell fractions were removed by centrifugation at ∼3000 g for 10 min and the supernatant were loaded into an Ni2+ resin column (Invitrogen). After binding for 1 h, the column was washed 15× with washing buffer (50 mM NaH2PO4, pH 8.0, 0.5 M NaCl, 50 mM immidazole) before being eluted with elution buffer (50 mM NaH2PO4, pH 8.0, 0.5 M NaCl, 300 mM immidazole). The concentration of recombinant proteins was determined by measuring OD280 with a NanoDrop (NanoDrop Technologies, Wilmington, DE). Purity of the protein samples was analysed by SDS-PAGE and gel staining. Purified protein Vp1656 was submitted to Monoclonal Antibody Center at Washington State University for immunization of two mice to raise polyclonal antibody against Vp1656.
The EMSA
DNA fragments of approximately 180 bp, corresponding to sequences upstream of vp1668 and vp1687 start codons, were amplified by PCR using primers promoter1668-FW/promoter1668-RE and promoter1687-FW/promoter1687-RE. Forward primer for fragements 1–8 in Fig. 5C is 1668FW_motif and the reverse primers are 1668RE_motif1–8 respectively. Fragment 9 in Fig. 5C was amplified with primers 1668FW_motif9 and 1668RE_motif1. Fragment 10 in Fig. 5C was amplified with primers 1668FW_motif and 1668RE_motif10. Forward primer for fragments 1–3 in Fig. 5D is promoter1687-FW and the reverse primers are EMSA1687motif1_RE, EMSA1687motif2_RE and EMSA1687motif3_RE respectively. Forward primer for fragments 4–6 in Fig. 5D is EMSA1687motif_FW and the reverse primers are EMSA1687motif4_RE, EMSA1687motif5_RE and EMSA1687motif6_RE respectively. Increasing amounts of the purified proteins were mixed with 200 ng of DNA probes (PCR products) in buffer containing 10 mM Tris (pH 7.5), 1 mM EDTA, 100 mM KCl, 0.1 mM DTT, 5% v/v glycerol and 0.01 mg ml−1 BSA, and binding reactions were incubated at room temperature for 30 min before loading onto a 6% native polyacrylamide gel in 0.5× TBE buffer (45 mM Tris-HCl, 45 mM boric acid, 1 mM EDTA, pH 8.0). DNA–protein complexes were electrophoresed for 70 min at 120 V followed by staining with ethidium bromide and imaging.
Preparation of proteins and Western blot analysis
To collect extracellular proteins, overnight culture grown in LB-S was diluted into LB-S (1:100) and DMEM (1:10) with addition of antibiotics as necessary. Diluted samples were grown for 2 and 4 h at 37°C with shaking. Protein expression was induced by the addition of IPTG to the final concentration of 1 mM. The cells were removed from 15 ml culture by centrifugation at ∼3000 g for 15 min and the supernatant for each sample was passed through a 0.22 μm syringe filter to exclude the remaining bacteria within the supernatant. Trichloroacetic acid was added to the supernatant to achieve a final concentration of 10% (v/v) and incubated on ice overnight to precipitate proteins. The samples were centrifuged at 4°C for 20 min at ∼12 000 g followed by washing once with ice-cold acetone and the protein pellets were re-suspended in 50 μl of 1× loading buffer (40 mM Tris/HCl pH 8.0; 5% mercaptoethanol; 1% SDS; 5% glycerin; 1% bromphenol blue). To collect the total proteins for detection of Vp1656, an overnight culture was diluted into LB-S or DMEM and grown for 2 or 4 h with addition of antibiotics and IPTG as needed. The bacterial cultures (15 ml) was centrifuged for 20 min at ∼3000 g and the pellets were re-suspended in 500 μl PBS, followed by sonication and the addition of equal volume of 2× loading buffer. To collect proteins for detection of recombinant 6xHis-tagged ExsA and ExsD, an overnight culture for each recombinant strain was diluted 1:100 into LB-S and grown for 6 h before collecting proteins as described above. To collect the proteins from infected HeLa samples, overnight culture was centrifuged and re-suspended into equal volume of DMEM and bacterial suspension (40 μl) was added to each well of 6-well plate containing ∼106 HeLa cells to achieve an m.o.i. of 25 cfu per cell. Plates were centrifuged at 600 g to synchronize bacteria–host cell contact. Two or four hours post infection, the infected cells were scraped from the well and re-suspended in 200 μl of PBS. After sonication, an equal volume of 2× loading buffer was added into each sample for SDS-PAGE and Western blot analysis. For Western blot analysis, all the samples were heated at 100°C for 5 min and loaded on a 12% (w/v) polyacrylamide gel. After electrophoresis, proteins were electrophoretically transferred to a nitrocellulose membrane for 12 h. The membrane was blocked with 5% skim milk in PBS containing 0.1% Tween 20 and probed with monoclonal anti-His antibody (1:2000) (Invitrogen) or polyclonal anti-Vp1656 (1:1000) for 1 h at room temperature. The secondary antibody was an anti-mouse immunoglobulin conjugated to horseradish peroxidase and was diluted in PBS with 5% milk at 1:5000. The blots were developed by using the Western blotting kit according to the manufacturer's instructions (Bio-Rad, Hercules, CA).
β-Galactosidase assay
Overnight culture for each strain was diluted 1:10 into DMEM and 1:100 into LB-S with addition of antibiotics as needed. After 4 h of incubation with shaking, 0.5 ml of the culture was centrifuged and the pellets were re-suspended in PBS (0.5 ml) and permeabilized by adding 50 μl of SDS (0.5%) before measuring OD600. To determine the β-galactosidase activity after contact with HeLa cells, overnight culture (0.5 ml) suspended in DMEM medium was added into a 25 m2 flask containing confluent adhered HeLa cells (∼107). The flask was centrifuged to synchronize bacteria–HeLa host cell contact. After 2 h incubation, nonadherent as well adherent bacteria were collected by carefully washing the HeLa cells and re-suspending in PBS (0.5 ml). The suspension of the bacteria was permeabilized by adding 50 μl of SDS (0.5%) before measuring OD600. O-nitrophenyl-beta-galactoside (4 mg ml−1) (200 μl; Sigma, St. Louis, MO) was added to the permeabilized samples and the reaction was incubated until sufficient yellow colour developed before adding stop solution (0.1 ml of 1 M Na2CO3). OD414 for each sample was recorded and β-galactosidase activity was calculated by using the following formula:
Statistical analysis
Statistical analysis was performed with anova and P-values < 0.05 were considered statistically significant. Pairwise comparisons were conducted using a Tukey–Kramer test and the analyses were conducted using ncss 2004 (Number Cruncher Statistical Systems, Kaysville, UT).
Acknowledgments
We acknowledge excellent technical assistance from Lisa Orfe, Stacey LaFrentz and Pat Friel and services provided through Washington State University Monoclonal Antibody Center. Dr Kathryn J. Boor provided the wild-type strain of V. parahaemolyticus (NY-4). Dr Anthony Garza provided assistance with implementation of our gel mobility shift assay. This project was supported in part by National Institute of Health, Department of Health and Human Services under the contract number No1-AI-30055 and by the Agricultural Animal Health Program, College of Veterinary Medicine, Washington State University.
Supplementary material
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/
j.1365-2958.2008.06326.x
(This link will take you to the article abstract).
Please note: Blackwell Publishing is not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abe H, Tatsuno I, Tobe T, Okutani A, Sasakawa C. Bicarbonate ion stimulates the expression of locus of enterocyte effacement-encoded genes in enterohemorrhagic Escherichia coli O157: H7. Infect Immun. 2002;70:3500–3509. doi: 10.1128/IAI.70.7.3500-3509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arricau N, Hermant D, Waxin H, Ecobichon C, Duffey PS, Popoff MY. The RcsB-RcsC regulatory system of Salmonella typhi differentially modulates the expression of invasion proteins, flagellin and Vi antigen in response to osmolarity. Mol Microbiol. 1998;29:835–850. doi: 10.1046/j.1365-2958.1998.00976.x. [DOI] [PubMed] [Google Scholar]
- Bahrani FK, Sansonetti PJ, Parsot C. Secretion of Ipa proteins by Shigella flexneri: inducer molecules and kinetics of activation. Infect Immun. 1997;65:4005–4010. doi: 10.1128/iai.65.10.4005-4010.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj V, Lucas RL, Hwang C, Lee CA. Co-ordinate regulation of Salmonella typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol Microbiol. 1996;22:703–714. doi: 10.1046/j.1365-2958.1996.d01-1718.x. [DOI] [PubMed] [Google Scholar]
- Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- Cornelis GR, Van Gijsegem F. Assembly and function of type III secretory systems. Annu Rev Microbiol. 2000;54:735–774. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- Dai JH, Lee YS, Wong HC. Effects of iron limitation on production of a siderophore, outer membrane proteins, and hemolysin and on hydrophobicity, cell adherence, and lethality for mice of Vibrio parahaemolyticus. Infect Immun. 1992;60:2952–2956. doi: 10.1128/iai.60.7.2952-2956.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta N, Lykken GL, Wolfgang MC, Yahr TL. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol. 2004;53:297–308. doi: 10.1111/j.1365-2958.2004.04128.x. [DOI] [PubMed] [Google Scholar]
- DiRita VJ. Co-ordinate expression of virulence genes by ToxR in Vibrio cholerae. Mol Microbiol. 1992;6:451–458. doi: 10.1111/j.1365-2958.1992.tb01489.x. [DOI] [PubMed] [Google Scholar]
- Dong YH, Zhang XF, Xu JL, Tan AT, Zhang LH. VqsM, a novel AraC-type global regulator of quorum-sensing signalling and virulence in Pseudomonas aeruginosa. Mol Microbiol. 2005;58:552–564. doi: 10.1111/j.1365-2958.2005.04851.x. [DOI] [PubMed] [Google Scholar]
- Ebel F, Deibel C, Kresse AU, Guzman CA, Chakraborty T. Temperature- and medium-dependent secretion of proteins by Shiga toxin-producing Escherichia coli. Infect Immun. 1996;64:4472–4479. doi: 10.1128/iai.64.11.4472-4479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan SM. Growing repertoire of AraC/XylS activators. J Bacteriol. 2002;184:5529–5532. doi: 10.1128/JB.184.20.5529-5532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enos-Berlage JL, Guvener ZT, Keenan CE, McCarter LL. Genetic determinants of biofilm development of opaque and translucent Vibrio parahaemolyticus. Mol Microbiol. 2005;55:1160–1182. doi: 10.1111/j.1365-2958.2004.04453.x. [DOI] [PubMed] [Google Scholar]
- Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, et al. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol. 1997;25:547–557. doi: 10.1046/j.1365-2958.1997.4891851.x. [DOI] [PubMed] [Google Scholar]
- Gallegos MT, Michan C, Ramos JL. The XylS/AraC family of regulators. Nucleic Acids Res. 1993;21:807–810. doi: 10.1093/nar/21.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev. 1997;61:393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendlina I, Gutman DM, Thomas V, Collins CM. Urea-dependent signal transduction by the virulence regulator UreR. J Biol Chem. 2002;277:37349–37358. doi: 10.1074/jbc.M203462200. [DOI] [PubMed] [Google Scholar]
- Gunn JS, Hohmann EL, Miller SI. Transcriptional regulation of Salmonella virulence: a PhoQ periplasmic domain mutation results in increased net phosphotransfer to PhoP. J Bacteriol. 1996;178:6369–6373. doi: 10.1128/jb.178.21.6369-6373.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke JM, Bassler BL. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J Bacteriol. 2004;186:3794–3805. doi: 10.1128/JB.186.12.3794-3805.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlady WG, Klontz KC. The epidemiology of Vibrio infections. Florida, 1981–93. J Infect Dis. 1996;173:1176–1183. doi: 10.1093/infdis/173.5.1176. [DOI] [PubMed] [Google Scholar]
- Hovey AK, Frank DW. Analyses of the DNA-binding and transcriptional activation properties of ExsA, the transcriptional activator of the Pseudomonas aeruginosa exoenzyme S regulon. J Bacteriol. 1995;177:4427–4436. doi: 10.1128/jb.177.15.4427-4436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MW, Silva-Herzog E, Plano GV. The ATP-dependent ClpXP and Lon proteases regulate expression of the Yersinia pestis type III secretion system via regulated proteolysis of YmoA, a small histone-like protein. Mol Microbiol. 2004;54:1364–1378. doi: 10.1111/j.1365-2958.2004.04353.x. [DOI] [PubMed] [Google Scholar]
- Joseph SW, Colwell RR, Kaper JB. Vibrio parahaemolyticus and related halophilic Vibrios. Crit Rev Microbiol. 1982;10:77–124. doi: 10.3109/10408418209113506. [DOI] [PubMed] [Google Scholar]
- Joshi MV, Bignell DR, Johnson EG, Sparks JP, Gibson DM, Loria R. The AraC/XylS regulator TxtR modulates thaxtomin biosynthesis and virulence in Streptomyces scabies. Mol Microbiol. 2007;66:633–642. doi: 10.1111/j.1365-2958.2007.05942.x. [DOI] [PubMed] [Google Scholar]
- Kodama T, Rokuda M, Park KS, Cantarelli VV, Matsuda S, Iida T, Honda T. Identification and characterization of VopT, a novel ADP-ribosyltransferase effector protein secreted via the Vibrio parahaemolyticus type III secretion system 2. Cell Microbiol. 2007;9:2598–2609. doi: 10.1111/j.1462-5822.2007.00980.x. [DOI] [PubMed] [Google Scholar]
- Lee CA, Falkow S. The ability of Salmonella to enter mammalian cells is affected by bacterial growth state. Proc Natl Acad Sci USA. 1990;87:4304–4308. doi: 10.1073/pnas.87.11.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Kumagai K, Baba K, Mekalanos JJ, Nishibuchi M. Vibrio parahaemolyticus has a homolog of the Vibrio cholerae toxRS operon that mediates environmentally induced regulation of the thermostable direct hemolysin gene. J Bacteriol. 1993;175:3844–3855. doi: 10.1128/jb.175.12.3844-3855.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch T, Livingstone S, Buenaventura E, Lutter E, Fedwick J, Buret AG, et al. Vibrio parahaemolyticus disruption of epithelial cell tight junctions occurs independently of toxin production. Infect Immun. 2005;73:1275–1283. doi: 10.1128/IAI.73.3.1275-1283.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Masuda N, Foster JW. Characterization of EvgAS-YdeO-GadE branched regulatory circuit governing glutamate-dependent acid resistance in Escherichia coli. J Bacteriol. 2004;186:7378–7389. doi: 10.1128/JB.186.21.7378-7389.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaw ML, Lykken GL, Singh PK, Yahr TL. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol Microbiol. 2002;46:1123–1133. doi: 10.1046/j.1365-2958.2002.03228.x. [DOI] [PubMed] [Google Scholar]
- Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, et al. Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V cholerae. Lancet. 2003;361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- Milton DL, Norqvist A, Wolf-Watz H. Cloning of a metalloprotease gene involved in the virulence mechanism of Vibrio anguillarum. J Bacteriol. 1992;174:7235–7244. doi: 10.1128/jb.174.22.7235-7244.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton DL, O'Toole R, Horstedt P, Wolf-Watz H. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol. 1996;178:1310–1319. doi: 10.1128/jb.178.5.1310-1319.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales VM, Backman A, Bagdasarian M. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene. 1991;97:39–47. doi: 10.1016/0378-1119(91)90007-x. [DOI] [PubMed] [Google Scholar]
- Mota LJ, Tavares P, Nogueira I. Mode of action of AraR, the key regulator of 1-arabinose metabolism in Bacillus subtilis. Mol Microbiol. 1999;33:476–489. doi: 10.1046/j.1365-2958.1999.01484.x. [DOI] [PubMed] [Google Scholar]
- Nagayama K, Oguchi T, Arita M, Honda T. Correlation between cell-associated mannose-sensitive hemagglutination by Vibrio parahaemolyticus and adherence to a human colonic cell line Caco-2. FEMS Microbiol Lett. 1994;120:207–210. doi: 10.1111/j.1574-6968.1994.tb07032.x. [DOI] [PubMed] [Google Scholar]
- Nakasone N, Iwanaga M. Pili of a Vibrio parahaemolyticus strain as a possible colonization factor. Infect Immun. 1990;58:61–69. doi: 10.1128/iai.58.1.61-69.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakasone N, Iwanaga M. The role of pili in colonization of the rabbit intestine by Vibrio parahaemolyticus Na2. Microbiol Immunol. 1992;36:123–130. doi: 10.1111/j.1348-0421.1992.tb01649.x. [DOI] [PubMed] [Google Scholar]
- Onaca C, Kieninger M, Engesser KH, Altenbuchner J. Degradation of alkyl methyl ketones by Pseudomonas veronii MEK700. J Bacteriol. 2007;189:3759–3767. doi: 10.1128/JB.01279-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Park KS, Ueta M, Iida T, Honda T. Identification of proteins secreted via Vibrio parahaemolyticus type III secretion system 1. Infect Immun. 2006;74:1032–1042. doi: 10.1128/IAI.74.2.1032-1042.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T. Functional characterization of two type III secretion systems of Vibrio parahaemolyticus. Infect Immun. 2004;72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge J, Pellegrini O. Expression of the chitobiose operon of Escherichia coli is regulated by three transcription factors: NagC, ChbR and CAP. Mol Microbiol. 2004;52:437–449. doi: 10.1111/j.1365-2958.2004.03986.x. [DOI] [PubMed] [Google Scholar]
- Puente JL, Bieber D, Ramer SW, Murray W, Schoolnik GK. The bundle-forming pili of enteropathogenic Escherichia coli: transcriptional regulation by environmental signals. Mol Microbiol. 1996;20:87–100. doi: 10.1111/j.1365-2958.1996.tb02491.x. [DOI] [PubMed] [Google Scholar]
- Raposo MP, Inacio JM, Mota LJ, de Sa-Nogueira I. Transcriptional regulation of genes encoding arabinan-degrading enzymes in Bacillus subtilis. J Bacteriol. 2004;186:1287–1296. doi: 10.1128/JB.186.5.1287-1296.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Castanie-Cornet MP, Foster JW, Crawford JA, Brinkley C, Kaper JB. An activator of glutamate decarboxylase genes regulates the expression of enteropathogenic Escherichia coli virulence genes through control of the plasmid-encoded regulator, Per. Mol Microbiol. 2001;41:1133–1150. doi: 10.1046/j.1365-2958.2001.02570.x. [DOI] [PubMed] [Google Scholar]
- Sochard MR, Colwell RR. Toxin isolation from a Kanagawa-phenomenon negative strain of Vibrio parahaemolyticus. Microbiol Immunol. 1977;21:243–254. doi: 10.1111/j.1348-0421.1977.tb00285.x. [DOI] [PubMed] [Google Scholar]
- van der Straaten T, Zulianello L, van Diepen A, Granger DL, Janssen R, van Dissel JT. Salmonella enterica serovar Typhimurium RamA, intracellular oxidative stress response, and bacterial virulence. Infect Immun. 2004;72:996–1003. doi: 10.1128/IAI.72.2.996-1003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley SC, Plano GV, Skrzypek E, Haddix PL, Fields KA. Regulation by Ca2+ in the Yersinia low-Ca2+ response. Mol Microbiol. 1993;8:1005–1010. doi: 10.1111/j.1365-2958.1993.tb01644.x. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Iida T, Naim R, Naykaya Y, Honda T. Chloride secretion induced by thermostable direct haemolysin of Vibrio parahaemolyticus depends on colonic cell maturation. J Med Microbiol. 2001;50:870–878. doi: 10.1099/0022-1317-50-10-870. [DOI] [PubMed] [Google Scholar]
- Thomas VJ, Collins CM. Identification of UreR binding sites in the Enterobacteriaceae plasmid-encoded and Proteus mirabilis urease gene operons. Mol Microbiol. 1999;31:1417–1428. doi: 10.1046/j.1365-2958.1999.01283.x. [DOI] [PubMed] [Google Scholar]
- Troisfontaines P, Cornelis GR. Type III secretion: more systems than you think. Physiology (Bethesda) 2005;20:326–339. doi: 10.1152/physiol.00011.2005. [DOI] [PubMed] [Google Scholar]
- Urbanowski ML, Lykken GL, Yahr TL. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc Natl Acad Sci USA. 2005;102:9930–9935. doi: 10.1073/pnas.0504405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallis AJ, Finck-Barbancon V, Yahr TL, Frank DW. Biological effects of Pseudomonas aeruginosa type III-secreted proteins on CHO cells. Infect Immun. 1999;67:2040–2044. doi: 10.1128/iai.67.4.2040-2044.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KA, Miller VL. Regulation of the Ysa type III secretion system of Yersinia enterocolitica by YsaE/SycB and YsrS/YsrR. J Bacteriol. 2004;186:4056–4066. doi: 10.1128/JB.186.13.4056-4066.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahr TL, Frank DW. Transcriptional organization of the trans-regulatory locus which controls exoenzyme S synthesis in Pseudomonas aeruginosa. J Bacteriol. 1994;176:3832–3838. doi: 10.1128/jb.176.13.3832-3838.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahr TL, Wolfgang MC. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol. 2006;62:631–640. doi: 10.1111/j.1365-2958.2006.05412.x. [DOI] [PubMed] [Google Scholar]
- Yahr TL, Hovey AK, Kulich SM, Frank DW. Transcriptional analysis of the Pseudomonas aeruginosa exoenzyme S structural gene. J Bacteriol. 1995;177:1169–1178. doi: 10.1128/jb.177.5.1169-1178.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung PS, Boor KJ. Epidemiology, pathogenesis, and prevention of foodborne Vibrio parahaemolyticus infections. Foodborne Pathog Dis. 2004;1:74–88. doi: 10.1089/153531404323143594. [DOI] [PubMed] [Google Scholar]
- Young BM, Young GM. YplA is exported by the Ysc, Ysa, and flagellar type III secretion systems of Yersinia enterocolitica. J Bacteriol. 2002;184:1324–1334. doi: 10.1128/JB.184.5.1324-1334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Jin S. aph(3′)-IIb, a gene encoding an aminoglycoside-modifying enzyme, is under the positive control of surrogate regulator HpaA. Antimicrob Agents Chemother. 2003;47:3867–3876. doi: 10.1128/AAC.47.12.3867-3876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.